
Abstract
Background and Objectives
Lorlatinib is approved (100 mg once daily [QD]) for the treatment of patients with anaplastic lymphoma kinase- (ALK) positive metastatic non-small cell lung cancer. This study evaluated the impact of varying degrees of renal impairment on the safety and pharmacokinetics of lorlatinib.
Methods
Participants were assigned to mild, moderate, and severe renal impairment groups and to a matching normal renal function group based on absolute estimated glomerular filtration rate (eGFR, based on the Modification of Diet in Renal Disease equation and adjusted for body surface area [BSA]) and were evaluated for pharmacokinetics and safety.
Results
A total of 29 participants (5 with severe renal impairment; 8 each with moderate and mild impairment and normal renal function) were enrolled and received a single dose of lorlatinib 100 mg. One of the participants with severe renal impairment had end-stage renal disease with a baseline absolute eGFR of 10.3 mL/min. No serious adverse events (AEs) were reported. Eighteen AEs, all mild or moderate in severity, were reported by 12 participants (5, 2, 4, and 1 in the normal, mild, moderate, and severe groups, respectively). Area under the plasma concentration–time profile from time zero extrapolated to infinity (AUCinf) for lorlatinib was increased by 4%, 19%, and 41% in the mild, moderate, and severe renal impairment groups, respectively, compared with the normal renal function cohort.
Conclusion
Lorlatinib 100 mg was well tolerated. As participants with mild and moderate renal impairment did not experience clinically meaningful increases in lorlatinib exposure, no lorlatinib dose adjustment is recommended in these populations. Patients with severe renal impairment are recommended to reduce the starting dose of lorlatinib from 100 mg QD to 75 mg QD.
Clinicaltrials.gov identifier
NCT03542305 (available May 31, 2018 on clinicaltrials.gov)
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Study participants with mild and moderate kidney dysfunction did not have very different results compared to those with normal kidney function, and no lorlatinib dose adjustments are recommended in these populations. |
Those with severe kidney dysfunction had 41% higher lorlatinib concentrations, and instead of the 100 mg QD lorlatinib starting dose, a lower starting dose of 75 mg QD is recommended in this population. |
1 Introduction
Renal impairment is a common comorbidity in oncology patients for multiple reasons such as the malignancy itself, toxicities from anticancer treatments, or other factors, including the older age of many cancer patients [1, 2]. With the increasing use of oral tyrosine kinase inhibitor (TKI) anticancer agents, renal impairment can have a substantial impact on the pharmacokinetic profiles of drugs, potentially resulting in an increased risk of experiencing adverse events [3]. Therefore, it is important to evaluate the impact of renal dysfunction on the safety and dispositions of anticancer drugs to ensure that appropriate recommendations are made for dosing in cancer patients with comorbid mild, moderate, and severe renal impairment.
Lorlatinib (PF-06463922; Lorbrena, Lorviqua) is a potent third-generation anaplastic lymphoma kinase/c-ros oncogene 1 (ALK)/ROS1 TKI that has broad coverage of acquired resistance mutations and is currently indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors are ALK-positive as detected by an FDA-approved test [4]. PF-06895751 is the most abundant human circulating metabolite of lorlatinib and is not pharmacologically active against ALK and ROS1 kinase targets. It was recently reported from an interim analysis of the CROWN phase III study that in patients with previously untreated, advanced ALK-positive NSCLC, progression-free survival based on blinded independent central review was significantly longer in those who received first-line lorlatinib compared with crizotinib (hazard ratio 0.28; 95% CI 0.19–0.41) [5].
The current approved starting dose of lorlatinib is 100 mg once daily (QD). On the basis of findings from nonclinical in vitro studies and in vivo metabolic profiling, it was determined that extensive metabolism of lorlatinib occurred via oxidation and conjugation [6]. In two human absorption, distribution, metabolism, and excretion (ADME) studies, also called mass balance studies, unchanged lorlatinib accounted for < 2% of the dose in urine, indicating minimal urinary excretion of the parent drug [6]. Therefore, renal impairment would not be expected to have a major effect on lorlatinib pharmacokinetics or safety. However, results from a population pharmacokinetic analysis demonstrated that baseline creatinine clearance (CLcr) was a statistically significant predictor of variability in lorlatinib plasma clearance. The median estimated single-dose lorlatinib clearance was 18% and 26% lower in NSCLC patients with mild and moderate renal impairment, respectively, in that analysis [7]. Consequently, it was considered important to formally evaluate the potential impact of varying degrees of renal impairment on the pharmacokinetics and safety of lorlatinib via a prospective study.
The objectives of this phase I study (B7461010; clinicaltrials.gov identifier NCT03542305) were to (1) evaluate the effect of renal impairment on the single-dose pharmacokinetics of lorlatinib in otherwise healthy participants, and (2) evaluate the safety and tolerability of a single dose of lorlatinib in participants with renal impairment. This study was also intended to provide definitive label dose recommendations for lorlatinib in patients with mild, moderate, and severe renal impairment.
2 Methods
The study was conducted in compliance with the principles in the Declaration of Helsinki and in compliance with International Conference on Harmonization Good Clinical Practice Guidelines. The protocol was approved by the Institutional Review Board at each study center. All participants provided written informed consent before undergoing any study procedures.
2.1 Study Design and Participants
This was a phase I, open-label, multicenter, single-treatment study in non-cancer participants with normal renal function or varying degrees of renal impairment who were otherwise healthy. Each participant received a single oral dose of lorlatinib administered in the fasted state.
Renal impairment group assignment was based on the average of two absolute estimated glomerular filtration rate (eGFR) values derived during screening and derived from two separate creatinine measurements, which were required to be within 25% of each other. The absolute eGFR estimation was based on the Modification of Diet in Renal Disease (MDRD) equation and adjusted for body surface area (BSA), as defined in the Kidney Disease Outcomes Quality Initiative guidelines (Table 1) [8].
A single oral dose of lorlatinib 100 mg was administered first in this study to participants with mild renal impairment (Group B). After a single oral dose of lorlatinib 100 mg was tolerated by at least three participants with mild renal impairment, participants with moderate renal impairment (Group C) were enrolled one at a time and administered a single 100 mg oral dose of lorlatinib. After dosing three participants with moderate renal impairment, the pharmacokinetics, safety, and tolerability were evaluated during an observation period of at least 1 week. Then, the remaining participants in Group C and the participants in Group D (severe renal impairment) were enrolled and dosed. Participants with normal renal function (Group A) were matched to the participants with renal impairment (Groups B, C, and D) by demographically pooled average age (± 10 years), weight (± 20 kg), and sex (ratio 1:1, ± 2 patients per sex). Therefore, enrollment in Group A began after all participants from Groups B, C, and D had completed the pharmacokinetic collection and safety assessments.
In this study, all participants received a single 100 mg dose of lorlatinib. Lorlatinib was administered as four 25 mg tablets that were swallowed whole with approximately 240 mL of water after an overnight fast of at least 10 h. All participants refrained from eating or drinking beverages other than water for 4 h after lorlatinib dosing.
2.2 Pharmacokinetic Assessments
Blood samples (6 mL) that were used to determine plasma concentrations of lorlatinib and its predominant circulating metabolite, PF-06895751, were collected in tubes containing dipotassium ethylenediaminetetraacetic acid at the following times: pre-dose (0 h) and 0.5, 1, 1.5, 2, 4, 6, 8, 12, 24, 48, 72, 96, and 120 h after lorlatinib dosing. A blank urine sample of at least 50 mL was collected prior to lorlatinib administration for each participant. Post-dose urine collections occurred at intervals of 0–24, 24–48, 48–72, 72–96, and 96–120 h.
Plasma and urine samples were analyzed for lorlatinib concentrations using validated, sensitive, and specific high-performance liquid chromatography–tandem mass spectrometric methods (HPLC-MS/MS) as previously described [9,10,11]. Plasma samples were analyzed for PF-06895751 concentrations by a validated LC-MS/MS method. The lower limit of quantification (LLOQ) for lorlatinib and PF-06895751 was 2.50 (plasma and urine assay) and 1.00 ng/mL (plasma assay), respectively. All samples were analyzed at Covance Bioanalytical Services (Shanghai, China).
2.3 Pharmacokinetic Analysis
The primary lorlatinib pharmacokinetic parameters of interest were area under the plasma concentration–time profile (AUC) from time zero extrapolated to infinity (AUCinf) and maximum observed plasma concentration (Cmax). Other pharmacokinetic endpoints of interest for lorlatinib included AUC from time zero to last quantifiable concentration (AUClast), time to Cmax (Tmax), terminal elimination half-life (t1/2), apparent oral clearance (CL/F), apparent volume of distribution (Vz/F), renal clearance (CLR), cumulative amount of drug recovered unchanged in urine from time zero to 120 h post dose (Ae), and Ae expressed as the fraction of the dose that is excreted unchanged in urine (Ae%). Pharmacokinetic parameters of interest for PF-06895751 included plasma AUCinf, AUClast (area under the plasma concentration versus time curve from time zero to time of last quantifiable concentration), Cmax, Tmax, t1/2, metabolite to parent ratio for Cmax (MRCmax), metabolite to parent ratio for AUCinf (MRAUCinf), and metabolite to parent ratio for AUClast (MRAUClast).
Plasma concentration–time data for lorlatinib and PF-06895751 were analyzed by noncompartmental methods using an internally validated software system (eNCA; Electronic Non-compartmental Analysis version 2.2.4) to estimate pharmacokinetic parameters for each participant. Plasma concentrations below the LLOQ were set to 0 ng/mL for the pharmacokinetic analysis. Actual sample collection times were available for the majority of samples and were used for the analysis; nominal time post dose was used for the remaining samples. The parameters AUCinf, CL/F, and Vz/F were not reported for cases where the terminal elimination half-life could not be reliably determined. For this study, a well-characterized elimination half-life was defined as one with at least three data points, r2 ≥ 0.9, and AUCextrap% ≤ 35% for both lorlatinib and PF-06895751.
2.4 Safety Analysis
Safety assessments in the study included evaluations of adverse events (AEs), physical exams/vital signs, safety laboratory tests, and electrocardiogram (ECG). Any AEs occurring following lorlatinib dosing or increasing in severity during the study were considered treatment emergent. All AEs were classified as mild, moderate, or severe in severity. Per protocol, a mild AE was one that did not interfere with the participant’s usual function, a moderate AE was one that interferened to some extent with the participant’s usual function, and a severe AE was one that significantly interfered with the participant’s usual function.
2.5 Statistical Methods
Approximately 8 participants were planned to be enrolled into each of the normal, mild, and moderate renal impairment groups, and 4–8 participants in the severe renal impairment group. The sample size for this study was empirically selected based on recommendations from the Food and Drug Administration (FDA) Guidance for Industry: Pharmacokinetics in Patients with Impaired Renal Function—Study Design, Data Analysis, and Impact on Dosing and Labeling [12], and was not based on statistical power calculation.
The natural-log-transformed plasma lorlatinib AUCinf, AUClast, and Cmax were analyzed using a mixed-effects model with renal function group as a fixed effect, participant as a random effect, and an unequal covariance structure. The mixed-effects model was implemented using SAS Proprietary Software v9.4 (TS1M5), SAS Institute Inc. (Cary, NC, USA). Estimates for the adjusted mean differences (test group − reference group) and the corresponding 90% confidence interval (CI) were obtained. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (test/reference) and 90% CIs for the ratios. The normal renal function group was the reference group, whereas the mild, moderate, and severe renal impairment groups were the test groups.
3 Results
3.1 Study Participants and Demographics
A total of 29 participants (5 with severe renal impairment, and 8 each with moderate impairment, mild impairment, and normal renal function) were enrolled. All participants completed the study with no important protocol deviations noted. Demographic and baseline characteristics were generally balanced among renal function groups; however, age and baseline body mass index were slightly higher in the moderate and severe renal impairment groups (Table 2). One participant in the severe renal impairment group was considered to have end-stage renal disease not requiring dialysis, with a baseline absolute eGFR value of 10.3 mL/min.
3.2 Lorlatinib and PF-06895751 Plasma Pharmacokinetics
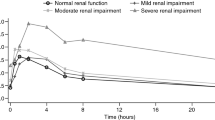
The mean plasma concentration–time profiles for lorlatinib and PF-06895751 by renal function group are presented in Fig. 1, with corresponding pharmacokinetic parameters summarized in Table 3. Box plots of the individual and geometric mean plasma AUCinf and Cmax values for lorlatinib by renal function group are provided in Fig. 2.

Plasma concentration–time profiles for lorlatinib and PF-06895751 by renal function group plotted on a semi-log scale. Data are mean ± standard deviation. The inset for lorlatinib shows the early profile (for the first 24 h)

Box plots of individual and geometric mean plasma AUCinf and Cmax values for lorlatinib by renal function group. Box plots are median and 25%/75% quartiles with whiskers to the last point within 1.5 times the interquartile range; stars represent geometric means and circles represent individual values. AUCinf area under the plasma concentration–time profile from time zero extrapolated to infinity, Cmax maximum observed plasma concentration
Following the administration of a single oral dose of lorlatinib 100 mg, median lorlatinib plasma concentrations increased marginally for participants in the moderate and severe renal impairment groups compared with the normal and mildly impaired renal function groups. Lorlatinib Cmax was reached at approximately the same time in all participants, with median Tmax values of 1.0–1.5 h post dose. Lorlatinib plasma elimination t½ was longer in the moderate and severe renal impairment groups, with mean ± standard deviation (SD) values of approximately 39 ± 7.1 and 42 ± 7.6 h, respectively, compared with 26 ± 4.8 and 28 ± 3.5 h in participants with normal renal function and mild renal impairment, respectively.
Geometric mean lorlatinib oral clearance was lower in participants with renal impairment than in participants with normal renal function, while geometric mean Vz/F was generally similar for all renal function groups. This implied that lorlatinib plasma exposure based on the geometric mean AUCinf value was slightly higher for participants in the moderate and severe renal impairment groups than for participants with normal renal function and mild renal impairment. Geometric mean Cmax values for lorlatinib were generally similar across all renal function groups. Variability in lorlatinib plasma exposure based on geometric %CV was similar across the renal function groups and ranged from 24 to 52% for Cmax and from 27 to 37% for AUCinf.
Results of the linear regression analyses of absolute eGFR and CLcr as estimated by the Cockcroft–Gault equation suggest correlations between worsening renal function and increasing lorlatinib oral clearance (Fig. 3).

Scatter plots show the correlations between absolute eGFR and lorlatinib CL/F (R2 = 0.1333 and p = 0.0515) (a) and between creatinine clearance and lorlatinib CL/F (R2 = 0.1024 and p = 0.0905) (b). The solid black lines represent linear regression, and the shaded areas are the 90% confidence regions. CLcr creatinine clearance, CL/F apparent oral clearance, eGFR estimated glomerular filtration rate
Compared to lorlatinib, PF-06895751 concentrations gradually declined for all renal function groups, and PF-06895751 Cmax was reached considerably later; the median Tmax was 24 h for the normal renal function and mild renal impairment groups and 72 h for the moderate and severe renal impairment groups. Mean plasma elimination t½ for PF-06895751 was longer in the normal renal function and mild renal impairment groups, with mean values of approximately 41 and 35 h, respectively, compared with the parent lorlatinib in the same renal function groups. However, it should be noted that the time span for collection of pharmacokinetic samples (120 h post dosing) was only approximately threefold higher than the estimated lorlatinib half life; hence, the lorlatinib terminal half-life in moderate and severe renal impairment subjects may not have have been precisely estimated. PF-06895751 plasma t½ could not be estimated accurately for moderate and severe renal impairment participants. The molar metabolite ratios based on AUClast and Cmax were generally similar across all renal function groups.
Although the lorlatinib Tmax for moderate and severe renal impairment subjects was delayed, in general, the overall plasma exposure based on the geometric mean PF-06895751 AUClast and Cmax values was similar across the renal function groups. Variability in PF-06895751 based on the geometric %CV was similar among normal renal function, mild renal impairment, and moderate renal impairment groups (ranges: 26–28% for Cmax and 18–28% for AUClast), but was higher in the severe renal impairment group (59% for Cmax and 58% for AUClast).
The adjusted geometric mean ratios (90% CIs) of lorlatinib plasma AUCinf for the renal impairment groups (test groups) versus the normal renal function group (reference group) were 104.25% (79.73%, 136.31%), 118.75% (91.43%, 154.24%), and 141.14% (97.82%, 203.66%) for the mild, moderate, and severe renal impairment groups, respectively (Table 4). The adjusted geometric mean ratios of lorlatinib plasma Cmax were close to 100% for the mild, moderate, and severe renal impairment groups.
3.3 Lorlatinib Urine Pharmacokinetics
Approximately 0.78–1.2% of the 100 mg lorlatinib dose was recovered in urine as unchanged lorlatinib (Ae%) across the renal function groups (Table 5). Geometric mean CLR was lower for participants in the moderate and severe renal impairment groups than for the normal and mildly impaired renal function groups.
3.4 Safety and Tolerability
A single dose of lorlatinib 100 mg was well tolerated across all renal function groups. No deaths, SAEs, severe AEs, or discontinuations from the study due to AEs were reported during the study. No clinically significant abnormalities in vital signs, safety laboratory tests, or ECG were found.
A total of 18 AEs were reported by 12 participants (5, 2, 4, and 1 participants in the normal renal function and mild, moderate, and severe renal impairment groups, respectively). Twelve AEs were mild in severity and 6 AEs were moderate in severity. Most (15) AEs occurred within 5 days of lorlatinib dosing. All AEs were resolved by the end of the study, and most (14) AEs lasted 4 days or less.
Eight AEs in 7 participants (3, 2, 1, and 1 participants in the normal renal function and mild, moderate, and severe renal impairment groups, respectively) were considered to be treatment related by the investigator. Increased diarrhea and blood pressure, each reported by 2 participants, were the only treatment-related AEs reported by more than 1 participant across all groups. The participant in the severe renal impairment group with end-stage renal disease did not report any AEs during the study.
4 Discussion
Historically, pivotal studies in the target patient population have excluded patients with renal impairment. A dedicated renal impairment study is important in informing dosing recommendations in patients who may present with varying degrees of renal impairment [13].
Although the renal contribution to lorlatinib elimination is minimal (< 2%), a population pharmacokinetic analysis found that baseline CLcr was a significant covariate of lorlatinib clearance [7]. The population pharmacokinetic analysis pooled data from six healthy participant studies as well as data from patients with NSCLC in the phase I/II study B7461001. In total, the analyzed population included 226 participants with normal renal function, 120 participants with mild renal impairment, 45 participants with moderate renal impairment, and 1 participant with severe renal impairment, classified based on CLcr as estimated using the Cockcroft–Gault equation. The median estimated single-dose lorlatinib clearance was 18% and 26% lower in the mild and moderate renal impairment groups (8.04 L/h and 7.22 L/h), respectively, than in those with baseline normal renal function (9.80 L/h), suggesting potentially higher lorlatinib exposure in the renally impaired subpopulations.
The results of this clinical study demonstrated an increase of approximately 19% and 41% in lorlatinib AUCinf in the moderate and severe renal impairment participants, respectively, compared with participants with normal renal function, corroborating the finding of decreased single-dose lorlatinib clearance in moderate renal impairment participants from the population pharmacokinetic analysis. Lorlatinib plasma exposures in the mild renal impairment group were similar to the plasma exposures of participants with normal renal function. Mean lorlatinib plasma elimination t½ increased and geometric mean CL/F decreased with increasing renal impairment severity, indicating that renal elimination of lorlatinib was impacted, although not substantially, by worsening renal function, particularly in the severe renal impairment group. As expected, renal impairment had a limited impact on drug absorption, as no clinically meaningful difference in lorlatinib plasma Cmax was observed between the renal function groups. Although changes in PF-06895751 exposure were also observed in renally impaired participants, these are not expected to be clinically meaningful because PF-06895751 is not pharmacologically active and only accounts for ~ 5.6% of the lorlatinib dose recovered in excreta [6].
There are a few mechanisms that have been reported in the literature to explain the reduced clearance observed with renal impairment for drugs primarily metabolized by the liver such as lorlatinib; these include the accumulation of uremic toxins following renal impairment, resulting in reduced function of CYP enzymes and transporters, as well as the accumulation of pro-inflammatory cytokines, leading to the downregulation of CYP enzymes and transporters [14]. This is corroborated by several clinical renal impairment studies of drugs that are non-renally cleared; these studies demonstrated increased plasma exposures concurrent with chronic renal impairment. This finding has been reported in renal impairment studies with lidocaine, nicardipine, propranolol, and sildenafil [15,16,17,18]. An initial hypothesis was that, for drugs that are primarily cleared through metabolism, those with a high hepatic extraction ratio are more likely to demonstrate a substantial decrease in clearance with renal impairment; lidocaine, nicardipine, propranolol, and sildenafil are all drugs with intermediate to high hepatic extraction ratios [19,20,21,22]. However, lorlatinib is a low extraction ratio drug (estimated as 12%) [23]. Hence, overall, it is not clear why increased plasma exposures following renal impairment are observed for some predominantly hepatically cleared drugs but not for others.
Although increased lorlatinib plasma exposure was observed in this study, particularly in moderate and severe renal impairment participants, no increase in AEs were observed in participants with worse renal function following administration of a single dose of lorlatinib. Currently, safety data following multiple-dose administration of lorlatinib in participants with severe renal impairment is limited. Thus, there could be a theoretical increased risk of those toxicities typically reported with continuous lorlatinib dosing. Therefore, reducing the dose of lorlatinib is recommended for patients with severe renal impairment, e.g., a starting dose modification from lorlatinib 100 mg QD to 75 mg QD. Since severe renal impairment in this study was associated with a 41% increase in AUC, patients with severe renal impairment who receive the 75 mg QD lorlatinib dose should achieve plasma exposures that are the equivalent of a 106 mg QD dose (75 mg × 1.41), which is close to the 100 mg QD recommended dose for lorlatinib. Given that plasma lorlatinib exposures in the mild impairment group were similar to those in the normal renal function group, and the 19% increase in lorlatinib AUCinf in the moderate impairment group is not expected to be clinically meaningful, this study provides additional data to support the 100 mg QD starting dose in mild and moderate renal impairment patients. Although the number of subjects in a typical renal impairment study is low, with 5–8 participants in each renal function group in the lorlatinib study, the results provided adequate information to inform lorlatinib dosing recommendations for mild, moderate, and severe renal impairment.
Crizotinib and brigatinib are two other ALK inhibitors for which dose reductions to approximately 50% of the approved dose are recommended in patients with severe renal impairment [24, 25]. Following a single dose of 250 mg, the AUCinf of crizotinib increased by 79% and its Cmax increased by 34%. Following a single 90 mg dose, unbound brigatinib AUCinf increased by 84%. The impact of severe renal impairment on the pharmacokinetic of alectinib, a second-generation ALK inhibitor, has not been reported [26]. The higher increases in AUCinf observed with crizotinib and brigatinib are likely due to the higher percentages of these drugs excreted in urine (22% and 25%, respectively) versus lorlatinib (< 2% urine excretion). No dose modifications of any of the ALK inhibitors are recommended in cases of mild or moderate renal impairment.
5 Conclusions
The single 100 mg lorlatinib dose administered to all participants in this study was well tolerated. Participants with mild and moderate renal impairment did not experience clinically meaningful increases in lorlatinib exposure; therefore, no lorlatinib dose adjustment is recommended in these populations. Patients with severe renal impairment are recommended to reduce the starting dose of lorlatinib from 100 mg QD to 75 mg QD.
Change history
08 March 2022
A Correction to this paper has been published: https://doi.org/10.1007/s13318-022-00759-8
References
Krens SD, Lassche G, Jansman FGA, et al. Dose recommendations for anticancer drugs in patients with renal or hepatic impairment. Lancet Oncol. 2019;20(4):e200–7.
Shahinian VB, Bahl A, Niepel D, et al. Considering renal risk while managing cancer. Cancer Manag Res. 2017;9:167–78.
Sgambato A, Casaluce F, Maione P, Gridelli C. Targeted therapies in non-small cell lung cancer: a focus on ALK/ROS1 tyrosine kinase inhibitors. Expert Rev Anticancer Ther. 2018;18(1):71–80.
Johnson TW, Richardson PF, Bailey S, et al. Discovery of (10R)-7- amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57(11):4720–44.
Shaw AT, Bauer TM, de Marinis F, CROWN Trial Investigators, et al. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N Engl J Med. 2020;383(21):2018–29.
Stypinksi D, Fostveldt L, Lam JL, et al. Metabolism, excretion, and pharmacokinetics of lorlatinib (PF-06463922) and evaluation of the impact of radiolabel position and other factors on comparability of data across 2 ADME studies. J Clin Pharmacol. 2020;60(9):1254–67.
Chen J, Houk B, Pithavala YK, Ruiz-Garcia A. Population pharmacokinetic model with time-varying clearance for lorlatinib using pooled data from patients with non-small cell lung cancer and healthy participants. CPT Pharmacometr Syst Pharmacol. 2021;10(2):148–60.
Inker LA, Astor BC, Fox CH, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am J Kidney Dis. 2014;63(5):713–35.
Stypinksi D, Fostvedt L, Lam JL, et al. Metabolism, excretion, and pharmacokinetics of lorlatinib (PF-06463922) and evaluation of the impact of radiolabel position and other factors on comparability of data across 2 ADME studies. J Clin Pharmacol. 2020;60(9):1254–67.
Patel M, Chen J, McGrory S, et al. The effect of itraconazole on the pharmacokinetics of lorlatinib: results of a phase I, open-label, crossover study in healthy participants. Investig New Drugs. 2020;38(1):131–9.
Chen J, Xu H, Pawlak S, et al. The effect of rifampin on the pharmacokinetics and safety of lorlatinib: results of a phase one, open-label, crossover study in healthy participants. Adv Ther. 2020;37(2):745–58.
US Food and Drug Administration (FDA). Guidance for Industry. Pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing. 2020. https://www.fda.gov/media/78573/download. Accessed 15 July 2021.
European Medicines Agency (EMA). Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf. Accessed 15 July 2021.
Momper JD, Venkataramanan R, Nolin TD. Nonrenal drug clearance in CKD: searching for the path less traveled. Adv Chronic Kidney Dis. 2010;17(5):384–91.
De Martin S, Orlando R, Bertoli M, et al. Differential effect of chronic renal failure on the pharmacokinetics of lidocaine in patients receiving and not receiving hemodialysis. Clin Pharmacol Ther. 2006;80(6):597–606.
Ahmed JH, Grant AC, Rodger RS, et al. Inhibitory effect of uraemia on the hepatic clearance and metabolism of nicardipine. Br J Clin Pharmacol. 1991;32(1):57–62.
Bianchetti G, Graziani G, Brancaccio D, et al. Pharmacokinetics and effects of propranolol in terminal uraemic patients and in patients undergoing regular dialysis treatment. Clin Pharmacokinet. 1976;1(5):373–84.
Muirhead GJ, Wilner K, Colburn W, Haug-Pihale G, Rouviex B. The effects of age and renal and hepatic impairment on the pharmacokinetics of sildenafil. Br J Clin Pharmacol. 2002;53(suppl 1):21S-30S.
Huet P-M, Lelorier J. Effects of smoking and chronic hepatitis B on lidocaine and indocyanine green kinetics. Clin Pharmacol Ther. 1990;28(2):208–15.
Raucoules-Aime M, Drici M, Goubaux B, et al. Intravenous nicardipine does not alter hepatic blood flow after orthotopic liver transplant. Intensive Care Med. 1996;22(5):420–5.
Weiss YA, Safar ME, Lehner JP, et al. (+)-Propranolol clearance, an estimation of hepatic blood flow in man. Br J Clin Pharmacol. 1978;5(5):457–60.
Hill KD, Sampson MR, Li SJ, et al. Pharmacokinetics of intravenous sildenafil in children with palliated single ventricle heart defects: effect of elevated hepatic pressures. Cardiol Young. 2016;26(2):354–62.
Hibma J, O’Gorman M, Nepal S, et al. Evaluation of the absolute oral bioavailability of the anaplastic lymphoma kinase/c-ROS oncogene 1 kinase inhibitor lorlatinib in healthy participants. Cancer Chemother Pharmacol. 2021;10(11):1395–404.
Takeda Pharmaceutical Company Limited. Alunbrig (brigatinib) [package insert]. Cambridge, MA: Takeda Pharmaceutical Company Limited. Revised May 2020. https://www.alunbrig.com/assets/pi.pdf. Accessed 15 July 2021.
Pfizer Inc. Xalkori (crizotinib) [package insert]. New York, NY: Pfizer Inc. http://labeling.pfizer.com/ShowLabeling.aspx?id=676#S2.4. Accessed 15 July 2021.
Genentech USA, Inc. Alecensa (alectinib) [package insert]. South San Francisco, CA: Genentech USA, Inc. Revised 2017. https://www.gene.com/download/pdf/alecensa_prescribing.pdf. Accessed 15 July 2021.
Acknowledgements
Editorial support was provided by Cynthia Pereira, CMPP, of CMC AFFINITY, McCann Health Medical Communications, and was funded by Pfizer Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Pfizer Inc.
Ethics approval
All procedures in this study were in accordance with the 1964 Helsinki Declaration and its amendments, and the Aspire Institutional Review Board and Salus IRBs, which approved the study at the two clinical sites.
Consent to participate
All versions of the IRB-approved informed consent documents used in the study are included in the sponsor’s trial master file. All participants signed informed consent documents prior to participation in the trial; the signed informed consent documents are maintained at the respective investigator sites to preserve the confidentiality of the participants.
Consent for publication
Not applicable.
Code availability
The proprietary software program SAS was used for the analysis reported here.
Conflict of interest
SL was an employee of Pfizer Inc. at the time of the study; JG is an employee of Pfizer Inc.; GCC and PW have no conflicts of interest; KP, RRL, KG, and YKP are employees of Pfizer Inc. and own stock in Pfizer.
Availability of data and material
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Author contributions
SL, JG, KP, RRL, and YKP contributed toward writing the manuscript; SL, JG, GCC, PW, KP, RRL, KG, and YKP designed the research, and SL, JG, GCC, PW, KP, RRL, KG, and YKP analyzed and interpreted the data.
Additional information
The original online version of this article was revised: Corrections to figure 2 updated.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lin, S., Gong, J., Canas, G.C. et al. A Phase I Study to Evaluate the Pharmacokinetics and Safety of Lorlatinib in Adults with Mild, Moderate, and Severe Renal Impairment. Eur J Drug Metab Pharmacokinet 47, 235–245 (2022). https://doi.org/10.1007/s13318-021-00747-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-021-00747-4