
Abstract
Background and Objectives
Alirocumab is a cholesterol-lowering monoclonal antibody targeting proprotein convertase subtilisin kexin type 9 (PCSK9) indicated in the prevention of cardiovascular risk and exhibiting target-mediated drug disposition (TMDD). The aim of this work was to develop an integrated pharmacokinetic–pharmacodynamic model to describe the interaction of alirocumab with PCSK9 and its impact on the evolution of low-density lipoprotein cholesterol (LDL-C) levels and explore labeling specification for subpopulations.
Methods
Using data collected from nine phase I/II/III clinical studies (n = 527, subcutaneous or intravenous administration), a TMDD model considering the quasi-steady-state approximation was developed to characterize the interaction dynamics of alirocumab and PCSK9, combined with an indirect pharmacodynamic model describing the inhibition of LDL-C by PCSK9 in a one-step approach using nonlinear-mixed effects modeling. A “full fixed effects modeling” strategy was implemented to quantify parameter–covariate relationships.
Results
The model captures the interaction between alirocumab and its target PCSK9 and how this mechanism drives LDL-C depletion, with an estimation of the associated between-subject variability of model parameters and the quantification of clinically relevant parameter–covariate relationships. Co-administration of statins was found to increase the central volume of distribution of alirocumab by 1.75-fold (5.6 L versus 3.2 L) and allow for a 14% greater maximum lipid-lowering effect (88% versus 74%), highlighting the synergy of action between anti-PCSK9 therapeutic antibodies and statins toward lowering LDL-C plasma levels. Baseline levels of PCSK9 were found to be related to the amplitude of LDL-C variations by increasing the concentration of free PCSK9 necessary to reach half its capacity of inhibition of LDL-C degradation.
Conclusion
The maximum effect of alirocumab is achieved when free PCSK9 concentration is close to zero, as seen mostly after 150 mg every 2 weeks (Q2W) or 300 mg every 4 weeks (Q4W), indicating that there would be no additional clinical benefit of increasing the dose higher than these recommended dosing regimens.
Similar content being viewed by others

A semi-mechanistic population pharmacokinetic–pharmacodynamic (PK–PD) relationship allowing one to describe, in a single step, the dynamic interaction between a drug (alirocumab) and its target (PCSK9) and how this physiologically drives pharmacodynamics (LDL-C depletion) was obtained. |
The dose administration simulations clarified dose recommendations and the need for dosage adjustment. |
Alirocumab dosage was confirmed: 75 mg Q2W or 300 mg Q4W, which can be increased up to 150 mg Q2W for patients requiring greater reduction of blood LDL-C levels or after insufficient response to treatment. |
1 Introduction
Alirocumab (Praluent) is a lipid-lowering monoclonal antibody approved for the treatment of adults with primary hyperlipidemia, including heterozygous familial hypercholesterolemia or mixed dyslipidemia. Alirocumab reduces low-density lipoprotein cholesterol (LDL-C) to reduce the risk of myocardial infarction, stroke, and unstable angina requiring hospitalization in adults with established cardiovascular disease, and as adjunct to diet, alone or in combination with other lipid-lowering therapies (e.g., statins, ezetimibe). Alirocumab targets proprotein convertase subtilisin kexin type 9 (PCSK9), a circulating protein involved in cholesterol metabolism by reducing the life cycle of low-density lipoprotein receptor (LDL-R), located on the surface of hepatocytes. PCSK9 binds to the extracellular domain of LDL-R and promotes its internalization and degradation, causing a decreased ability to remove LDL-C particles from the circulation, leading to higher serum LDL-C levels [1]. By blocking PCSK9 from binding to LDL-R, alirocumab increases the number of LDL-R available to remove LDL-C from circulation. Hence, alirocumab is an effective treatment to reduce LDL-C levels and, thus, to reduce the risk for cardiovascular disease.
Pharmacokinetics and pharmacodynamics of alirocumab have been well characterized [2]. Owing to its high specificity and affinity to its circulating target, alirocumab exhibits target-mediated drug disposition (TMDD). The pharmacokinetic effect on LDL-C is indirect, depending on free PCSK9 concentrations, until target saturation is reached. Once the binding to PCSK9 is saturated, an additional increase in the concentration of alirocumab does not lead to a further reduction in LDL-C levels. However, we can observe a prolongation of drug effect [3, 4]. Clinical studies have shown that treatment with alirocumab reduces LDL-C levels drastically (up to 62.7%) when added to background statin therapy [5,6,7,8]. To support the clinical development of alirocumab, three empirical to stepwise semi-mechanistic population pharmacokinetic models have been developed and submitted to regulatory agencies. The first model, a population model (PopPK), aimed to characterize the pharmacokinetic profile of alirocumab and identify the covariates affecting individual patient exposures. Free PCSK9 was integrated as a time-varying covariate impacting Michaelis–Menten constant (Km), therefore impacting the nonlinear clearance. This model characterized the nonlinear pharmacokinetics of alirocumab using a Michaelis–Menten approximation [9] and thus not capturing the resulting variations in PCSK9.
Second, the pharmacokinetic–pharmacodynamic (PK–PD) indirect response model allowed us to relate alirocumab concentrations to LDL-C levels and to estimate derived pharmacodynamic parameters (e.g., maximum decrease in LDL cholesterol values from baseline) [10].
The third model, a PopPK-TMDD [11] model, considered the antibody-specific mechanistic TMDD process by integrating all the information regarding the target (PCSK9) and the alirocumab–PCSK9 complex. This model was more mechanistic and allowed the best concomitant estimation of the alirocumab and PCSK9 data on the target population.
However, the modeling strategy detailed above has one main limitation: the PK–PD model is an indirect response model, considering that alirocumab concentrations (instead of PCSK9) were directly impacting LDL-C levels. The target, PCSK9, was incorporated in the model as a time-varying covariate to support a better fit of the LDL-C concentrations instead of describing its kinetics and demonstrating how it controls LDL-C fluctuation.
In this analysis, we aimed to capture a semi-mechanistic population PK–PD relationship allowing us to describe, in a single step, using the population approach, the dynamic interaction between alirocumab and its target PCSK9 as well as how PCSK9 kinetics drives LDL-C depletion and to quantify the between-subject variability at each level of the mechanism. This model would allow one to simulate different dosing regimens and to study the resulting effect.
2 Methods
2.1 Clinical Studies
The model was developed using data from nine clinical trials (phases I–III) carried out in accordance with the principles of the Declaration of Helsinki for medical research on human subjects.
The main characteristics of these clinical studies are described in Table 1. Alirocumab was administered subcutaneously (SC) either in a single dose or in repeated doses. Only one phase I study involved single-dose intravenous (IV) administration. Doses of alirocumab ranged from 0.3 to 12 mg/kg in the IV study and from 50 to 300 mg in the SC studies. In the repeated-dose studies, administrations were spaced by 2–4 weeks over a period of up to 104 weeks (2 years).
2.2 Dataset
Total alirocumab (corresponding to free alirocumab plus alirocumab–PCSK9 complex), total PCSK9 (free PCSK9 plus alirocumab–PCSK9 complex) and circulating LDL-C levels were obtained from serum samples by ELISA assays. The lower/upper limits of quantification (LLOQ/ULOQ) were 0.078/5 µg/mL for total alirocumab and 0.156/10 µg/mL for total PCSK9 in undiluted human serum. Serum lipid samples were assayed in compliance with the Centers for Disease Control and Prevention.
Subjects from clinical studies who received a placebo were excluded in the analysis. From the remaining subjects receiving alirocumab, data were excluded from the dataset if concentrations (alirocumab, PCSK9, and LDL-C) were missing, or values were below the LLOQ. Missing covariate values were replaced with the last known values for the same individual or interpolated for continuous covariates varying over time. In the case of baseline values (i.e., before first administration), the median calculated over the entire dataset was used as a substitute.
2.3 Nonlinear Mixed Effect Model
The modeling analysis was carried out using the NONMEM software version 7.4.1 (Icon Development Solutions, ICON, MD, USA) to develop a nonlinear mixed effects model. The stochastic approximation expectation–maximization (SAEM) algorithm, known for being suited for complex PK–PD problems described by differential equations with many parameters [12], was used to estimate the model parameters. The SAEM algorithm was followed by a few iterations of a final importance sampling (IMP) step for a more precise estimation of the objective function value (OFV) and standard errors. R programming [13] with RStudio version 1.2.5033 (Integrated Development for R. RStudio, PBC, Boston, MA) software was used for the preparation of the analyses (data mining and exploration) and for postprocessing of model runs. At each stage of development, the models were evaluated according to different graphical (goodness-of-fit plots) and numerical diagnostic criteria (biological plausibility of the estimated parameter values, precision of estimates) as well as data adjustment (bias, precision). Estimate precision was evaluated by the relative standard error (RSE) obtained by the method of asymptotic standard deviations implemented in NONMEM with the $COVARIANCE procedure. Bias or mean prediction error (MPE) and precision or root mean square error (RMSE) were calculated relative to the mean of the observations, according to Eqs. 1 and 2:
\(C_{{{\rm pred}_{i} }}\) and \(C_{{{\rm obs}_{i} }}\) are the individual predicted and observed concentrations, respectively, \(\overline{{C_{\rm obs} }}\) is the mean observed concentration, and N is the number of observations.
2.4 Structural model
The PK–PD structural model consists of a combination of two distinct submodels: a PK–TMDD model to describe the concentrations of total alirocumab and total PCSK9 as a function of time, and a pharmacodynamic model to describe how the drug impacts LDL-C levels as a function of free PCSK9 concentrations. To avoid overparameterization issues in the nonlinear mixed-effects modeling framework, “quasi-steady-state” (QSS)-approximated TMDD was considered instead of a full TMDD [14]. The analysis was carried out according to a one-step strategy allowing for the simultaneous estimation of pharmacokinetic, target, and pharmacodynamic parameters of the model. Additive, proportional, and combined residual error models were tested [15].
2.5 Covariate Model
For this analysis, the search for the covariate model was carried out according to a “full fixed effects model” (FFEM) approach [16]. From the structural model, the potential covariate–parameter relationships were added to the model—using fixed effects—on the basis of knowledge about the pathology, treatment, clinical interest, or even mechanistic plausibility.
The selection of covariate–parameter relationships is based on clinical relevance criteria that must be defined a priori. The statistical inference can then be established on the basis of the estimated fixed effects (θ) and their confidence intervals (i.e., uncertainty/precision of estimate), the impact of these relationships on model parameters, pharmacokinetic values [area under the curve (AUC) of concentrations, maximum concentration, or residual concentration], and pharmacodynamic values (level of a biomarker of interest) can be quantified.
From the variance–covariance matrix, 1000 parameter vectors containing all the fixed effects of the model were randomly drawn from a multinormal distribution, and then used to perform simulations and to compute the pharmacokinetic and pharmacodynamic parameters of clinical interest. Simulations were performed at the 75 mg initiation dosage every 2 weeks to estimate the AUC in the 2-week interval intake following alirocumab administration at week 10 (considered close to steady state) and the decrease from baseline in LDL-C levels at week 12.
A covariate–parameter relationship can then be categorized as clinically relevant, not clinically relevant, or not providing enough information. This approach also allows one to quantify the probability for a covariate to induce a significant clinical effect in cases of intermediate clinical relevance. Here, covariate–parameter relationships were considered as clinically relevant when impacting alirocumab exposure (± 20% change in AUC) or treatment efficacy (variation in the LDL-C decrease of ± 20%) and integrated into the model (Supplementary Material, Fig. S1).
According to the parsimony principle, a phase of reduction of the covariate model was then carried out to exclude effects that are both not statistically significant (confidence interval including the null hypothesis: no variation in the parameter of interest) and not clinically relevant (confidence interval fully included in the predefined interval as “clinically non-important”). At this step, the model was considered final.
2.6 External Validation of the Model
The final model was then validated using an external dataset obtained from four other clinical studies: one phase II and three phase III studies (Table 1) following sparse sampling collection strategies. In this dataset, individual profiles of subjects were estimated by maximum a posteriori (MAP) Bayesian method using the final model, and its predictive performance was evaluated using usual diagnostic methods (goodness-of-fit plots, bias, and accuracy).
3 Results
3.1 Available Data
The initial modeling dataset consisted of 13,835 observations including 4408 concentrations of total alirocumab, 4983 measurements of total PCSK9, and 4444 of LDL-C. Among those, 104 measurements (27 from alirocumab, 3 from PCSK9, and 74 from LDL-C) had a value lower than the LLOQ and were excluded from the analysis. The final dataset included 13,731 observations with 4381 concentrations of total alirocumab, 4980 measurements of total PCSK9, and 4370 of LDL-C from 527 individuals. The validation dataset included 29,015 observations with 9458 concentrations of total alirocumab, 9643 total PCSK9, and 9914 LDL-C from 2273 patients. Descriptive statistics of the demographic and biological characteristics of the available patients are summarized in Table 2.
3.2 Pharmacokinetic–Pharmacodynamic Model
3.2.1 Structural Model
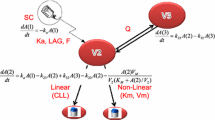
On the basis of known information on the nonlinear pharmacokinetics of alirocumab and the role of PCSK9 in LDL-cholesterol metabolism, the PK–PD model developed to describe the alirocumab/PCSK9 pharmacokinetic interaction and its lipid-lowering effect combines: (i) a PK-TMDD model considering the “quasi-steady-state” (QSS) approximation, assuming the instantaneous formation of complex between alirocumab and PCSK9, as we previously described [11], and (ii) an indirect pharmacodynamic model capturing the depleting effect of free PCSK9 on LDL-C levels. Thus, the model describes a mechanism for inhibiting the degradation of the response (i.e., the LDL-C level), or “type II” according to the classification of Sharma and Jusko [17], as a function of the concentration of free PCSK9 (Fig. 1). The model is described by a system of differential equations in Table 3.

Schematic representation of the semi-mechanistic population pharmacokinetic–pharmacodynamic model describing alirocumab, PCSK9 and LDL-C. PCSK9, proprotein convertase subtilisin kexin type 9 LDL-C; LDL-C, low-density lipoprotein cholesterol; kcp/kpc, transfer constants between the central and peripheral compartments; ka, rate constant of absorption, if subcutaneous administration; In(t), infusion rate, when administered intravenously; kel, free alirocumab elimination rate constant; SC, subcutaneous; IV, intravenous; ksyn, PCSK9 zero-order rate of synthesis; kdeg, PCSK9 first-order rate of degradation; kon, association rate constant of the drug–target complex; koff, complex dissociation rate constant; kclear, rate constant for complex clearance; Imax, maximal capacity of inhibition of LDL-C degradation; IC50, concentration of free PCSK9 necessary to reach half of Imax; γ, Hill coefficient; kout, first-order LDL-C degradation rate constant during treatment; kout(0), first-order LDL-C degradation rate constant before alirocumab
To describe the process of extravascular absorption of alirocumab, two additional parameters were necessary: absolute bioavailability (F) and lag time between the administration time and the appearance of concentrations (LAG).
All the parameters of the structural model were estimated, except peripheral volume (Vp) and complex formation constant rate (kon). These parameters were set to a fixed value following a sensitivity analysis, Vp set at 2.61 L—corresponding to a usual value of peripheral volume of distribution for a monoclonal antibody [18]—and the kon was set at 559/nM/day consistent with previous findings [11]—corresponding to very fast drug–target binding.
All parameters follow log-normal distributions except F and maximal capacity of inhibition of LDL-C degradation by PCSK9 (Imax), which represent percentages from 0% to 100% and require logit-normal distributions to constrain their estimation within this interval.
Interindividual variability was estimated for clearance, rate constant for complex clearance (kclear), PCSK9 first-order rate of degradation (kdeg), central compartment volume of distribution (Vc), rate constant of absorption (ka), F, first-order LDL-C degradation rate constant during treatment (kout), concentration of free PCSK9 necessary to reach half of Imax (IC50), and Imax.
To model the three entities (alirocumab, PCSK9, and LDL-C) simultaneously, three independent error models were used. Combined error models (additive error and proportional error) allowed best description of the residual variability.
Estimation precision (RSEs) of population parameters characterizing the structural model were less than 30% for all parameters, suggesting very precise estimation. Biases (MPE) and precisions (RMSE) of adjustment between observations and population predictions (PRED) or individual predictions (IPRED) confirm the good predictive performance of the structural model. Graphical comparisons of observations and predictions show good fitting quality of the structural model and absence of major systematic deviation, confirmed by the plots of residuals. Population parameters characterizing the structural model, biases, precisions, and goodness-of-fit plots obtained during the internal evaluation are presented in Supplementary Material (Tables S1–S2, Figs. S2–S3).
3.2.2 Covariate model
On the basis of the mechanism of action of alirocumab, the role of PCSK9 in cholesterol metabolism, and leveraged information from previously developed models for alirocumab [10, 11], the following covariate–parameter relationships were integrated a priori into the model (Eqs. 3–7):
-
Effect of statins on Vc:
$$V_{{\text{C}}} = \Theta_{{V_{{\text{c}}} }} \cdot \Theta_{{V_{{{\text{c}}_{{{\text{STATIN}}}} }} }}^{{{\text{STATIN}}}} $$(3)where \(\Theta_{{V_{{{\text{c}}_{{{\text{STATIN}}}} }} }}\) is the typical value of the effect, STATIN = 1 in case of co-administration of statin, STATIN = 0 otherwise.
-
Effect of statins on kout:
$$k_{{{\text{out}}}} = \Theta_{{k_{{{\text{out}}}} }} \cdot \Theta_{{k_{{{\text{out}}_{{{\text{STATIN}}}} }} }}^{{{\text{STATIN}}}} $$(4)where \(\Theta_{{k_{{{\text{out}}_{{{\text{STATIN}}}} }} }}\) is the typical value of the effect, STATIN = 1 in case of co-administration of statin, STATIN = 0 otherwise.
-
Effects of statins and sex on Imax :
$$I_{{{\text{max}}}} = \Theta_{{I_{{{\text{max}}}} }} + \Theta_{{I_{{{\text{max}}_{{{\text{STATIN}}}} }} }} \cdot {\text{STATIN}} + \Theta_{{I_{{{\text{max}}_{{{\text{SEX}}}} }} }} \cdot {\text{SEX}}$$(5)where \(\Theta_{{I_{{{\text{max}}_{{{\text{STATIN}}}} }} }}\) is the typical value of the effect of static co-administration (in case of co-administration of statin, STATIN = 0 otherwise), \(\Theta_{{I_{{{\text{max}}_{{{\text{SEX}}}} }} }}\) is the typical value of the effect of female (SEX = 1 for women, SEX = 0 for men).
-
Effect of baseline total PCSK9 on IC50:
$$ {\text{IC}_{50}} = \Theta_{{{\text{IC}_{50}}}} \cdot \left( {\frac{{{\text{TBSPCSK}}}}{6.99}} \right)^{{\Theta_{{{\text{IC}_{50}}_{{{\text{TBSPCSK}}}} }} }} , $$(6)where \(\Theta_{{{\text{IC}}_{50_{\text{TBSPCSK}}}}}\) is the typical value of the effect and TBSPCSK9 is the baseline total PCSK9 level (standardized to the median value in the dataset: 6.99 nM).
-
Effect of baseline total PCSK9 on kdeg):
$$k_{{{\text{deg}}}} = \Theta_{{k_{{{\text{deg}}}} }} \cdot \left( {\frac{{{\text{TBSPCSK}}}}{6.99}} \right)^{{\Theta_{{k_{{{\text{deg}}_{{{\text{TBSPCSK}}}} }} }} }} $$(7)
where \(^{{\Theta_{{k_{{{\text{deg}}_{{{\text{TBSPCSK}}}} }} }} }}\) is the typical value of the effect and TBSPCSK9 is the baseline total PCSK9 (standardized to the median value in the dataset: 6.99 nM).
Analysis of the effects of the inclusion of covariate–parameter relationships on alirocumab exposure (effect on pharmacokinetics) and on the decrease in LDL-C level (effect on pharmacodynamics) is summarized in Fig. 2. Among the six initially included covariate–parameter relationships, three were highlighted as potentially clinically relevant.

Effects of the inclusion of covariate–parameter relationships on alirocumab exposure and on the reduction of the LDL-C level. a Effect of co-administration of statins on Imax, kout, and V. b Effect of baseline total PCSK9—less than the 25th percentile (p25) or higher than the 75th percentile (p75)—on IC50 and kdeg. c Effect of sex on Imax. V, volume of distribution; kout(0), first-order LDL-C degradation rate constant before alirocumab; Imax, maximal capacity of inhibition of LDL-C degradation by PCSK9; kout, first-order LDL-C degradation rate constant during treatment; kdeg, PCSK9 first-order rate of degradation; kon, association rate constant of the drug–target complex; IC50, concentration of free PCSK9 necessary to reach half of Imax
The relationship between the co-administration of statins and the volume of distribution highlights a derived significant decrease in exposure to alirocumab (approximately −45% AUC) as well as a 42% probability of observing a resulting decrease in the lipid-lowering effect of more than 20%.
The relationship between the co-administration of statins and Imax highlights a 48% probability of observing an increase in the lipid-lowering effect of more than 20%.
The relationship between baseline PCSK9 and the concentration of free PCSK9 allowing 50% of Imax (i.e., IC50) to be reached indicates that patients with a baseline level greater than or equal to 9.10 nM (equivalent to the 75th percentile in the population and representing the hardest-to-treat population) will, on average, have a lipid-lowering effect reduced by approximately 60% compared with patients with the median rate (6.99 nM).
The relationships between sex and Imax, co-administration of statins and baseline PCSK9, and kdeg did not show any significant changes in terms of pharmacokinetics or pharmacodynamics.
Thus, only the effects of co-administration of statins on Vc and Imax and the effect of baseline PCSK9 on IC50 were retained in the final model.
3.2.3 Final Model
Population parameters of the final model are reported in Table 4. The final model integrates three parameter–covariate relationships: co-administration of statins intervening at the same time on the (i) volume of distribution and (ii) on the capacity of maximum inhibition of degradation of LDL-C (Imax) and (iii) total baseline PCSK9 acting on the concentration of free PCSK9 allowing half of the maximum effect (IC50) to be reached.
Vc is estimated to be 3.2 L in subjects treated with alirocumab alone and 5.6 L when combined with a statin. Imax is estimated at 74% in monotherapy with alirocumab and 88% in combination with a statin.
IC50 increases with total baseline PCSK9. Addition of these covariates made it possible to reduce the inter-individual variability of the parameters Vc and IC50 respectively from 31% to 26% and from 31% to 7%. In addition, a decrease of interindividual variability of clearance from 83% to 56% was obtained. RSEs of parameters were all below 30%, demonstrating good confidence in their estimation.
The additive parts of residual variability were 0.426 nM for alirocumab, 1.07 nM for PCSK9 and 5.71 mg/dL for LDL-C, corresponding to very low values compared with their respective average concentrations of 82.7 nM, 29.7 nM, and 84.4 mg/dL. The proportional parts were 25.5%, 27.9%, and 14.2% respectively. Diagnostic plots of the final model are reported in Fig. 3. The performances of the final model were evaluated by visual predictive check, stratified by study (Supplementary Material, Fig. S4).

Comparison of observations and (individual) predictions of total alirocumab (a), total PCSK9 (b), and LDL-C (c) of final model in a logarithmic scale. d Subplot of conditional weighted residuals (CWRES) versus population predictions for total alirocumab (focused on concentrations < 250 nM) of the final model. PRED population predictions, IPRED individual predictions, PCSK9 proprotein convertase subtilisin kexin type 9 LDL-C, LDL-C low-density lipoprotein-cholesterol
3.3 External Validation of the Model
The final model was then applied to the validation dataset to predict concentrations using MAP Bayesian estimation to evaluate its ability to predict data coming from external studies. Diagnostic plots show good correspondence between observed and predicted data (Supplementary Material, Fig. S5). Bias (MPE) and precision (RMSE) were evaluated for PRED/IPRED. For alirocumab MPE were −6.36%/−2.77% and RMSE were 72.1%/31.6%; for total PCSK9 MPE were −6.04%/−9.45% and RMSE 42.0%/25.6%; for LDL-C MPE were −6.35%/1.54% and RMSE 29.4%/15.7%. MPE and RMSE were close to those obtained with the modeling dataset, confirming the ability of the model to predict the concentrations of alirocumab, PCSK9, and LDL-C from an external dataset.
3.4 Simulations
Dose administration simulations on the unbound PCSK9 and, consequently, on the LDL-C lowering are presented in Fig. 4. The administration of a single dose of 150 mg of alirocumab is simulated considering a typical V for alirocumab alone (red, V = 3.2 L) or co-administered with a statin (blue, V = 5.6 L), all other model parameters being identical. The maximum LDL-C reduction is typically obtained after 150 mg Q2W or 300 mg Q4W. This maximum effect is achieved when free PCSK9 concentration is close to zero, as seen mostly after 150 mg Q2W or 300 mg Q4W.

Simulation for the effect of dose on the unbound PCSK9 and consequently on the LDL-C lowering. a Simulation of alirocumab concentration (nM) versus time (day). b Simulation of free PCSK9 concentration (nM) versus time (day). c LDL change from baseline (%) after: 75 mg subcutaneous administration, every 14 days (red line), 150 mg subcutaneous administration, every 14 days (green line), and 300 mg subcutaneous administration, every 28 days (blue line). PCSK9 proprotein convertase subtilisin kexin type 9 LDL-C, LDL-C low-density lipoprotein cholesterol
4 Discussion
From nine clinical studies, a semi-mechanistic PK–PD model of alirocumab, its interaction with PCSK9 and its LDL-C-lowering effect was developed using a population-based approach. This combines a pharmacokinetic model of the TMDD-QSS type to characterize the alirocumab–PCSK9 interaction with a turnover pharmacodynamic model of inhibition of the degradation of the response to describe the impact of circulating PCSK9 levels on the decline of LDL-C.
The major advantage of the TMDD-QSS model compared with the Michaelis–Menten model [9] is that it makes possible to predict the pharmacokinetics of a monoclonal antibody considering the quantitative information of its target by integrating them into the same physiological system describing the mechanism of their interactions: binding, dissociation, and complex internalization. Lipid-lowering effect of alirocumab has been dynamically described by TMDD: decreasing concentration of free PCSK9 promotes the degradation of LDL-C.
Unlike usual indirect PK–PD models that use the concentration of an administered drug to influence the pharmacodynamic system, this model captures the dynamics of an endogenous effector (PCSK9) with a non-null initial concentration (i.e., pretreatment) that is used to drive the effect. The model allowed an accurate description of the concentrations of all entities.
There are two ways of jointly analyzing pharmacokinetic and pharmacodynamic observations, either simultaneously or capturing sequentially: first the drug concentrations with a pharmacokinetic model, then the response measurements with a pharmacodynamic model without estimating the parameters (population parameters and individual parameters). The simultaneous approach reduces the biases associated with sequential model estimation, considers simultaneously the uncertainty of all the data, and allows drug effect (pharmacodynamics) to influence its pharmacokinetic [10]. The sequential approach, which does not consider the uncertainty of estimation of the pharmacokinetic parameters when estimating pharmacodynamics, is faster and yields results usually close to the simultaneous method. It is thus the most frequently used for PK–PD modeling.
For this study, to avoid bias in estimating the parameters of the response linked to a preconditioning of the model by an adjustment of pharmacokinetics alone, the simultaneous approach was applied. The complexity of our model leads to a major increase in computation duration compared with a sequential approach. That is why this approach was possible only because pharmacokinetics and pharmacodynamics of alirocumab had already been studied minutely.
Previously, effects of covariates were tested on pharmacokinetic parameters [11]. The only covariate–pharmacokinetic parameter relationship retained according to statistical criteria, in the final model was the effect of statin co-administration (correlated with disease state: healthy volunteers or patients) on Vc. Considering the previous knowledge about the pharmacokinetics [10, 11], the high number of covariate–parameter (pharmacokinetic or pharmacodynamic) combinations and the runtime of several hours for each run; therefore, we did not reassess the covariate model for the TMDD part of our integrated model and kept the final model previously published.
Owing to the size of the dataset, model complexity, and the very long time needed to reach the convergence, a FFEM approach was chosen for the search of the covariate model rather than a conventional step-by-step approach (or “stepwise covariate modeling,” SCM). It is particularly effective when the objective of the model is to predict data, but it frequently presents drawbacks, making it difficult to generalize the results beyond the dataset [16, 19]. In addition, it requires testing many models and therefore significant computation and postprocessing times. The FFEM method requires only two modeling steps: a first one for the complete model and a second one for the reduced model. It is therefore much faster and advantageous for models that have many parameters to estimate and/or that take a long time to converge to the solution, as for this analysis. A limitation of the FFEM approach is its lack of serendipity: only parameter–covariate relationships already anticipated or highly probable (based on a mechanistic hypothesis) are evaluated and have a chance to be included in the final model. On the basis of prior knowledge, mechanistic plausibility, and clinical relevance, the FFEM method provides a more precise and useful inference from the covariate–parameter relationships. It is therefore a method of choice during advanced stages of development that helps to clarify the practical applications of treatments: dose recommendation, dosage adjustment, or labeling specification for subpopulations. Mechanistic modeling of biological processes brings the advantage of requiring fewer explanatory covariates than more empirical approaches: as compared with the 13 covariates previously identified [9,10,11], the current model allowed the discrimination of three relevant covariate–parameter relationships explaining part of the interindividual variability in pharmacokinetics and response of the drug with a potentially significant clinical impact.
Co-administration of statins has been linked to an increase in the volume of distribution Vc already described [11, 20] and an increase in the inhibitory effect of PCSK9 on the degradation of LDL-C. Baseline total PCSK9 level was linked to an increase in IC50. These covariate–parameter relationships can be explained by a well-documented biological mechanism: stimulation of the synthesis of PCSK9 in response to treatment with statins. Statins, as inhibitors of HMG-CoA reductase involved in the synthesis of cholesterol, decrease the hepatic production of cholesterol that activates SREBP2 protein (sterol regulatory element-binding protein 2) and results in increasing the synthesis of PCSK9 and LDL-R [21]. Thus, treatment with statins can influence the response to anti-PCSK9 drugs and lead to a synergy between the two drug classes: alirocumab neutralizes the increase in expression of PCSK9 induced by statins and potentiates the action of the latter.
In the pharmacodynamic model, the lower the PCSK9, the lower the LDL-C. Imax and IC50 are two pharmacodynamic parameters that express the amplitude of LDL-C variations. The positive effects of co-administration of statins on Imax (88% versus 74% without statins) and of baseline PCSK9 on IC50 (increasing linear relationship) can be explained by overexpression of PCSK9 induced by statin treatment: internalization activity of LDL-R is strengthened, leading to less LDL-R available on hepatocytes’ surface and increases in the LDL-C level. Thus, the inhibitory effect on the degradation of LDL-C (Imax) is increased. Similarly, activation of the synthesis of PCSK9 in response to the effect of statins mechanically increases IC50. Although the dynamic pharmacodynamic model previously developed for alirocumab is different and less mechanistic (i.e. estimation of LDL-C degradation by alirocumab concentrations), the parameters describing the amplitude of the lipid-lowering effect Emax and EC50 [10] increased with the initial level of PCSK9 and the concomitant use of statins. This model also reported effects of statins on kout and sex on the maximum lipid-lowering effect (Emax). The search for covariates by FFEM approach of this study allowed to establish that effects of statins on kout and sex on Imax (whose meaning is close to Emax in this model) were not sufficient to detect an impact on the efficiency of the treatment, evaluated by the drop in LDL-C after 12 weeks.
5 Conclusion
Alirocumab is indicated in the prevention of cardiovascular risk treatment, initially at a dosage of 75 mg Q2W or 300 mg Q4W, which can be increased up to 150 mg Q2W for patients requiring greater reductions of blood LDL-C or after insufficient response to treatment, irrespective of body weight, baseline LDL-C level, or statin co-therapy. Our model allowed an accurate description of the concentrations of all entities and simulations supported this labeling. As demonstrated by the simulations the maximum LDL-C reduction is typically obtained after 150 mg Q2W or 300 mg Q4W. This maximum effect is achieved when free PCSK9 concentration is close to zero, as seen mostly after 150 mg Q2W or 300 mg Q4W, indicating that there would be no additional clinical benefit of increasing the dose higher than these recommended dosing regimens.
References
Lambert G, Sjouke B, Choque B, Kastelein JJP, Hovingh GK. The PCSK9 decade. J Lipid Res. 2012;53:2515–24.
Lunven C, Paehler T, Poitiers F, Brunet A, Rey J, Hanotin C, et al. A randomized study of the relative pharmacokinetics, pharmacodynamics, and safety of alirocumab, a fully human monoclonal antibody to PCSK9, after single subcutaneous administration at three different injection sites in healthy subjects. Cardiovasc Ther. 2014;32:297–301.
European Medecines Agency. Praluent. https://www.ema.europa.eu/en/medicines/human/EPAR/praluent. Accessed 1 Jun 2022.
FDA USF and DA. Drug Approval Package: Praluent (alirocumab). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/125559Orig1s014TOC.cfm. Accessed 1 Jun 2022.
Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99.
Roth EM, Taskinen M-R, Ginsberg HN, Kastelein JJP, Colhoun HM, Robinson JG, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized Phase 3 trial. Int J Cardiol. 2014;176:55–61.
Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–107.
Kastelein JJP, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36:2996–3003.
Martinez J-M, Brunet A, Hurbin F, DiCioccio AT, Rauch C, Fabre D. Population pharmacokinetic analysis of alirocumab in healthy volunteers or hypercholesterolemic subjects using a Michaelis–Menten approximation of a target-mediated drug disposition model-support for a biologics license application submission: part I. Clin Pharmacokinet. 2019;58:101–13.
Nicolas X, Djebli N, Rauch C, Brunet A, Hurbin F, Martinez J-M, et al. Population pharmacokinetic/pharmacodynamic analysis of alirocumab in healthy volunteers or hypercholesterolemic subjects using an indirect response model to predict low-density lipoprotein cholesterol lowering: support for a biologics license application submission: part II. Clin Pharmacokinet. 2019;58:115–30.
Djebli N, Martinez J-M, Lohan L, Khier S, Brunet A, Hurbin F, et al. Target-mediated drug disposition population pharmacokinetics model of alirocumab in healthy volunteers and patients: pooled analysis of randomized phase I/II/III studies. Clin Pharmacokinet. 2017;56:1155–71.
Bauer RJ. NONMEM tutorial part II: estimation methods and advanced examples. CPT Pharmacomet Syst Pharmacol. 2019;8:538–56.
R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2009. https://www.r-project.org/. Accessed 1 Jun 2022.
Gibiansky L, Gibiansky E. Target-mediated drug disposition model: approximations, identifiability of model parameters and applications to the population pharmacokinetic–pharmacodynamic modeling of biologics. Expert Opin Drug Metab Toxicol. 2009;5:803–12.
Proost JH. Combined proportional and additive residual error models in population pharmacokinetic modelling. Eur J Pharm Sci. 2017;109:S78-82.
Gastonguay MR. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies. https://www.page-meeting.org/?abstract=2229. Accessed 1 Jun 2022.
Sharma A, Jusko WJ. Characteristics of indirect pharmacodynamic models and applications to clinical drug responses. Br J Clin Pharmacol. 1998;45:229–39.
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:633–59.
Wählby U, Jonsson EN, Karlsson MO. Comparison of stepwise covariate model building strategies in population pharmacokinetic–pharmacodynamic analysis. AAPS PharmSci. 2002;4:E27.
Gibbs JP, Doshi S, Kuchimanchi M, Grover A, Emery MG, Dodds MG, et al. Impact of target-mediated elimination on the dose and regimen of evolocumab, a human monoclonal antibody against proprotein convertase subtilisin/kexin type 9 (PCSK9). J Clin Pharmacol. 2017;57:616–26.
Guo Y-L, Zhang W, Li J-J. PCSK9 and lipid lowering drugs. Clin Chim Acta Int J Clin Chem. 2014;437:66–71.
Acknowledgements
We thank Guilhem Darche from Sanofi R&D Montpellier for dataset creation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Sanofi.
Conflicts of interest
The authors have no conflicts of interest.
Ethics approval
All procedures of studies were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Clinical study transparency was based on high ethical standards and norms and was consistent with all relevant national and regional regulations and laws and declared with following official number: NCT01026597, NCT01074372, NCT01161082, NCT01448317, NCT01723735, NCT01288443, NCT01288469, NCT01266876, NCT01644474, NCT01623115, NCT01644188, NCT01812707, and NCT01507831.
Consent to participate
Subjects provided written informed consent in all included studies.
Consent for publication
Not applicable.
Code availability
On demand (email: David.Fabre@sanofi.com).
Availability of data
On demand (email: David.Fabre@sanofi.com).
Author contributions
P.N. was responsible for the literature review, data analysis, and data interpretation. P.N. and S.K. drafted the manuscript, tables, and the figures. N.D. and D.F. were responsible for the review of PK analyses. N.D., D.F., A.B., and S.K. were responsible for the final review of the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Nolain, P., Djebli, N., Brunet, A. et al. Combined Semi-mechanistic Target-Mediated Drug Disposition and Pharmacokinetic–Pharmacodynamic Models of Alirocumab, PCSK9, and Low-Density Lipoprotein Cholesterol in a Pooled Analysis of Randomized Phase I/II/III Studies. Eur J Drug Metab Pharmacokinet 47, 789–802 (2022). https://doi.org/10.1007/s13318-022-00787-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-022-00787-4