
Abstract
Background and Objectives
Tramadol is commonly prescribed to manage chronic pain in older patients. However, there is a gap in the literature describing the pharmacokinetic parameters for tramadol and its active metabolite (O-desmethyltramadol [ODT]) in this population. The objective of this study was to develop and evaluate a population pharmacokinetic model for tramadol and ODT in older patients.
Methods
Twenty-one patients who received an extended-release oral tramadol dose (25–100 mg) were recruited. Tramadol and ODT concentrations were determined using a validated liquid chromatography/tandem mass spectrometry method. A population pharmacokinetic model was developed using non-linear mixed-effects modelling. The performance of the model was assessed by visual predictive check.
Results
A two-compartment, first-order absorption model with linear elimination best described the tramadol concentration data. The absorption rate constant was 2.96/h (between-subject variability [BSV] 37.8%), apparent volume of distribution for the central compartment (V1/F) was 0.373 l (73.8%), apparent volume of distribution for the peripheral compartment (V2/F) was 0.379 l (97.4%), inter-compartmental clearance (Q) was 0.0426 l/h (2.19%) and apparent clearance (CL/F) was 0.00604 l/h (6.61%). The apparent rate of metabolism of tramadol to ODT (kt) was 0.0492 l/h (78.5%) and apparent clearance for ODT (CLm) was 0.143 l/h (21.6%). Identification of Seniors at Risk score (ISAR) and creatinine clearance (CrCL) were the only covariates included in the final model, where a higher value for the ISAR increased the maximum concentration (Cmax) of tramadol and reduced the BSV in Q from 4.71 to 2.19%. A higher value of CrCL reduced tramadol Cmax and half-life (T1/2) and reduced the BSV in V2/F (from 148 to 97.4%) and in CL/F (from 78.9 to 6.61%).
Conclusion
Exposure to tramadol increased with increased frailty and reduced CrCL. Prescribers should consider patients frailty status and CrCL to minimise the risk of tramadol toxicity in such cohort of patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A two-compartment, first-order absorption model with linear elimination best described the tramadol concentration data |
Prescribers should consider patients frailty (ISAR) and CrCL when prescribing tramadol for their elderly patients, as a higher value for the ISAR increased Q and reduced AUC for tramadol, whereas a higher CrCL decreased tramadol Cmax and T1/2 |
Risk of ODT toxicity should be evaluated in frail older patients as a high variability in tramadol and ODT exposure was observed among participants |
1 Introduction
Tramadol is a weak, centrally acting opioid analgesic which is widely prescribed to manage chronic pain in older patients [1]. It has a unique mechanism of action and its analgesic properties are related to both opioid and non-opioid pathways [2]. Tramadol has a multimodal mechanism as it acts on serotonin, adrenergic and opioids receptors [1, 3]. It is rapidly absorbed after oral administration and distributed throughout the body. Tramadol is metabolised in the liver through cytochrome P-450 (CYP) enzymes to produce active and inactive metabolites that are eliminated through urine [4]. CYP2D6 is responsible for the conversion of tramadol to its active metabolite, O-desmethyltramadol (ODT). ODT is mainly responsible for the opioid-related analgesia of tramadol, and its binding affinity to the mu (µ) opioid receptor is 300-fold higher than that of tramadol [5]. On the other hand, tramadol is converted into an inactive metabolite, N-desmethyltramadol (NDT), by CYP3A4 if CYP2D6 metabolic activity is low.
Chronic pain is common in older patients and this is related to various chronic conditions [1]. Clinical evidence to support evidence-based practice for managing chronic pain is limited in this population [6]. Older patients have many characteristics which may impact the pharmacokinetic (PK) and pharmacodynamic (PD) parameters of medications, including advancing age, polypharmacy and the presence of various geriatric syndromes [7]. Polypharmacy is common in older patients and is linked to an increased risk of drug-drug interactions (DDIs) and adverse drug reactions (ADRs) [8,9,10], and frailty can also impact drug pharmacokinetics and pharmacodynamics [11]. Frail patients have a reduced skeletal mass, which can impact drug distribution, and frailty increases physiological heterogeneity among older patients, particularly regarding the therapeutic and adverse effects associated with the use of medications [1].
Population pharmacokinetic (PopPK) modelling enables parameter estimation and concentration time-course prediction for both the study population and individual subjects simultaneously [12]. Understanding the pharmacokinetics and pharmacodynamics of tramadol and ODT after an extended release oral dosing may aid appropriate use in older patients regarding appropriate drug selection and dosing [13]. Numerous PopPK models for tramadol and its active metabolites have been reported where pharmacokinetic parameters have been described in different cohorts of patients [14,15,16], but none have focussed on older patients.
To contribute to this understanding, the aims of this study were to develop and evaluate a PopPK model for tramadol and ODT for older patients, describe the relationship between patients’ characteristics and the tramadol and ODT concentration-time profile and determine the influence of patients’ characteristics on tramadol and ODT pharmacokinetics.
2 Methods
2.1 Study Population
Patients presenting to the Royal Adelaide Hospital (RAH) between 15 June and 31 November 2019 were considered for inclusion. The inclusion criteria were patients aged ≥ 65 years and prescribed an extended-release oral tramadol dose of between 25 and 100 mg. Patients in intensive care units or those deemed unable to provide informed consent were not considered for inclusion.
2.2 Study Design and Data Collection
Once consent was collected, up to five finger prick blood samples were collected using volumetric absorptive micro-sampling (VAMS) devices from each participant at different time points not exceeding 12 h after the dose administration. The precise dosing and sampling times were collected. VAMS were left open in air for 4 h to dry before storing at − 80 °C until analysis.
The following patient data were collected from the electronic patient administration system or paper-based medical records: demographic information including age, sex, body weight and height; admission reason and comorbidities; concurrent prescribed medications; clinical biochemistry data including liver enzymes and serum creatinine concentration. Creatinine clearance (CrCL) was calculated using the Cockcroft-Gault formula [17]. Charlson Comorbidity Index (CCI) was calculated to predict participants’ 1-year mortality risk [18], and the Identification of Seniors At Risk (ISAR) tool was used to estimate participants’ frailty level [19]. The total number of prescribed medications (NPM) was determined as the total number of pre-admission or recently administered medications. Medications known to induce or inhibit the activity of cytochrome P450 CYP3A4 and CYP2D6 were identified based on available literature [20,21,22]. The ratio of ODT to tramadol concentration at steady state was determined by collecting a sample at trough (before next dose) from patients who were on long-term tramadol therapy.
Tramadol dose was standardised to 25 mg to allow a better visual presentation and comparison of tramadol and ODT concentration over time between participants, but modelling used the actual doses. Participants were allocated into different groups based on their defined characteristics. Patients were categorised according to their ISAR as fit (ISAR score 0–2) or frail (3 and above), mortality risk was categorised as moderate (CCI 0–5) and high (CCI ≥ 6), and polypharmacy was defined as present (prescribed five or more medications) or absent [23]. Graphical presentation of tramadol and ODT exposure and correlation between tramadol/ODT concentration ratio at steady state and patient characteristics were presented using (ggplot2) and (GGally) packages in R, where correlation was considered strong (r ≥ 0.5), weak (r = 0.2–0.5) and not correlated (r < 0.2). Outliers were assessed through visual presentation before the model-building step and from the weighted residuals of the model fit. A conditional weighted residual (CWRES) of > 6 or < − 6 was set to determine an outlier, and when one was identified it removed and the model refitted [24].
2.3 Tramadol and O-desmethyltramadol Assay
Tramadol and ODT were quantified using a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) method [25, 26]. In brief, the assay includes VAMS extraction with methanol and protein precipitation by 100% acetonitrile. High-pressure liquid chromatography (HPLC) was utilized, as analytes were separated using C-18 column with trimethylsilyl end-capping (Acquity). Analytes were detected and measured using a triple quadrupole mass spectrometer (Shimaszu 8030). Deuterated tramadol (tramadol-D6) was used as an internal standard. The ion transitions were tramadol (264.1 > 58.1), O-desmethyltramadol (250.1 > 58.1) and tramadol—D6 (270.1 > 64.1). Tramadol and ODT concentrations were determined by back calculation from standard curves (range from 10 to 750 ng/ml) with linear regression and double-weighting factor. The limit of quantitation was 10 ng/ml and the mean recovery from plasma was within a range of 7% and 19% for tramadol and ODT, respectively. The inter- and intra-assay precision and accuracy were assessed over five occasions with six replicates, using quality-controlled samples prepared in advance. The intra-assay accuracy was within 13% range for both analytes, where the intra-assay precision was within 4% and 9% range for tramadol and ODT, respectively. The inter-assay accuracy was within 8% and 13% range, where the inter-assay precision was within 13% and 12% range for tramadol and ODT, respectively.
VAMS were validated by two steps: correlating the concentration in blood samples to plasma samples and then determining the extractions coefficient of VAMS samples to blood samples. The ratio differences of tramadol and ODT concentration in blood samples to plasma samples were measured to be within a range of 12% and 13%, respectively, where the extraction coefficients of VAMS samples to blood samples were within a range of 18% and 19% for tramadol and ODT, respectively.
2.4 Population Pharmacokinetic Modelling
Population pharmacokinetic (PopPK) analysis was performed using the non-linear mixed effect modelling software Monolix, 2020, which utilises the stochastic approximation expectation maximisation algorithm (SAEM). The model-building process involved identification and development of a structural base model describing tramadol concentration data (step 1), development of a joint model for tramadol and ODT after fixing tramadol parameters obtained from step 1, addition of statistically significant patient characteristics as covariates and finally evaluation and validation of the model.
2.4.1 Base Structural Model Development for Tramadol
One- and two-compartment models with zero and first-order absorption, with and without a lag time (Tlag), and linear and non-linear elimination pharmacokinetic models were tested. The pharmacokinetic models estimated the following parameters: apparent clearance (CL/F), apparent central volume of distribution (V1/F), first-order absorption rate constant (ka), zero-order absorption rate constant (k0), inter-compartmental clearance (Q) and apparent peripheral volume of distribution (V2/F). For the residual unexplained variability, constant, proportional and combined models were evaluated. Between-subject variability (BSV) included in the model was assumed to have log-normal distribution (Eq. 1)
where \({\theta }_{i}\) is the individual parameter value for the \(\mathfrak{i}\)th individual, \({\theta }_{\mathrm{pop}}\) is the population parameter value, and \({\eta }_{i}\) is an independent random variable with a mean of zero and variance of \({\omega }^{2}\).
The selection criteria for the structural models were a lower Akaike information criterion (AIC), the precision of the parameter estimation expressed as the relative standard error, the final parameter estimate, physiological plausibility and goodness of fit (GOF) plots that included the observed versus predicted concentration, residual plots and visual predictive check (VPC) [27].
2.4.2 Joint Model for Tramadol and O-desmethyltramadol Development
A joint model for parent-metabolite pharmacokinetics was used as proposed by Bertrand et al. [28] and as presented in Fig. 1. In the two-compartment model the dose was absorbed into the parent compartments (central or peripheral) at a first-order rate constant, and the parent drug tramadol was either eliminated from the system with a clearance CL or transformed with a clearance (kt) into a metabolite, ODT, which was eliminated from the system with a clearance CLm. A similar approach was reported in previous PopPK models which investigated PopPK of nelfinavir and its metabolite and zidovudine and its metabolite [29, 30]. Since tramadol was orally administered, only first-order transformation rate constant, V/F and CL/F were identifiable. For identifiability purposes, the fraction of dose available after absorption (F) was set to 1.

Scheme of the model used to describe the tramadol and O-desmethyltramadol plasma concentration-time profiles. Ka first-order absorption constant, V1 volume of distribution in centeral compartment, V2 volume of distribution of peripheral compartment, Q inter-compartmental clearance, CL clearance, Kt rate of biotransforimg parent to metabolite, CLm clearance of metabolite
Notably, all pharmacokinetic parameters could not be estimated in the present study, since only oral administration of the parent drug was performed. Estimating all pharmacokinetic parameters requires that both the parent drug and the metabolite be given by the intravenous route in addition to the oral administration of the parent compound [31]. For ODT, since no urinary concentration data were available, and because no literature data were used, only volume of distribution (Vm/(Fkm)) and clearance (CLm/(Fkm)) could be determined. The fraction of tramadol metabolised to ODT (Fm = km/k) does not appear explicitly in these equations and was not identifiable, in which km is defined as the rate of metabolite formation and k is the rate of absorption for the parent drug [32, 33].
The model could be re-parameterised using Fm, whereby the corresponding identifiable parameters for ODT were Vm/(Fkm) and CLm/(Fkm). PopPK parameters of tramadol obtained from the base model were fixed in the joint model to avoid shifting of the parent values as an effect of fitting the metabolite. The residual unexplained variability and subject variability (BSV) were modelled as per the parent drug.
2.4.3 Covariate Model
Once a base parent-metabolite joint model was established, demographic and clinical variables were screened as potential covariates. The tested covariates included age, sex, body weight, NPM, ISAR, CCI, CrCL and concurrent use of CYP3A4 and CYP2D6 inhibitors or inducers.
The covariate screening process was performed using both visual and numerical approaches. For the visual assessment, covariate versus parameter scatter plots were used for continuous variables, whereas box plots were used for the categorical variables. The − 2 log-likelihood (− 2LL) was computed and used to discriminate between models. A P-value of < 0.05 was considered statistically significant for inclusion of covariates in the model by stepwise addition, and a P-value change of < 0.01 was used for backward deletion of covariates from a full final model [27].
2.4.4 Model Evaluation
GOF plots were first used as an indicator of suitability, including the representation of model-based individual predictions and population predictions versus the observed concentration. VPC was constructed using the 10th, 50th and 90th percentiles of the observed data to evaluate the simulation performance of the final model.
2.5 Simulation of Tramadol Exposure
Predicted tramadol exposure was simulated using the final model parameters using MIxplore 2020 to examine the influence of key covariates.
2.6 Compliance with Ethical Standards
This study was approved by the Human Research Ethics Committee of the Central Adelaide Local Health Network (R20190105) and the Human Ethics Committee at University of South Australia (202316). All participants were deidentified or anonymous.
3 Results
3.1 Patient Characteristics
The study included 21 patients who were taking tramadol at a dose of 25 mg (n = 3), 50 mg (n = 6), 75 mg (n = 1) and 100 mg (n = 11). Patient characteristics are presented in Table 1. All participants presented with normal liver function tests, including alanine transaminase, aspartate transaminase and gamma glutamyl transpeptidase. The most reported admission reasons were infections, falls and musculoskeletal pain.
3.2 Tramadol and ODT Exposure
Ninety-nine blood samples were included in this study—all samples were above the lower limit of quantification of the assay. Figure 2 shows variability in tramadol exposure between participants, where two patients were notably exposed to higher ODT concentrations compared to the remainder of the cohort.

Observed individual concentrations of tramadol and O-desmethyltramadol. Tramadol dose was standardised to 25 mg. Sex: • = female, ▲ = Male. Each colour represents an individual participant’s data
Higher exposure to tramadol was associated with an increased ISAR, reduced CrCL and concurrent intake of a CYP3A4 inhibitor (Fig. 3). Figure 4 shows that exposure to ODT was higher in those who were concurrently using a CYP3A4 inhibitor.

Relationships between participants’ characteristics and tramadol exposure. Frailty: fit; 0–2 Identification of Senior At Risk (ISAR), frail; ISAR ≥ 3. Mortality risk: moderate; CCI 0–5, high; CCI ≥ 6. Polypharmacy: not present; < 5 medications, present; ≥ 5 medications. Tramadol dose was standardised to 25 mg. CCI Charlson Comorbidity Index

Relationships between participants’ characteristics and O-desmethyltramadol exposure. Frailty: fit; 0–2 Identification of Senior At Risk (ISAR), frail; ISAR ≥ 3. Mortality risk: moderate; CCI 0–5, high; CCI ≥ 6. Polypharmacy: not present: < 5 medications, present; ≥ 5 medications. Tramadol dose was standardised to 25 mg. CCI Charlson Comorbidity Index
Trough samples were collected from 17 patients and the observed ODT/tramadol concentration ratio at steady state was 0.26 (0.08–0.82) (Table 1). Positive correlations between CCI and NPM (r = 0.52, P = 0.02), weight and CrCl (r = 0.66, P = 0.001), age and CCI (r = 0.62, P = 0.003) and age and NPM (r = 0.46, P = 0.04), but negative correlations between ISAR and ratio (r = − 0.51, P = 0.02) and ISAR and CrCL (r = − 0.45, P = 0.04) were apparent (Fig. 5).

Correlation between participant characteristics. Ratio ODT/tramadol concentration ratio at steady state, CrCL creatinine clearance, ISAR Identification of Senior at Risk, CCI Charlson Comorbidity Index, NPM Number of prescribed medications, X3A4.inh CYP3A4 inhibitor, X2D6.inh CYP2D6 inhibitor
3.3 Population Pharmacokinetic Parameters of Tramadol and O-desmethyltramadol
The development of the structural model suggested that the pharmacokinetics of an extended-release oral tramadol dose was best described by a model with first-order absorption, two compartments and linear elimination. This was confirmed by the GOF plots and a statistically significant drop in the AIC with respect to zero-order absorption with/without a Tlag, a one-compartment and/or nonlinear elimination models (Supplementary Materials). A model with a first-order metabolism rate constant, one compartment and linear elimination was selected for ODT. Proportional error models were used for residual variability for tramadol and a combined residual model for ODT.
After covariate testing (Supplementary Materials), CrCL and ISAR were the only covariates which demonstrated a significant relationship with the PK parameters (Table 2).
The final estimated pharmacokinetic parameters for tramadol and ODT in older patients are presented in Table 3. A relationship between ISAR and tramadol inter-compartmental clearance and between CrCL and CL/F and V2/F was observed, as a higher value of ISAR increased tramadol inter-compartmental clearance and reduced the BSV for tramadol inter-compartmental clearance. A higher value of CrCL increased CL/F and V2/F and reduced the BSV for CL/F and V2/F (Table 3). The shrinkage values for the estimated parameters were between − 5.39 and 12.4%, whereas the condition number for the final model was 210 (Supplementary Materials).
3.3.1 Model Evaluation
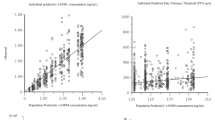
Except for the first-order absorption rate constant, the relative standard error (RSE), reported as a percentage, revealed that all parameters were precisely estimated (Table 3). A higher RSE for the first-order absorption rate constant was accepted as the data had limited sampling in the absorption phase. In addition, the VPC revealed a good correspondence between the percentile intervals obtained by simulation in the final model with those of the observed data (Fig. 6). Diagnostic GOF plots for the final covariate joint model are shown in Figs. 7, 8 and 9 and demonstrate that the final pharmacokinetic model described the measured concentrations adequately.

Visual predictive check (VPC) for tramadol (upper) and O-desmethyltramadol (lower) concentration versus time based on 1000 Monte Carlo simulations. The solid blue lines represent the 10th, 50th and 90th percentiles of the observed data. The shaded regions represent the 90% confidence intevals around the 10th, 50th and 90th percentiles of the simulated data. The blue circles are observed concentration

Goodness-of-fit (GOF) plots of observed versus predicted concentrations obtained from the final model for tramadol (top) and O-desmethyltramadol (bottom). (Right) Individual predicted concentrations versus observed concentration. (Left) Population predicted concentration versus observed concentrations. The yellow curve represents the spline line of the plot

Goodness-of-fit (GOF) plots of the residual obtained from the final model for tramadol. (Top) Residuals versus time with on left population residuals and on right individual residuals; (bottom) residuals versus predicted tramadol concentrations with on left population residuals and on right individual residuals. The yellow curve represents the spline line of the plot

Goodness-of-fit (GOF) plots of the residual obtained from the final model for O-desmethyltramadol. (Top) Residuals versus time with on left population residuals and on right individual residuals; (bottom) residuals versus predicted O-desmethyltramadol concentrations with on left population residuals and on right individual residuals. The yellow curve represents the spline line of the plot
3.4 Simulation of Tramadol Exposure versus ISAR and CrCL
Figure 10 clearly shows that increasing ISAR (frailty) was associated with a reduced tramadol Cmax, which could be explained by an increasing tramadol inter-compartmental clearance. Figure 11 demonstrates that decreasing CrCL is associated with increased tramadol maximum concertation as a result of decreased CL/F. The figure also shows that half-life (T1/2) was reduced with increasing CrCL.

Simulated exposure of tramadol with different doses at different range of ISAR (fit = 0, frail = 3 and severe frail = 6). Tramadol doses are presented with line colour; red = 100 mg, black = 50 mg. ISAR Identification of Seniors at Risk

Simulated exposure of tramadol with different doses at different range of creatinine clearance (CrCL 30, 75 and 120 ml/min). Tramadol doses are presented with line colour; red = 100 mg, black = 50 mg
4 Discussion
This study has presented a PopPK model to describe the concentration of tramadol and its active metabolite (ODT) following administration of an extended-release oral dose in older patients. A two-compartment model with first-order absorption and linear elimination was the best to fit to the tramadol concentration data, where a first-order metabolism rate constant (kt), one-compartment and linear elimination model was the best to fit ODT concentration data. ISAR and CrCL were the only covariates included in the final model, where a higher value for the ISAR decreased tramadol Cmax and reduced the BSV in inter-compartmental clearance. A higher value of the CrCL reduced tramadol Cmax and T1/2 and reduced the BSV in V2/F and CL/F.
The two-compartment model that was used to describe the tramadol data is similar to previously reported models describing tramadol pharmacokinetic parameters in a paediatric population [14]. However, other models have used between one and five compartments to describe the tramadol concentration data [15, 16]. These referenced models investigated the pharmacokinetics of tramadol in breast milk or the effect of CYP2D6 genotyping on tramadol pharmacokinetic parameters. A possible reason for this difference could be related to the limited sampling time schedules before and immediately after the maximum concentration of tramadol in blood in the present study, which may mask full determination of more complex multi-compartmental distribution.
A first-order absorption constant best described tramadol absorption in this model. The estimated value in this study is considered higher compared to previous published models [15, 16, 34]. One explanation for this difference could be related to the age of the cohort included in this study. With advancing age, absorption may be reduced as result of altered gastric acidity, delayed gastric emptying, longer intestinal transit time and reduced blood flow to the small and large intestines [35]. Another explanation could be related to the limited number of samples collected during the absorption phase, which could limit the ability to accurately determine the absorption rate constant. In addition, the RSE% value for the first-order absorption rate constant and its BSV were high (63.7% and 87.2%), suggesting that there was limited information in the dataset to estimate these parameters. However, this study involved testing different models with first- or zero-order absorption rate constant with/without a Tlag during structural base model development. A first-order absorption rate constant best described the tramadol concentration data as the AIC value was the lowest compared to the other tested models.
The V/F for the central and peripheral compartments for tramadol and the inter-compartmental tramadol clearance were smaller in this study compared to the values reported in a previously published model [36]. The bioavailability of tramadol was determined in a previous study to be between 68 and 84%, as tramadol is subject to first pass metabolism [37]. This is important to consider when prescribing tramadol for older patients, as these patients often possess many physiological changes and take more concurrent medications than younger adults, which could influence tramadol disposition. Polypharmacy and reduced liver size could interfere with the absorption and bioavailability of tramadol [7]. Frailty could also be a marker of greater physiological heterogeneity between older patients. It is important to note that frailty (as measured by ISAR) had a relationship with the inter-compartmental tramadol clearance in the presently reported model. This is important to consider, as the simulated tramadol exposure showed that tramadol Cmax was reduced with increasing ISAR as result of increasing inter-compartmental tramadol clearance. Another reason for the low estimated values for tramadol distribution volumes could be related to the two-compartment model, as one compartment could present a higher value for the volume of distribution [16].
Frailty in older patients develops as result of advancing age and the presence of multiple comorbidities, combined with polypharmacy, which alters homeostasis and results in increased risk to stress and adverse effects such as falls and delirium. Generally, 10% of older persons aged ≥ 65 years are defined as frail, and this percentage raises to nearly half of all people > 85 years [11, 38]. For example, gentamicin clearance was reduced in frail older patients compared to fit older patients [39]. It was recommended that gentamicin prescribing guidelines should address frailty status when prescribing gentamicin to reduce the risk of its toxicity. As this study identified that ISAR (frailty) alters tramadol pharmacokinetic, prescribers should pay close attention when prescribing tramadol for this cohort of patients to avoid risk of tramadol toxicity.
The condition number of the model is considered slightly high, suggestive of over-parameterisation, but the parameter correlation coefficients were all < 0.69 and the precision of the parameter estimates was good. This suggests the parameter estimates are reliable for the intended purpose. Note that a high condition number does not affect the simulation performance of a model and hence the major conclusions of this part of the work.
This study showed a highly variable tramadol exposure amongst participants. Toxicity with tramadol is common in clinical practice, and most of the pharmacological activity of tramadol is accounted for by its active metabolite ODT [3]. An advantage of this model compared to the previous reported models is the ability to predict ODT pharmacokinetic parameters. Tramadol (and ODT) inhibits the reuptake of noradrenaline and serotonin, stimulates dopamine receptors and inhibits gamma amino butyric acid release in the central nervous system [1, 3]. In older patients, this is crucial to understand as polypharmacy is common and can potentiate the risk of DDIs and ADRs. Therefore, concurrent intake of CYP2D6 inhibitors or inducers could potentiate or block tramadol conversion into ODT and this could increase or decrease efficacy/ADRs of the prescribed tramadol. Literature describing the minimal tramadol or ODT concentrations associated with ADRs is scarce, but the high variability seen here may, particularly in frail participants, contribute to the risk of tramadol or ODT-related ADRs in some patients.
The CL/F value estimated in this model was low compared to the previously reported models [15, 16, 34], which may point to a higher risk of tramadol toxicity and ADRs in this frail, elderly cohort. Since CrCL was associated with V2/F and CL/F in the final model, those with poorer renal function (as determined by CrCL) may have even higher tramadol exposure, higher Cmax and extended T1/2, potentially increasing the risk of ADRs further.
The rates of tramadol metabolism (kt) to ODT and CLm/F for ODT were 0.0492 L/h (RSE 21.8%) and 0.143 l/h (11.9%), respectively. The BSV value for CLm represents a relatively low variability among participants; however, a high variability was reported for kt in this cohort. This has implications when considering the risk of toxicity mediated by ODT. A previous study reported that the therapeutic serum concentration of ODT required to produce analgesia was 84 µg/l [5]. This is important to consider in this study as ODT concentration was around 100 µg/l (± 10) in patients received 100 mg of tramadol.
The observed ODT/tramadol concentration ratio at steady state had high variability among participants (0.26 ± 0.2). This is the first study reporting this ratio in such a cohort of frail, elderly patients. Metabolite/parent ratio is one way to assess the toxicity risk of a drug, as a ratio of ≥ 0.1 demonstrates a high risk of toxicity [40]. The reported value in this study raises a concern regarding toxicity associated with tramadol, not only because the ratio is double the accepted level but also considering that ODT is 100 times more potent than tramadol. In addition, the model did not identify any participants characteristics which could account for this high variability. Another potential reason for this high variability could be related to the effect of aging and co-administered medicines on drug transporters. ODT is a substrate of the organic cation transporter 1 (OCT1). Polymorphisms in OCT1 are associated with significant changes in the concentration of ODT in the plasma of healthy participants [41]. Robertson et al. reported a high variability in ODT plasma concentration in older participants, and they proposed this variability was related to age effects on OCT1 expression [42].
Strengths of the present study are the inclusion of an older population that included a broad range of characteristics. This provides insights regarding tramadol and ODT exposure, the relationship between exposure and participants characteristics and the relationship between these characteristics and the pharmacokinetic parameters of tramadol and ODT. This study also is the first to report ODT/tramadol concentration ratio at steady state in older patients.
However, data regarding ADRs were not available, which limits the full understanding of the observed high variability in tramadol and ODT exposure and the risk of ADRs. A higher number of participants could improve the ability to characterise relationships between participant characteristics and tramadol and the pharmacokinetic parameters of tramadol and ODT. Due to the nature of the sample collection, samples during the early absorption phase were not obtained for some patients.
The reduced CL/F, V1/F and V2/F of tramadol may be important to consider for clinical practice. Tramadol is hepatically metabolised to active (ODT, via CYP2D6) and inactive (via CYP3A4) metabolites and renally cleared. So far, the effect of aging on hepatic clearance is yet not fully determined, but it is clear that aging reduces overall renal function [7]. Moreover, reduced CrCL was associated with increased tramadol exposure, Cmax and T1/2, where increased frailty reduced exposure and increased inter-compartmental tramadol clearance. It is important to consider these characteristics when prescribing tramadol for older patients to minimise the risk of toxicity and ADRs.
Future studies should have expanded recruitment criteria and a larger sample size and include additional samples during the absorption and distribution phase and data on efficacy and ADRs. A pharmacokinetic and pharmacodynamic population (PopPK/PD) model is required to determine the concentration-analgesic response relationship, which will help to more precisely determine the risk:benefit profile of oral sustained release tramadol in frail, elderly patients.
5 Conclusions
In conclusion, this study described the pharmacokinetic parameters of tramadol and its active metabolite (ODT) in older patients. A two-compartment model with first-order absorption and linear elimination was the best model to describe the observed tramadol concentrations, where a one-compartment model with first-order metabolism with a linear elimination model best described ODT concentrations. CrCL and frailty were the only covariates included in the final model, as increased frailty reduced tramadol exposure and reduced CrCL increased tramadol exposure. A relatively high variability among participants was reported regarding tramadol exposure and ODT/tramadol ratio at steady state, which warns us about the risk of unpredictable toxicity in frail elderly patients.
References
McLachlan AJ, Bath S, Naganathan V, Hilmer SN, Le Couteur DG, Gibson SJ, et al. Clinical pharmacology of analgesic medicines in older people: impact of frailty and cognitive impairment. Br J Clin Pharmacol. 2011;71(3):351–64.
Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an’atypical’opioid analgesic. J Pharmacol Exp Ther. 1992;260(1):275–85.
Rahimi HR, Soltaninejad K, Shadnia S. Acute tramadol poisoning and its clinical and laboratory findings. J Res Med Sci. 2014;19(9):855.
Scott LJ, Perry CM. Tramadol: a review of its use in perioperative pain. Drugs. 2000;60(1):139–76.
Grond S, Meuser T, Uragg H, Stahlberg HJ, Lehmann KA. Serum concentrations of tramadol enantiomers during patient-controlled analgesia. Br J Clin Pharmacol. 1999;48(2):254–7.
Chung J. Geriatric clinical pharmacology and clinical trials in the elderly. Transl Clin Pharmacol Therapeut. 2014;22(2):64–9.
Sadean MR, Glass PS. Pharmacokinetics in the elderly. Best Pract Res Clin Anaesthesiol. 2003;17(2):191–205.
Hajjar ER, Cafiero AC, Hanlon JT. Polypharmacy in elderly patients. Am J Geriatr Pharmacother. 2007;5(4):345–51.
Al-Qurain AA, Gebremichael LG, Khan MS, Williams DB, Mackenzie L, Phillips C, et al. Prevalence and factors associated with analgesic prescribing in poly-medicated elderly patients. Drugs Aging. 2020;37(4):291–300.
Al-Qurain AA, Gebremichael LG, Khan MS, Williams DB, Mackenzie L, Phillips C, et al. Opioid prescribing and risk of drug-opioid interactions in older discharged patients with polypharmacy in Australia. Int J Clin Pharm. 2020;2020:18.
Clegg A, Young J, Iliffe S, Rikkert MO, Rockwood K. Frailty in elderly people. Lancet. 2013;381(9868):752–62.
Hughes JH, Phelps MA, Upton RN, Reuter SE, Gao Y, Byrd JC, et al. Population pharmacokinetics of lenalidomide in patients with B-cell malignancies. Br J Clin Pharmacol. 2019;85(5):924–34.
Prostran M, Vujovic KS, Vuckovic S, Medic B, Srebro D, Divac N, et al. Pharmacotherapy of pain in the older population: the place of opioids. Front Aging Neurosci. 2016;8:144.
Bressolle F, Rochette A, Khier S, Dadure C, Ouaki J, Capdevila X. Population pharmacokinetics of the two enantiomers of tramadol and O-demethyl tramadol after surgery in children. Br J Anaesth. 2009;102(3):390–9.
Salman S, Sy S, Ilett KF, Page-Sharp M, Paech MJ. Population pharmacokinetic modeling of tramadol and its O-desmethyl metabolite in plasma and breast milk. Eur J Clin Pharmacol. 2011;67(9):899–908.
Lee J, Yoo H, Bae J, Lee S, Shin K. Population pharmacokinetic analysis of tramadol and O-desmethyltramadol with genetic polymorphism of CYP2D6. Drug Des Dev Ther. 2019;13:1751–61.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373–83.
Dendukuri N, McCusker J, Belzile E. The identification of seniors at risk screening tool: further evidence of concurrent and predictive validity. J Am Geriatr Soc. 2004;52(2):290–6.
Flockhart D. Drug interactions: cytochrome P450 drug interaction table. 2007. https://drug-interactions.medicine.iu.edu/MainTable.aspx. Accessed 2010.
Medicine So. P450 drug interactions, abbreviated "Clinically Relevant" Table. 2018. https://drug-interactions.medicine.iu.edu/Clinical-Table.aspx. Accessed 1 Feb 2019.
Handbook AM. Australian medicines handbook Australian Medicines Hand. 2017.
Wallace J, Paauw DS. Appropriate prescribing and important drug interactions in older adults. Med Clin North Am. 2015;99(2):295–310.
Mould D, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development—part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometr Syst Pharmacol. 2013;2(4):1–14.
Al-Qurain AA, Williams DB, Mackenzie L, Roberts MS, Wiese MD. Simultaneous LC-MS/MS quantification of oxycodone, tramadol and fentanyl and their metabolites (noroxycodone, oxymorphone, O-desmethyltramadol, N-desmethyltramadol, and norfentanyl) in human plasma and whole blood collected via venepuncture and volumetric absorptive micro sampling. J Pharm Biomed Anal. 2021;2021:114171.
Shah VP, Midha KK, Findlay JW, Hill HM, Hulse JD, McGilveray IJ, et al. Bioanalytical method validation—a revisit with a decade of progress. Pharm Res. 2000;17(12):1551–7.
Al-Qurain AA, Upton R, Williams DB, Mackenzie L, Phillips C, Russell PT, et al. Population pharmacokinetic model of subcutaneous fentanyl in older acute care patients. Eur J Clin Pharmacol. 2021;2021:1–12.
Bertrand J, Laffont CM, Mentre F, Chenel M, Comets E. Development of a complex parent-metabolite joint population pharmacokinetic model. AAPS J. 2011;13(3):390–404.
Panhard X, Goujard C, Legrand M, Taburet AM, Diquet B, Mentre F, et al. Population pharmacokinetic analysis for nelfinavir and its metabolite M8 in virologically controlled HIV-infected patients on HAART. Br J Clin Pharmacol. 2005;60(4):390–403.
Mentre F, Escolano S, Diquet B, Golmard JL, Mallet A. Clinical pharmacokinetics of zidovudine: inter and intraindividual variability and relationship to long term efficacy and toxicity. Eur J Clin Pharmacol. 1993;45(5):397–407.
Cheng H, Jusko WJ. Pharmacokinetics of reversible metabolic systems. Biopharm Drug Dispos. 1993;14(9):721–66.
Houston JB. Drug metabolite kinetics. Pharmacol Ther. 1981;15(3):521–52.
Rowland M, Tozer TN. Clinical pharmacokinetics/pharmacodynamics: Lippincott Williams and Wilkins Philadelphia; 2005.
Murthy BP, Skee DM, Danyluk AP, Brett V, Vorsanger GJ, Moskovitz BL. Pharmacokinetic model and simulations of dose conversion from immediate-to extended-release tramadol. Curr Med Res Opin. 2007;23(2):275–84.
Hutchison LC, O’Brien CE. Changes in pharmacokinetics and pharmacodynamics in the elderly patient. J Pharm Pract. 2007;20(1):4–12.
Allegaert K, Holford N, Anderson BJ, Holford S, Stuber F, Rochette A, et al. Tramadol and o-desmethyl tramadol clearance maturation and disposition in humans: a pooled pharmacokinetic study. Clin Pharmacokinet. 2015;54(2):167–78.
Faria J, Barbosa J, Moreira R, Queirós O, Carvalho F, Dinis-Oliveira RJ. Comparative pharmacology and toxicology of tramadol and tapentadol. Eur J Pain. 2018;22(5):827–44.
van Deudekom FJ, van de Ruitenbeek M, Te Water W, Smit JM, van Munster BC. Frailty index and frailty phenotype in elderly patients with cancer. Acta Oncol. 2016;55(5):644–6.
Hilmer SN, Tran K, Rubie P, Wright J, Gnjidic D, Mitchell SJ, et al. Gentamicin pharmacokinetics in old age and frailty. Br J Clin Pharmacol. 2011;71(2):224–31.
Obach RS, Lin J, Kimoto E, Duvvuri S, Nicholas T, Kadar EP, et al. Estimation of circulating drug metabolite exposure in human using in vitro data and physiologically based pharmacokinetic modeling: example of a high metabolite/parent drug ratio. Drug Metab Dispos. 2018;46(2):89–99.
Tzvetkov MV, Saadatmand AR, Lötsch J, Tegeder I, Stingl JC, Brockmöller J. Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin Pharmacol Ther. 2011;90(1):143–50.
Robertson SS, Mouksassi MS, Varin F. Population pharmacokinetic/pharmacodynamic modeling of O-desmethyltramadol in young and elderly healthy volunteers. Drugs Aging. 2019;36(8):747–58.
Acknowledgements
The authors thank the medical and nursing staff at the Royal Adelaide Hospital, particularly Dr. Patrick Russel, for their endless support and help while recruiting patients and collecting samples. The authors also thank Dr. Lorraine Mackenzie, Dr. Craig Phillips, Dr. Desmond Williams and Dr. Ahmad Abuhelwa for their help and support through early stages of model development and paper drafting.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Authors’ contributions
AA, AlQurain: Planning, Data collection, Sample analysis, Model development, writing first draft, Evaluate and modify final manuscript. RN. Upton: Model development, Evaluating subsequent drafts and the final manuscript. R. Tadros: Patient recruitment, Evaluating final manuscript. MS, Roberts: Planning, Evaluating the final manuscript. MD, Wiese: Planning, Sample analysis, Writing and Evaluating the subsequent drafts and the final manuscript, Supervisor.
Conflict of interest
The authors have no potential conflicts of interest that might be relevant to the contents of this manuscript.
Code availability
Not applicable.
Consent for publication
Not applicable.
Funding
No source of funding was used to conduct this study or prepare the manuscript.
Ethical approval and informed consent
The study was approved by the Central Adelaide Local Health Network Ethics Committee and University of South Australia Human Ethics Committee. All participants were de-identified or anonymous.
Availability of data and material
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Al-Qurain, A.A., Upton, R.N., Tadros, R. et al. Population Pharmacokinetic Model for Tramadol and O-desmethyltramadol in Older Patients. Eur J Drug Metab Pharmacokinet 47, 387–402 (2022). https://doi.org/10.1007/s13318-022-00756-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-022-00756-x