
Abstract
Background and Objectives
In vitro and in vivo studies were performed with the novel, selective, nonsteroidal mineralocorticoid receptor antagonist finerenone to assess the relevance of inhibitory effects on the transporters breast cancer resistance protein (BCRP), organic anion transporting polypeptide 1B1 (OATP1B1), and OATP1B3. These transporters are involved in the disposition of a number of drugs, including statins. Statins are also a frequent comedication in patients receiving finerenone. Therefore, inhibitory effects on BCRP and OATPs are of potential clinical relevance.
Methods
The effect on the transport of specific substrates of BCRP and OATP1B1/1B3 was assessed in cell-based in vitro assays with finerenone or its metabolites. A fixed-sequence crossover study in 14 healthy male volunteers investigated the effects of finerenone (40 mg once daily) on the pharmacokinetics of the index substrate rosuvastatin (5 mg) administered alone, simultaneously with, or approximately 4 h before finerenone. The effect of finerenone on the endogenous OATP substrates coproporphyrin I and III was also assessed.
Results
Based on in vitro findings and threshold values proposed in regulatory guidelines, finerenone appeared to be a potentially relevant inhibitor of all three transporters. Relevant inhibition could also not be ruled out for the finerenone metabolites M1a (OATP1B1) and M3a (OATP1B1 and OAT1B3), which prompted an investigation into the relevance of these findings in vivo. After administration on a background of finerenone 40 mg, all point estimates of area under the curve ratios (114.47% [rosuvastatin], 99.62% [coproporphyrin I; simultaneous], and 105.28% [rosuvastatin; 4 h separation]) and maximum concentration ratios (111.24% [rosuvastatin], 101.22% [coproporphyrin I], 89.14% [coproporphyrin III; simultaneous], and 96.84% [rosuvastatin; 4 h separation]) of the investigated substrates were within 80.0–125%. In addition, the 90% confidence intervals of the ratios were within the conventional no-effect boundaries of 80.0% and 125% for rosuvastatin after temporally separated administration, and for coproporphyrin I and III.
Conclusion
Administration of finerenone 40 mg once daily confers no risk of clinically relevant drug–drug interactions with substrates of BCRP, OATP1B1, or OATP1B3. The potential for relevant inhibition of these transporters suggested by in vitro findings was not confirmed in vivo.
Plain Language Summary
Finerenone is a drug that is used to reduce the risk of adverse kidney and cardiovascular outcomes in patients with chronic kidney disease associated with type 2 diabetes mellitus. Taking more than one medicine, and often several at the same time, is common in these patients; therefore, it is important to investigate the drug–drug interaction potential of finerenone. These studies were carried out to assess the interaction potential of finerenone and its metabolites in both laboratory experiments and healthy volunteers. Initial laboratory experiments indicated that finerenone and its metabolites could inhibit transporters used by other drugs. A study in healthy volunteers was performed and demonstrated that finerenone is not associated with any clinically meaningful changes to drugs that are substrates for the transporters. The study in healthy volunteers demonstrates that medications that are substrates of these transporters can be safely co-administered with finerenone.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A potential pharmacokinetic interaction was identified in vitro for finerenone and its metabolites as perpetrators with sensitive substrates of breast cancer resistance protein (BCRP), organic anion transporting polypeptide 1B1 (OATP1B1), and OATP1B3 transporters. |
A phase I study in healthy volunteers was performed to investigate the clinical relevance of this potential drug–drug interaction (DDI). Simultaneous co-administration of rosuvastatin (a reference substrate of both BCRP and OATPs) with finerenone indicated no evidence of a clinically relevant DDI in vivo. Administration of finerenone 4 h after rosuvastatin was also not associated with a meaningful change in rosuvastatin exposure. Finally, there was no effect of finerenone on the exposures of endogenous substrates of OATP1B1 and OATP1B3 (coproporphyrin I and coproporphyrin III). |
As there was no clinically relevant effect of finerenone on substrates of both BCRP and OATPs in vivo, the results of this DDI study indicate that medications that are substrates of these transporters can be safely co-administered. |
1 Introduction
Finerenone is a novel, selective, and nonsteroidal antagonist of the mineralocorticoid receptor (MR) that blocks the binding of the MR ligands aldosterone and cortisol more potently than spironolactone and eplerenone [1]. Finerenone reduced the urinary albumin-to-creatinine ratio in phase II studies in patients with chronic kidney disease (CKD) while having smaller effects on serum potassium levels than spironolactone [2, 3]. A phase III study (FIDELIO-DKD) in patients with CKD and type 2 diabetes mellitus (T2DM) met both its composite primary renal endpoint and its composite key secondary cardiovascular endpoint [4]. In a second phase III study (FIGARO-DKD), finerenone improved cardiovascular outcomes in patients with T2DM and stage 2–4 CKD with moderately elevated albuminuria or stage 1 or 2 CKD with severely elevated albuminuria as compared with placebo [5]. Finerenone has been approved in the United States to reduce the risk of a sustained estimated glomerular filtration rate decline, end-stage kidney disease, cardiovascular death, nonfatal myocardial infarction, and hospitalization for heart failure in adults with CKD associated with T2DM [6]. Currently, the drug is also approved in several other countries for the treatment of CKD, while regulatory assessments of the indication for heart failure are ongoing in these regions.
The pharmacokinetics of finerenone in phase I clinical studies were described in healthy volunteers [7,8,9,10,11,12] and in subjects with renal or hepatic impairment [13, 14]. In addition, the pharmacokinetics in patients and their relationships with pharmacodynamic parameters or clinical endpoints were assessed in phase II and III studies [15, 16].
In summary, finerenone is rapidly and completely absorbed after oral administration (time to maximum plasma concentration [Tmax] of 0.5 to 1.25 h), has dose-linear pharmacokinetics, and is rapidly eliminated (terminal half-life of 2 to 3 h). About 80% of the administered dose is excreted in urine (<1% is unchanged) and approximately 20% in feces (< 0.2% is unchanged). Finerenone’s absolute bioavailability is 43.5% due to first-pass metabolism in the gut wall and liver. The metabolites of finerenone are not pharmacologically active. Cytochrome P450 (CYP) 3A4 is responsible for about 90% of the metabolic clearance of finerenone, with the remainder attributable to CYP2C8. Mild, moderate, or severe renal impairment and mild or moderate hepatic impairment have no relevant effect on the pharmacokinetics of finerenone.
The phase I program included studies investigating finerenone as a victim [8] or potential perpetrator [10] of drug–drug interactions mediated by enzymes of the CYP system. The systemic exposure of finerenone as a victim of drug–drug interactions is increased by inhibitors of CYP3A4. From the perspective of being a potential perpetrator, finerenone has no relevant effect on CYP enzymes and the pharmacokinetics of substrates metabolized by these enzymes.
In vitro experiments suggested that finerenone could inhibit the activities of the drug transporters breast cancer resistance protein (BCRP), organic anion transporting polypeptide 1B1 (OATP1B1), and OATP1B3, which are involved in the absorption and disposition of potential comedications, such as statins [17]. Subsequently, an in vivo study was performed to assess the clinical relevance of these findings. The results of both the in vitro experiments and the in vivo study are presented in this paper.
2 Methods
2.1 In Vitro Investigations
The assessment of the inhibitory potential of finerenone and its metabolites towards drug transporters was conducted using cell-based in vitro assay systems. For the detailed assay protocols, please see Supplementary Methods S1.
2.2 Clinical Study
The study protocol was reviewed and approved by the Independent Ethics Committee of the North-Rhine Medical Council (Düsseldorf, Germany) and by the Federal Institute for Drugs and Medical Devices (Bonn, Germany). Informed consent was obtained from all participants before study commencement. The studies were conducted in accordance with the Declaration of Helsinki and adhered to the International Conference on Harmonization Good Clinical Practice guideline, European Union Clinical Trial Directive 2001/20/EC, and German drug law.
2.2.1 Participants
Participants were eligible for inclusion if they were healthy Caucasian males 18–45 years of age with a body mass index of 18.0–29.9 kg/m2, a systolic blood pressure (BP) of 100–140 mmHg, a diastolic BP of 60–90 mmHg, and a resting heart rate (HR) of 50–90 beats per minute.
Key exclusion criteria were: relevant diseases evident from medical histories or pre-study examinations; contraindications for rosuvastatin; a relevant deviation from the normal range in clinical chemistry, hematology, or urinalysis parameters; history of severe allergies, non-allergic drug reactions, or multiple drug allergies; regular use of therapeutic or recreational drugs; regular consumption of more than 20 g of alcohol per day; use of foods or medications (especially those affecting CYP3A4) within the 2 weeks or 5 half-lives (whichever was longer) preceding the study that could interfere with finerenone metabolism; participation in another clinical trial within the previous 3 months; history of coronavirus disease 2019 (COVID-19) or contact with a patient who tested positive for COVID-19 within the past 4 weeks or a positive severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) polymerase chain reaction (PCR) test.
2.2.2 Study Drug
Both study drugs—finerenone 40 mg (Bayer AG, Leverkusen, Germany) and rosuvastatin 5 mg (Crestor®, AstraZeneca GmbH, Wedel, Germany)—were administered as a film-coated tablet.
2.2.3 Safety and Tolerability
The safety of finerenone 40 mg and the co-administered drug rosuvastatin was monitored by physical examinations and regular measurements of systolic/diastolic BP, HR in the supine position, and the 12-lead electrocardiogram (ECG) after a rest of 15 min before and 1, 2, 3, 4, 6, 12, and 24 h after same-time dosing and 1, 2, 4, 5, 6, 12, and 24 h after the temporally separated administration of finerenone and rosuvastatin. BP, HR, and ECG were measured before and 1, 2, 4, 5, and 6 h after the dosing of rosuvastatin alone. Blood samples for the analysis of standard hematological and clinical chemistry laboratory parameters, including serum electrolytes, were taken the morning before and 24 and 48 h after dosing (but not for temporally separated dosing). Tolerability was assessed by asking subjects regularly about their well-being and any adverse events (AEs) using non-leading questions from pre-dose to follow-up 1, 2, 4, 6, and 12 h after rosuvastatin alone, 1, 2, 4, 6, and 12 h after same-time treatment, and 1, 2, 4, 6, and 12 h after separated finerenone and rosuvastatin treatment. The intensity of AEs (mild [not interfering with everyday activities], moderate [sufficient discomfort to interfere with normal everyday activities], or severe [prevents normal everyday activities]) and any causal relationship between study drugs and AEs were assessed by the investigator, based on criteria predefined in the study protocol.
2.2.4 Pharmacokinetic Sampling
Blood samples for the determination of plasma concentrations of the respective analyte(s) were taken pre-administration and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 15, 24, 36, 48, 60, and 71.5 h thereafter (rosuvastatin; 3.0 mL per sample) or 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 15, and 23.5 h after administration (finerenone and its metabolites M1, M2, and M3; 1.5 mL per sample). Blood samples for the determination of coproporphyrin I (CP-I) and CP-III (1.2 mL) were collected across an interval of 24 h at relative times of 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 15, and 24 h, where the ‘0 h’ sample was taken at approximately 8 a.m., immediately before the administration of rosuvastatin. Plasma was prepared by centrifugation at approximately 2000×g for 10 min at ambient temperature or at approximately 4 °C (rosuvastatin). Blood and plasma samples were not exposed to sunlight. Samples were stored in temperature-controlled freezers at ≤ − 20 °C (finerenone and metabolites, and rosuvastatin) and at − 85 ± 10 °C (CP-I and CP-III), respectively.
2.2.5 Study Design and Treatment
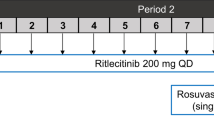
The effect of finerenone on BCRP and OATP1B1 and OATP1B3 was investigated in a single-center, open-label, fixed-sequence interaction study with three subsequent periods. Single doses of rosuvastatin 5 mg in the fasted state were administered on three occasions, alone (period 1) and on a background of finerenone at steady state (periods 2 and 3). The first dose of rosuvastatin was administered alone on day 1 of period 1, followed by the collection of pharmacokinetic samples over 71.5 h. On day 4, after collection of the last pharmacokinetic sample for rosuvastatin, a regimen of multiple doses of finerenone 40 mg once daily for 10 days was started. On the third day of administration of finerenone, the second dose of rosuvastatin 5 mg was given (at the same time as finerenone), again followed by the collection of pharmacokinetic samples for rosuvastatin over 71.5 h (period 2). On the eighth day of the finerenone once-daily regimen, the third single dose of rosuvastatin was administered (temporally separated, 4.25 h before finerenone), and pharmacokinetic samples for rosuvastatin were collected over the following 71.5 h (period 3).
In period 1 (rosuvastatin alone) and period 2 (rosuvastatin and finerenone simultaneously), pharmacokinetic samples were also collected over 24 h to determine CP-I and CP-III plasma concentrations.
Following the last (tenth) dose of finerenone, pharmacokinetic samples were collected over 24 h to determine finerenone and metabolites. On the following day, the subjects were discharged from the ward. Follow-up procedures were performed not less than 7 days after the last dose of finerenone. The duration of the in-house period was 15 days, with 14 overnight stays.
The study protocol included an optional study part 2 investigating the effect of the lower dose of finerenone 20 mg once daily according to the above design. However, the study was closed after the completion of part 1, as predefined criteria to conduct part 2 (i.e., an increase in rosuvastatin area under the concentration–time curve from zero to infinity [AUC] or maximum plasma concentration [Cmax] by > 25% after administration of finerenone 40 mg) were not met.
2.2.6 Bioanalysis
Rosuvastatin was determined in plasma after the addition of a stable-isotope–labeled internal standard (triple-deuterated rosuvastatin) and subsequent liquid–liquid extraction with methyl tert-butyl ether. Separation and detection were achieved by high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS). Calibrators (0.0200–10.0 µg/L [lower to upper limit of quantification (LLOQ–ULOQ)]) and quality control (QC) samples (0.0600–7.50 µg/L) were analyzed with study samples in a batchwise fashion. QC samples were determined with an accuracy and precision of 98.9–100.3% and 2.47–4.55%, respectively (n = 32 per QC concentration level).
Determination of CP-I and CP-III concentrations in plasma was conducted using HPLC–MS/MS after protein precipitation with methanol containing the internal standards (fourfold 15N-labeled CPs). The calibration range of the procedure was 0.100 µg/L (LLOQ) to 100 µg/L (ULOQ) for both analytes (CP-I and CP-III). QC samples (0.300–75.0 µg/L) were determined with an accuracy and precision of 95.0–98.0% and 3.3–7.6% (CP-I) and 93.0–96.7% and 3.4–5.6% (CP-III) (n = 10–12 per QC level), respectively.
Plasma concentrations of finerenone and its metabolites M1, M2, and M3 (the structures of the metabolites are given in [9]) were determined using HPLC–MS/MS, based on [11]. The respective stable-isotope–labeled compounds were used as internal standards for the quantification of the four analytes. The working range was 0.100 µg/L (finerenone) or 0.500 µg/L (metabolites) to 500 µg/L (all analytes). QC samples contained analyte concentrations of 0.300 µg/L to 375 µg/L (finerenone) and 1.50 µg/L to 375 µg/L (metabolites). The accuracy and precision for the QC samples were 93.3–99.5% and 1.7–2.2% (finerenone; n = 7–8 per QC level), 96.5–100.3% and 2.6–6.7% (M1; n = 9–10), 97.9–104.3% and 4.7–6.6% (M2; n = 5–6), and 102.4–107.8% and 3.4–7.1% (M3; n = 5–6), respectively.
All method validations and analyses of the study samples were performed in compliance with the pertinent guidelines on bioanalytical method validation [18, 19]. Assays were sensitive and selective for the respective analytes, as stated above, and were fully validated. No mutual effects on ionization or chromatographic interference between analytes and/or internal standards were observed in validation experiments conducted prior to starting the analysis of study samples.
2.2.7 Pharmacokinetic and Statistical Evaluation
Pharmacokinetic parameters were calculated by non-compartmental analysis using Phoenix WinNonlin 8.1 (Certara, Princeton, NJ, USA). They are presented as geometric mean values and the geometric coefficient of variation, except for the time to reach Cmax (Tmax), for which the median and range are reported. Summary statistics of plasma concentration values at any time were calculated if at least two-thirds of the individual data points were measured and were greater than the LLOQ. Values below the LLOQ were substituted by 0.5 × LLOQ if this condition was met. The parameters T1/2 (T1/2,md after multiple dose administration) and CL/F denote the half-life associated with the terminal slope and the apparent oral clearance, respectively. The areas under the curve from zero to infinity (AUC), from zero to the time of the last quantifiable concentration (AUC(0–t)) (both after single dose administration), and within a dosing interval (AUCτ,md) (after multiple dose administration) and the maximum plasma concentration (Cmax [single dose], and Cmax,md [multiple dose]) were analyzed assuming log-normally distributed data. The logarithms of pertinent pharmacokinetic parameters for rosuvastatin and CP-I and CP-III were subject to analysis of variance (ANOVA), and treatments were compared by calculating point estimates (least-squares [LS] means) and explorative 90% confidence intervals (CIs) for treatment ratios after re-transformation of the logarithmic data using the intraindividual standard deviation of the ANOVA. The pharmacokinetic parameters of finerenone and its metabolites were summarized by descriptive statistics.
N = 15 subjects were included to meet the required minimum sample size of subjects valid for pharmacokinetics, N = 12. Based on previous knowledge of the intraindividual pharmacokinetic variability of rosuvastatin, and assuming a point estimate for the treatment ratio of rosuvastatin AUC and Cmax of unity, this sample size was adequate to show no interaction within the no-effect boundaries (90% CI) of 80.0–125%.
3 Results
3.1 In Vitro Assessment of Inhibitory Potential Towards Drug Transporters
The potential of finerenone to act as a perpetrator for various transporter-mediated drug–drug interactions (DDIs) was investigated in vitro. The results for finerenone, both atropisomers of metabolite M1 (M1a and M1b), and the a-series of atropisomers for M2 and M3 [9] regarding the inhibition of BCRP, OATP1B1, and OATP1B3 are summarized in Table 1 and will be discussed here.
Finerenone inhibited BCRP-mediated transport with a half-maximal inhibitory concentration (IC50) of 17.4 µM. Furthermore, it was identified as an inhibitor of OATP1B1, with an IC50 of 3.2 µM. Both atropisomers of M1, M1a, and M1b did not relevantly inhibit BCRP; however, inhibition of OATP1B1 and OATP1B3 was observed with IC50 values of 3.8 µM and 3.9 µM for OATP1B1 and 7.6 µM and 7.3 µM for OATP1B3, respectively. For M2a, no inhibition > 50% was apparent for any of the tested transporters, whereas, for M3a, an IC50 of 3.4 µM was established for OATP1B3. If no exact IC50 value could be determined for a certain transporter (less than 50% inhibition at the highest tested assay concentration), the IC50 is indicated to be above the highest tested assay concentration in Table 1 (e.g., > 20 µM).
3.2 Calculation of the DDI Risk Based on In Vitro Results
The assessment of whether the in vitro results on transporter inhibition would necessitate clinical studies (i.e., the evaluation of the DDI risk from finerenone and its metabolites towards sensitive BCRP, OATP1B1, and OATP1B3 substrates) was performed according to pertinent regulatory guidelines (as issued by the US Food and Drug Administration [FDA] and European Medicines Agency [EMA]) [20, 21]. Input parameters for, and the results of, the calculations are summarized in Table 2. If no IC50 could be determined, the highest tested assay concentration was used for calculations, which represents the most conservative approach to DDI risk assessment.
Applying a cutoff value of [I]gut (the intestinal luminal concentration)/IC50 ≥ 10, a risk for a clinically relevant interaction towards intestinal BCRP was identified for finerenone, as [I]gut/IC50 amounted to 24.3 for a 40-mg dose. By contrast, a risk for inhibition of systemic BCRP by finerenone and its metabolites did not emerge when taking cutoff values of Cmax,tot (total mean Cmax)/IC50 ≥ 0.1 (FDA) or Cmax,u (unbound mean Cmax)/IC50 ≥ 0.02 (EMA) into consideration.
For hepatic uptake transporters OATP1B1 and OATP1B3, regulatory guidelines describe varying approaches to assess a potential DDI risk. The 2020 FDA DDI guideline [21] states that a detailed investigation of the clinical DDI risk is warranted once [I]inlet,max,u/IC50 ≥ 0.1 (stated as R = 1 + [I]inlet,max,u/IC50 ≥ 1.1), where [I]inlet,max,u is the estimated maximum unbound plasma inhibitor concentration at the inlet to the liver. The EMA guideline [20] applies a stricter threshold, requiring further investigation of the risk of potential DDIs once the inhibitory constant (Ki) or IC50 ≤ 25 × [I]inlet,max,u (which can also be expressed as [I]inlet,max,u/IC50 ≥ 0.04). For finerenone-mediated inhibition of OATP1B1 and OATP1B3, [I]inlet,max,u/IC50 amounted to 0.182 for OATP1B1 and < 0.058 for OATP1B3. Hence, guideline cutoffs were exceeded for both transporters.
For metabolites, Cmax,u serves as relevant inhibitory concentration and guidelines recommend using Cmax,u/IC50 ≥ 0.1 (FDA) [21] and Cmax,u/IC50 ≥ 0.02 (EMA) [20] as thresholds. Metabolites M1a and M3a also displayed a potential for relevant transporter inhibition. Calculations of Cmax,u/IC50 resulted in values of 0.025 for M1a towards OATP1B1 and < 0.023 and 0.068 for M3a for OATP1B1 and OATP1B3, respectively. By contrast, no relevant inhibition of OATP1B1 or OATP1B3 was predicted for the remaining metabolites investigated.
3.3 Clinical Study
3.3.1 Patient Enrollment
Thirty-four subjects were enrolled and 15 successfully completed screening, one of whom withdrew from the study prior to any study medication due to family problems. Fourteen subjects were treated with rosuvastatin alone and in combination with finerenone and completed the treatment sequence and follow-up visit as planned. The mean (standard deviation) age, weight, and height of the 14 subjects were 29.6 years (5.4 years), 82.4 kg (7.8 kg), and 179.9 cm (4.9 cm), respectively.
3.3.2 Safety and Tolerability
The safety and tolerability of the study treatments were compared with baseline measurements by physical examinations and regular monitoring of systolic/diastolic BP and HR, measurements of ECGs, and blood sampling for hematology, clotting, and clinical laboratory parameters, including serum electrolyte levels. These parameters showed no clinically relevant changes in individual and mean values after dosing of finerenone 40 mg administered alone or in combination with rosuvastatin. Repeated finerenone doses were well tolerated and there were no drug-specific or mode-of-action-related clinically relevant side effects. In general, no serious or severe AEs were observed with finerenone 40 mg, no relevant changes in potassium levels were seen, and no premature terminations due to study-drug-related side effects occurred. Two cases of headache as an AE were, according to criteria predefined in the study protocol, judged to be treatment related, with one case occurring after the combination and the other after rosuvastatin alone. As these are events that were previously and are generally observed in phase I studies, they were not judged to be related to the drug’s mode of action. Overall, the study treatments were well tolerated.
3.3.3 Pharmacokinetics
Plasma concentration vs. time profiles of rosuvastatin are shown in Fig. 1, and results of the pharmacokinetic analysis are presented in Table 3. Plasma concentration vs. time profiles of the endogenous OATP substrate CP-I are presented in Fig. 2, and pharmacokinetic parameters for both CP-I and CP-III are presented in Table 4. Concentration vs. time profiles for CP-III are not shown, owing to a large number of values below the LLOQ. The results of the statistical analysis (ANOVA) comparing treatment ratios for rosuvastatin, CP-I, and CP-III are given in Table 5.

Concentration of rosuvastatin (µg/L) in plasma by treatment (geometric mean/SD; pharmacokinetics set, N = 14). LLOQ = 0.02 μg/L. LLOQ lower limit of quantification, SD standard deviation

Concentration of CP-I (µg/L) in plasma by treatment (geometric mean/SD; pharmacokinetics set, N = 14). LLOQ = 0.1 μg/L. LLOQ lower limit of quantification, SD standard deviation
The ANOVA demonstrated small increases in the exposure (AUC +14% [geometric coefficient of variation (CV) = 17%] and Cmax +11% [geometric CV = 23%], based on point estimates) of rosuvastatin when administered simultaneously with finerenone (40 mg at steady state) compared with the administration of rosuvastatin alone. The 90% CI of the rosuvastatin Cmax ratio included unity.
When rosuvastatin was administered 4.25 h before finerenone, there was no effect on its AUC and Cmax compared with rosuvastatin alone. The point estimates for AUC and the Cmax ratios were close to unity, and their corresponding 90% CIs fell within the conventional no-effect boundary (80.0–125%).
Finerenone did not show an effect on Cmax,md and AUCτ,md of CP-I and Cmax,md of CP-III. The point estimates for AUCτ,md and the Cmax,md ratios for CP-I and the Cmax,md ratio for CP-III were close to unity, and the corresponding 90% CIs of all ratios fell within 80.0–125%. The low plasma concentrations of CP-III, with observations frequently below the LLOQ, precluded an assessment of AUCτ,md for this analyte. Therefore, the assessment for CP-III is based on Cmax,md alone.
Derived pharmacokinetic parameters for finerenone and its pharmacologically inactive metabolites M1, M2, and M3 are presented in Table 6.
4 Discussion
Finerenone has recently been approved by the FDA and EMA for the treatment of adults with CKD associated with T2DM [22] at doses of 10 mg and 20 mg. Currently, finerenone 40 mg is the highest dose investigated in patients with heart failure and left ventricular ejection fraction ≥ 40% in an ongoing trial [23].
In vitro data, together with prediction algorithms, identified a potential DDI risk with sensitive substrates of BCRP, OATP1B1, and OATP1B3 for finerenone and its metabolites M1a and M3a at the highest clinically relevant dose. More specifically, finerenone was identified as a potentially relevant inhibitor of all three transporters, whereas M1a exceeded cutoff values for OATP1B1 and M3a did so for both OATP transporters. It is noteworthy that the cutoff values currently stipulated in regulatory guidelines were not exceeded by a large margin (Table 2), raising the question as to whether these results would translate into a relevant effect in vivo.
For example, the threshold for finerenone-mediated inhibition of intestinal BCRP of [I]gut/IC50 ≥ 10 was exceeded at 24.3. Despite some successful attempts [24,25,26], there is no standardized procedure to quantitatively predict the exposure change of sensitive substrates for DDIs affecting intestinal efflux transporters. However, the threshold applied is set with the intention of avoiding false-negative predictions [27], which implies that it is rather conservative (a claim that was also substantiated by others; e.g., Costales et al. [25]).
The current FDA in vitro DDI guideline [21] utilizes the basic model [28, 29] to assess the potential for, and extent of, transporter-mediated DDIs at the systemic level, including hepatic uptake transporters OATP1B1 and OATP1B3. The model allows the prediction of an increase in AUC (as the AUC ratio [AUCR]), according to R = 1 + [I]inlet,max,u (or Cmax,u)/IC50. As indicated in Table 2, AUCRs of 1.182, 1.025, and 1.068 were calculated for the inhibition of OATP1B1 or OATP1B3 for finerenone, M1a, and M3a, respectively. These calculations imply that the clearance of the hypothetical substrate is mediated only by the transporter of interest, which renders the substrate maximally sensitive to inhibition. Therefore, it appeared somewhat questionable as to whether the inhibitory effect of finerenone and its metabolites could lead to an exposure increase beyond the no-effect boundary for clinically relevant OATP substrates such as statins, which do possess multiple clearance mechanisms [30].
A clinical phase I study was conducted to investigate finerenone as a potential inhibitor of BCRP and/or OATPs following the findings of the inhibition of these targets in vitro. Rosuvastatin was used as an index substrate of both BCRP and OATPs [17], similar to numerous previous studies with modulators of these drug transporters. Cyclosporine [31], an inhibitor of both BCRP and OATPs [17], increased rosuvastatin exposure in patients, with AUC and Cmax ratios of 7.1 and 10.6, respectively, compared with historic control data for rosuvastatin alone. Less pronounced effects on rosuvastatin (AUC and Cmax ratio of 1.55 and 2.03, respectively) were observed with eltrombopag [32], a BCRP inhibitor [17]. The OATP inhibitor gemfibrozil [17] increased the rosuvastatin AUC and Cmax 1.88- and 2.21-fold, respectively [33]. In order to discern the mechanism behind potential effects of finerenone on rosuvastatin exposure, CP-I and CP-III as endogenous substrates of OATP1B1 and OATP1B3 were also measured in the present study, both in the absence and in the presence of finerenone. The utility of CP-I and CP-III in this context was confirmed in a study with the OATP inhibitor rifampicin and rosuvastatin in which the effect on rosuvastatin (AUC ratio of 5.0) was paralleled by increased exposure of CP-I and CP-III (AUC ratios of 4.0 and 3.3, respectively) [34]. It was concluded from studies with other OATP inhibitors that the magnitude of increase in plasma exposure of CP-I and CP-III is proportional to the strength of the applied inhibitor [35]. Thus, while increased rosuvastatin exposure could be the result of BCRP and/or OATP inhibition, the plasma concentrations of CP-I and CP-III would be a selective indicator for effects on OATP.
In order to maximize the potential for a systemic interaction, the substrate rosuvastatin was administered after finerenone and its metabolites had reached a steady state in plasma. The investigation of the effect of finerenone on CP-I and CP-III exposure was performed on a background of rosuvastatin administration, which has been shown to have no effect of its own on CP-I or CP-III exposure [34]. This study also assessed the possibility of avoiding an interaction during absorption by temporally separating the involved medications. Simultaneous co-administration was intended to separate the Tmax values of finerenone and rosuvastatin, while temporal separation of intake (finerenone approximately 4 h after rosuvastatin, with the Tmax of rosuvastatin expected at approximately 5 h [36]) was utilized for Tmax alignment between the potential inhibitor and the substrate. Conversely, the potential for an effect of finerenone metabolite M3 (Tmax of approximately 6 h, almost coinciding with that of rosuvastatin) on OATP1B3 was expected to be enhanced after simultaneous intake of finerenone and rosuvastatin.
After simultaneous co-administration with finerenone, the point estimate of the increase in rosuvastatin exposure (AUC +14%, Cmax +11%) was < 25%; i.e., below the threshold of ‘weak inhibition’ defined in the context of metabolic pathways [37]. When rosuvastatin was administered approximately 4 h before finerenone, the point estimates for the AUC and the Cmax ratios, together with their corresponding 90% CIs, fell within the no-effect boundary (80.0–125%). The complete absence of increased CP I and CP III exposure in both conditions suggests that finerenone does not affect OATP1B1 and OATP1B3. A previous study focusing on CYP2C8-mediated metabolism [10] had shown no effect of finerenone (20 mg) on repaglinide, which is also an OATP substrate [17]. The observed not clinically relevant change in rosuvastatin AUC with finerenone (40 mg) is therefore believed to be due to an effect on BCRP.
Finerenone 40 mg is the highest investigated dose in patients [23] and was used to study the effect on rosuvastatin. An optional second part of the study with finerenone 20 mg, the highest approved dose in CKD associated with T2DM [6], was not performed after part 1 had shown no relevant effect of the 40-mg dose. The systemic exposure (AUCτ,md) of finerenone and metabolites in the present study following a dose of 40 mg once daily was consistent with published data (AUC) in healthy volunteers (20 mg [10, 11] for finerenone and 10 mg for the control group [13], finerenone and metabolites), applying dose-linear pharmacokinetics [7, 12].
5 Conclusions
In vitro investigations on the inhibitory potential of finerenone and its metabolites identified a potential DDI risk towards BCRP (for finerenone), OATP1B1 (for finerenone and M1a), and OATP1B3 (for finerenone, M1a, and M3a) by exceeding cutoff values established in pertinent regulatory guidelines. Accordingly, a clinical study was conducted to address these concerns by using rosuvastatin as the victim drug and substrate of all three transporters. Upon simultaneous co-administration with finerenone 40 mg, a 14% AUC and an 11% Cmax increase was observed for rosuvastatin, indicating no clinically relevant effect in vivo, despite the potential risk indicated in vitro. When rosuvastatin was administered 4 h prior to finerenone to synchronize the maximum plasma exposure of rosuvastatin and the potential inhibitor M3a, the resulting effect fell completely within the no-effect boundary, demonstrating that M3a is also not a clinically relevant inhibitor of BCRP or OATP transporters. To identify the site of a potential interaction between finerenone and rosuvastatin, levels of CP-I and CP-III, which are endogenous substrates of OATP1B1 and OATP1B3, were measured. No effect on the exposure of CP-I or CP-III was observed, consistent with the claim that finerenone and its metabolites are not relevant inhibitors of OATP1B1 and OATP1B3. This finding also suggests that the observed minor increase in rosuvastatin exposure is likely due to an effect on BCRP rather than OATP transporters. In summary, we conclude that finerenone 40 mg is safe to be administered with substrates of BCRP and/or OATP1B1 and OATP1B3. As previously published by others (e.g., by Costales et al. [25]), the data presented here indicate that current regulatory guidelines apply rather strict cutoffs above which it is recommended that clinical transporter interaction studies should be conducted. This avoids false-negative findings on the one hand, but, conversely, prompts additional in vivo studies in human subjects.
References
Kolkhof P, Baerfacker L. 30 years of the mineralocorticoid receptor: Mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol. 2017;234(1):T125–40.
Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, Remuzzi G, Rossing P, Schmieder RE, Nowack C, Kolkhof P, Joseph A, Pieper A, Kimmeskamp-Kirschbaum N, Ruilope LM. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA. 2015;314(9):884–94.
Pitt B, Kober L, Ponikowski P, Gheorghiade M, Filippatos G, Krum H, Nowack C, Kolkhof P, Kim SY, Zannad F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94–8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur Heart J. 2013;34(31):2453–63.
Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, Kolkhof P, Nowack C, Schloemer P, Joseph A, Filippatos G. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med. 2020;383(23):2219–29.
Pitt B, Filippatos G, Agarwal R, Anker SD, Bakris GL, Rossing P, Joseph A, Kolkhof P, Nowack C, Schloemer P, Ruilope LM. Cardiovascular events with finerenone in kidney disease and type 2 diabetes. N Engl J Med. 2021;385:2252–63.
Bayer HealthCare Pharmaceuticals Inc. KERENDIA (finerenone) tablets, for oral use: US prescribing information. 2021. https://labeling.bayerhealthcare.com. Accessed 18 Jan 2022.
Lentini S, Heinig R, Kimmeskamp-Kirschbaum N, Wensing G. Pharmacokinetics, safety and tolerability of the novel, selective mineralocorticoid receptor antagonist finerenone—results from first-in-man and relative bioavailability studies. Fundam Clin Pharmacol. 2016;30(2):172–84.
Heinig R, Gerisch M, Engelen A, Nagelschmitz J, Loewen S. Pharmacokinetics of the novel, selective, non-steroidal mineralocorticoid receptor antagonist finerenone in healthy volunteers: Results from an absolute bioavailability study and drug-drug interaction studies in vitro and in vivo. Eur J Drug Metab Pharmacokinet. 2018;43(6):715–27.
Gerisch M, Heinig R, Engelen A, Lang D, Kolkhof P, Radtke M, Platzek J, Lovis K, Rohde G, Schwarz T. Biotransformation of finerenone, a novel nonsteroidal mineralocorticoid receptor antagonist, in dogs, rats, and humans, in vivo and in vitro. Drug Metab Dispos. 2018;46(11):1546–55.
Heinig R, Gerisch M, Bairlein M, Nagelschmitz J, Loewen S. Results from drug-drug interaction studies in vitro and in vivo investigating the effect of finerenone on the pharmacokinetics of comedications. Eur J Drug Metab Pharmacokinet. 2020;45(4):433–44.
Rohde G, Loewen S, Heinig R. Determination of finerenone—a novel, selective, nonsteroidal mineralocorticoid receptor antagonist—in human plasma by high-performance liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study in venous and capillary human plasma. J Chromatogr B. 2021;1172:122643.
Heinig R, Nagelschmitz J, Loewen S. Results from phase I studies investigating the dose linearity of finerenone tablets and the influence of food or pH-modifying comedications on its pharmacokinetics in healthy male volunteers. Eur J Drug Metab Pharmacokinet. 2022;47(4):549–59.
Heinig R, Kimmeskamp-Kirschbaum N, Halabi A, Lentini S. Pharmacokinetics of the novel nonsteroidal mineralocorticoid receptor antagonist finerenone (BAY 94–8862) in individuals with renal impairment. Clin Pharmacol Drug Dev. 2016;5(6):488–501.
Heinig R, Lambelet M, Nagelschmitz J, Alatrach A, Halabi A. Pharmacokinetics of the novel nonsteroidal mineralocorticoid receptor antagonist finerenone (BAY 94–8862) in individuals with mild or moderate hepatic impairment. Eur J Drug Metab Pharmacokinet. 2019;44(5):619–28.
Snelder N, Heinig R, Drenth HJ, Joseph A, Kolkhof P, Lippert J, Garmann D, Ploeger B, Eissing T. Population pharmacokinetic and exposure-response analysis of finerenone: Insights based on phase IIb data and simulations to support dose selection for pivotal trials in type 2 diabetes with chronic kidney disease. Clin Pharmacokinet. 2020;59(3):359–70.
van den Berg P, Ruppert M, Mesic E, Snelder N, Seelmann A, Heinig R, Joseph A, Garmann D, Lippert J, Eissing T. Finerenone dose-exposure-response for the primary kidney outcome in FIDELIO-DKD phase III: Population pharmacokinetic and time-to-event analysis. Clin Pharmacokinet. 2022;61(3):439–50.
US Food & Drug Administration. Drug development and drug interactions—table of substrates, inhibitors and inducers. Table 5-1. 2020. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table5-1. Accessed 05 Feb 2022.
US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM). Bioanalytical method validation, guidance for industry. 2018. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf. Accessed 19 Apr 2022.
European Medicines Agency. Guideline on bioanalytical method validation. 2011. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed 19 Apr 2022.
European Medicines Agency and Committee for Human Medicinal Products (CHMP). Guideline on the investigation of drug interactions. 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf. Accessed 19 Apr 2022.
US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research (CDER). In vitro drug interaction studies—Cytochrome P450 enzyme- and transporter-mediated drug interactions, guidance for industry. 2020. https://www.fda.gov/media/134582/download. Accessed 19 Apr 2022.
Frampton JE. Finerenone: first approval. Drugs. 2021;81(15):1787–94.
US National Library of Medicine. Study to evaluate the efficacy (effect on disease) and safety of finerenone on morbidity (events indicating disease worsening) & mortality (death rate) in participants with heart failure and left ventricular ejection fraction (proportion of blood expelled per heart stroke) greater or equal to 40% (FINEARTS-HF). 2020. https://clinicaltrials.gov/ct2/show/NCT04435626. Accessed 5 Feb 2022.
Sane R, Cheung KWK, Kovács P, Farasyn T, Li R, Bui A, Musib L, Kis E, Plise E, Gáborik Z. Calibrating the in vitro-in vivo correlation for OATP-mediated drug-drug interactions with rosuvastatin using static and PBPK models. Drug Metab Dispos. 2020;48(12):1264–70.
Costales C, Lin J, Kimoto E, Yamazaki S, Gosset JR, Rodrigues AD, Lazzaro S, West MA, West M, Varma MVS. Quantitative prediction of breast cancer resistant protein mediated drug-drug interactions using physiologically-based pharmacokinetic modeling. CPT Pharmacomet Syst Pharmacol. 2021;10(9):1018–31.
Hanke N, Frechen S, Moj D, Britz H, Eissing T, Wendl T, Lehr T. PBPK models for CYP3A4 and P-gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet Syst Pharmacol. 2018;7(10):647–59.
Zhang L, Zhang YD, Strong JM, Reynolds KS, Huang SM. A regulatory viewpoint on transporter-based drug interactions. Xenobiotica. 2008;38(7–8):709–24.
Rowland M, Matin SB. Kinetics of drug-drug interactions. J Pharmacokinet Biopharm. 1973;1(6):553–67.
Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201–8.
Elsby R, Hilgendorf C, Fenner K. Understanding the critical disposition pathways of statins to assess drug-drug interaction risk during drug development: it’s not just about OATP1B1. Clin Pharmacol Ther. 2012;92(5):584–98.
Simonson SG, Raza A, Martin PD, Mitchell PD, Jarcho JA, Brown CD, Windass AS, Schneck DW. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004;76(2):167–77.
Allred AJ, Bowen CJ, Park JW, Peng B, Williams DD, Wire MB, Lee E. Eltrombopag increases plasma rosuvastatin exposure in healthy volunteers. Br J Clin Pharmacol. 2011;72(2):321–9.
Schneck DW, Birmingham BK, Zalikowski JA, Mitchell PD, Wang Y, Martin PD, Lasseter KC, Brown CD, Windass AS, Raza A. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2004;75(5):455–63.
Lai Y, Mandlekar S, Shen H, Holenarsipur VK, Langish R, Rajanna P, Murugesan S, Gaud N, Selvam S, Date O, Cheng Y, Shipkova P, Dai J, Humphreys WG, Marathe P. Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug-drug interactions mediated by organic anion transporting polypeptide inhibition. J Pharmacol Exp Ther. 2016;358(3):397–404.
Kunze A, Ediage EN, Dillen L, Monshouwer M, Snoeys J. Clinical investigation of coproporphyrins as sensitive biomarkers to predict mild to strong OATP1B-mediated drug-drug interactions. Clin Pharmacokinet. 2018;57(12):1559–70.
AstraZeneca UK Limited. Crestor 10 mg film-coated tablets—summary of product characteristics. 2021. https://www.medicines.org.uk/emc/product/7559/smpc. Accessed 19 Apr 2022.
US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Clinical drug interaction studies—Cytochrome P450 enzyme- and transporter-mediated drug interactions, guidance for industry. 2020. https://www.fda.gov/media/134581/download. Accessed 19 Apr 2022.
Acknowledgements
We thank Julia Lemmen for her valuable contribution in designing and conducting the in vitro studies described in this publication and Mahesh Kharmale for planning, conducting, and reporting the statistical analysis. The authors and study sponsor are indebted to the healthy volunteers and their families, as well as to the investigators and sites participating in the studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by Bayer AG, Wuppertal, Germany. Editorial support, including fact checking, referencing, figure preparation, formatting, proofreading, and submission was provided by Moamen Hammad, PhD, and Ian Norton, PhD, of Scion, London, UK, supported by Bayer AG, Wuppertal, Germany, according to Good Publication Practice guidelines.
Conflicts of interest
RH, JN, RF, and SW are employees of Bayer AG. SL is an employee of Chrestos Concept GmbH & Co. KG, which received funding for this analysis from Bayer AG. In addition, RH, JN, RF, and SW have stock in Bayer AG, but are not paid in stock or stock options.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Consent to participate
Informed consent was obtained from all individual participants included in the studies before study commencement.
Consent for publication
Not applicable.
Availability of data
Availability of the data underlying this publication will be determined later according to Bayer’s commitment to the EFPIA/PhRMA principles for responsible clinical trial data sharing. This pertains to the scope, time point, and process of data access. As such, Bayer commits to sharing, upon request from qualified scientific and medical researchers, participant-level clinical trial data, study-level clinical trial data, and protocols from clinical trials in participants for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 1, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymized participant-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the study sponsor’s section of the portal. Data access will be granted to anonymized participant-level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that participant privacy is safeguarded.
Code availability
Not applicable.
Author contributions
RH, JN, and SL participated in the study concept and design of the phase I study and were involved in the acquisition, analysis, and interpretation of the respective data. RF was involved in the study concept and design of in vitro studies, their data acquisition, and analysis. RF and SW were involved in the interpretation of the in vitro results. All authors participated in preparing the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Heinig, R., Fricke, R., Wertz, S. et al. Results From Drug–Drug Interaction Studies In Vitro and In Vivo Investigating the Inhibitory Effect of Finerenone on the Drug Transporters BCRP, OATP1B1, and OATP1B3. Eur J Drug Metab Pharmacokinet 47, 803–815 (2022). https://doi.org/10.1007/s13318-022-00794-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-022-00794-5