
Abstract
Background and Objectives
Advanced estrogen receptor-positive (ER+) breast cancer is currently treated with endocrine therapy. Elacestrant is a novel, nonsteroidal, selective estrogen receptor degrader with complex dose-related ER agonist/antagonist activity that is being developed as a treatment option for ER+ breast cancer.
Methods
Two first-in-human phase 1 studies of elacestrant in healthy postmenopausal women (Study 001/Study 004) were conducted to determine its pharmacokinetic and pharmacodynamic profile as well as its safety and maximum tolerated dose.
Results
In total, 140 postmenopausal subjects received at least one dose of study drug (114 received elacestrant and 26 received placebo). Single-ascending dose and multiple-ascending dose assessments showed that doses up to 1000 mg daily were safe and well tolerated, and the maximum tolerated dose was not reached. Oral administration of elacestrant had an absolute bioavailability of 10% and a mean half-life ranging from 27 to 47 h, reaching steady state after 5–6 days. Mean occupancy of the ER in the uterus after seven daily doses was 83% for 200 mg and 92% for 500 mg daily. The median ratio of elacestrant concentrations in the cerebral spinal fluid vs. plasma was 0.126% (500 mg dose) and 0.205% (200 mg dose). Most adverse events were related to the upper gastrointestinal tract.
Conclusions
These data demonstrate that elacestrant has good bioavailability when administered orally with a half-life that supports once-daily administration. Engagement of the ER and some ability to cross the blood-brain barrier was demonstrated in addition to an acceptable safety profile.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Elacestrant, an oral SERD, is safe and well tolerated at oral doses up to 500 mg per day |
Robust ERα occupancy (75–90%) is observed at elacestrant doses of 200 mg to 500 mg daily |
Elacestrant’s bioavailability and long t1/2 support a once-daily oral dosing |
1 Introduction
Breast cancer continues to be the most commonly diagnosed malignancy among women [1], and approximately 75% of all breast cancers are estrogen receptor-positive and human epidermal growth factor type-2-negative (ER+/HER2−) [2]. For patients with ER+/HER2− advanced or metastatic breast cancer (MBC), endocrine therapies such as aromatase inhibitors, selective ER modulators and selective ER degraders (SERDs) remain the cornerstone of treatment [3]. One of the most commonly used and effective endocrine therapies is fulvestrant. Fulvestrant is the only SERD currently approved for MBC in postmenopausal women [4] and has demonstrated therapeutic efficacy as monotherapy and in combination with targeted therapies, such as CDK4/6 inhibitors [5,6,7,8,9]. However, fulvestrant is limited by its poor oral bioavailability and the requirement for intramuscular injection [4].
Elacestrant is a novel nonsteroidal SERD [10] that has activity against ER+ breast cancer in both in vitro and in vivo models, including xenografts derived from heavily pretreated patients [11,12,13]. Importantly, elacestrant has antitumor activity in models resistant to fulvestrant and CDK4/6 inhibitors, including those harboring ESR1 Y537S and D538F mutations [11, 14,15,16]. In a phase 1 trial, single-agent oral elacestrant 400 mg daily showed an overall response rate of 19.4% and a median progression-free survival of 4.5 months in 50 heavily pretreated patients with ER+/HER2− MBC. Subjects in this trial had a median of three prior lines of therapy, including 52% with prior CDK4/6 inhibitors and 50% with prior fulvestrant, and 51% had tumors that harbored ESR1 mutations [17].
The pharmacokinetics, pharmacodynamic profile, oral bioavailability, and maximum tolerated dose (MTD) of elacestrant in healthy postmenopausal women have not been reported yet. We summarize the results from two first-in-human phase 1 studies that were undertaken to characterize the pharmacokinetic characteristics (including oral bioavailability and food effect), pharmacodynamic profile (ERα occupancy), safety and tolerability and to establish the MTD of elacestrant in healthy postmenopausal women volunteers to guide dose selection in the clinical development of elacestrant in ER+/HER− MBC.
2 Methods
2.1 Study Design
Two phase 1, randomized, placebo-controlled studies (Study 001 [EudraCT number: 2007-006547-41] and Study 004 [EudraCT number: 2014-001699-67]) were conducted in healthy postmenopausal women. Both studies were conducted with the capsule formulation of elacestrant, which has since been replaced with a tablet formulation in all ongoing trials.
Study 001 consisted of a single ascending dose (SAD) component (single oral doses of 1–200 mg) and a multiple ascending dose (MAD) component (oral 10–200 mg daily for 7 days). Study 001 also examined the oral bioavailability of elacestrant and potential food effect. Treatment groups are shown in Fig. 1. The starting oral dose of 1 mg was selected based on preclinical animal and toxicology studies.

Subject disposition in Study 001. aSubject withdrew informed consent after period 1; IV intravenous, MAD multiple ascending dose, PBO placebo, po orally, qd once daily, SAD single ascending dose
Study 004 was a dose-escalation study designed to assess the MTD of elacestrant at oral doses of 200–1000 mg once daily for 7 days. The planned treatment groups are shown in Fig. 2. In addition to safety and pharmacokinetics, this study included a cohort to determine the pharmacodynamic parameter of ERα occupancy in the uterus and pituitary as determined by 16α-[18F-]fluoroestradiol (FES) positron emission tomography (PET) imaging and lumbar puncture to obtain cerebrospinal fluid (CSF) to determine elacestrant's blood-brain barrier (BBB) penetration and CSF concentrations. The starting dose of 200 mg once daily for 7 days was based on the tested maximum single and repeated daily dose in Study 001. The dose-escalation components of both studies used a standard ascending dose tolerance design where investigators and the sponsor evaluated safety and tolerability following the completion of each dose cohort.

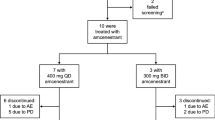
Subject disposition in Study 004. PBO placebo, po orally, qd once daily. aIn Cohort 4, only nine of the ten planned subjects enrolled because of logistical considerations. All withdrawals from the study were due to adverse events
The clinical study protocols and informed consent forms were reviewed and approved by an independent ethics committee, and all subjects provided written informed consent. Both studies were conducted in accordance with the principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonisation (ICH) E6 Guideline for Good Clinical Practice (GCP) (Committee for Proprietary Medicinal Products (CPMP) guideline CPMP/ICH/135/95) and with the EU CTD: Directive 2001/20/EC.
2.2 Procedures
Screening took place within 30 days before the first administration of study drug in both studies. The main inclusion criteria included age 40–75 years, body mass index (BMI) 18–30 kg/m2, postmenopausal status defined as ≥ 12 months of amenorrhea and FSH concentrations within the postmenopausal range. Key exclusion criteria included a history of significant medical conditions, pregnancy or lactation, use of any concomitant medications except acetaminophen within 14 days of study drug administration, and smoking or use of any tobacco products within 60 days of study drug administration.
In the SAD part of Study 001, subjects were enrolled into one of four cohorts and randomized to receive a single dose of elacestrant (1–200 mg) or placebo in oral capsules in each of two study periods. Cohorts 1 and 2 received two different oral doses of elacestrant in the fasted state in the two periods (Fig. 1). Cohort 3 evaluated the same oral dose (50 mg) in both periods, one under fasted and the other under fed conditions. Cohort 4 received 100 mg fasted oral dose in the first period and 1 mg intravenous dose in the second period. The washout between the two study periods was at least 1 week. Both study periods included pharmacokinetic assessments for 72 h after study drug administration, after which participants were discharged on the 4th day. In the MAD part, subjects were enrolled in one of five cohorts and randomized to receive oral doses of elacestrant 10–200 mg daily or placebo for 7 days, and the pharmacokinetic profile was assessed for 24 h after the day 1 dose and for 48 h following the last dose on day 7. Subjects were discharged on the morning of the 10th day. All patients in Study 001 had a follow-up visit 7–10 days after their discharge.
In all of the SAD and MAD cohorts in Study 001, except for period 2 of Cohort 3, oral elacestrant was administered between 9 and 10 h after a 10-h fast. Participants remained fasted until 4 h after drug administration. In period 2 of the SAD part of Cohort 3, which evaluated the food effect on pharmacokinetic parameters, elacestrant was administered after a high-fat breakfast per the Food and Drug Administration (FDA) industry guidelines [18].
In Study 004, Cohorts 1 through 5 were designed to evaluate the MTD of elacestrant while Cohort 6 assessed its ERα occupancy and BBB penetration. The elacestrant dose in Cohort 5 was not predefined but was to be based on an interim analysis of elacestrant exposure by dose relative to the incidence of treatment-emergent adverse events (TEAEs) in Cohorts 1 through 4 as well as ERα occupancy data in Cohort 6. In Cohorts 1 through 4, subjects were randomized to receive 200 to 1000 mg elacestrant or placebo (Fig. 2). For all cohorts, elacestrant was administered as oral capsules in the fed state, following a light meal, once daily for 7 days. The assigned elacestrant dose was achieved by a combination of either 100 mg or 150 mg capsules: 200 mg (2 × 100 mg capsules), 500 mg (2 × 100 mg capsules plus 2 × 150 mg capsules), 750 mg (5 × 150 mg capsules) or 1000 mg (1 × 100 mg capsule plus 6 × 150 mg capsules). Pharmacokinetic plasma concentrations were assessed over 24 h and then at 48, 72, 96, 144 and 192 h post-dose on day 7. Trough concentrations were sampled pre-dose on days 5 and 6. Samples were also obtained at outpatient follow-up visits on days 11, 13, 15 and day 19 ± 2.
In Study 004 Cohort 6, subjects received oral 200 or 500 mg elacestrant capsules daily for 7 days without randomization to placebo and underwent lumbar puncture 2–4 h after dosing on day 7, corresponding to the observed tmax in Study 001 (mean tmax ranged from 1.64 to 3.93 h, Table 2) to determine elacestrant concentrations in the CSF. The plasma sample corresponding to the CSF sampling time point was obtained immediately after the lumbar puncture procedure. As the study was originally designed, subjects in Cohort 6 were to be assessed with dynamic and static 18F-FES-PET imaging to determine ERα occupancy in the brain (in particular, the pituitary) and uterus, respectively. The tracer 18F-FES was produced according to a previously described method [19]. On day − 1 and 4 h after oral intake of elacestrant on day 6, 18F-FES (200 MBq) was administered intravenously, and a 90-min dynamic PET scan of the brain was performed immediately after tracer administration. During the dynamic PET scan, arterial blood sampling was performed to generate a plasma radioactivity input curve to enable pharmacokinetic modeling of the PET data. The plasma curve was corrected for the presence of radioactive metabolites of the tracer, as determined by thin-layer chromatography analysis [20]. Immediately after completion of the dynamic scan of the brain, a 5-min static PET scan of the uterus was started. The scans included a low-dose computed tomography (CT) of the brain and the uterus to correct for attenuation and scatter of radioactivity.
As a result of emerging data from Cohort 6 showing limited ERα occupancy in the brain (with the exception of pituitary) and low elacestrant concentrations in the CSF, the dynamic PET scan was eliminated, and the remaining subjects in Cohort 6 only underwent a 5-min static PET scan of the uterus and the brain 90 min after tracer injection on day − 1 and day 6.
Based on the ERα occupancy data at 200 mg and 500 mg in Cohort 6, as well as an interim analysis of elacestrant exposure by dose relative to the incidence of treatment-emergent adverse events (TEAEs) in Cohorts 1 to 4, it was determined that doses > 1000 mg daily were unlikely to be pursued in future studies. Hence, Cohort 5 was eliminated.
Adverse events, vital signs, ECGs and laboratory tests were monitored throughout both studies to assess the safety and tolerability of elacestrant or placebo.
Concentrations of elacestrant in plasma, urine and cerebrospinal fluid (CSF) were determined with validated liquid chromatography with tandem mass spectrometry detection (Sciex 5000 mass spectrometer coupled with an Acquity UPLC system) (LC–MS/MS) methods at PRA Health Sciences Bioanalytical Laboratory (Assen, The Netherlands). Characteristic ion dissociation transitions were m/z 459.35 → 268.15 for elacestrant and 463.35 → 272.23 for the internal standard (deuterated elacestrant-d4). Assessments were conducted via Acquity UPLC BEH Shield RP18 column (50 × 2.1 mm, 1.7 μm) (Waters Corp., Milford, MA, USA) with a blended mobile phase of acetonitrile and 0.1% formic acid in water using a varying gradient program. Flow rate was 0.8 ml/min. The assay was validated for specificity, precession, accuracy, sensitivity, stability and linearity over a concentration range of 0.05–100 ng/ml in all matrices. Samples were prepared using a liquid–liquid extraction procedure as follows: 300 µl 1% formic acid in water was added to 150 µl plasma, vortex-mixed for 10 s then transferred to a well of a SPE plate, which had been conditioned with 200 μl methanol and 200 μl 1% formic acid in water. After washing with 200 μl 1% formic acid in water and 200 μl methanol, the SPE-plate was eluted with 100 μl elution solvent (acetonitrile: methanol: ammonia [25%] [49:49:2, v/v/v]) into deep-well polypropylene collection plates. The sample was evaporated to dryness under nitrogen at approximately 60 °C at a gas flow of ca. 40 CFH in 10 min, and the residue was redissolved in 200 μl reconstitution solvent (acetonitrile: water: formic acid [20:80:0.2, v/v/v]) by vortex mixing for 1 min, and an aliquot of 5 μl was injected into the chromatographic system. All analytical runs were performed with QC samples of 0.15, 7.5, and 80 ng/ml, and the accuracy (% Bias) and precision (% CV) for the assay standards and QC samples had to be < 15% (or < 20% for the lower limit of quantitation [0.05 ng/ml]). Samples with concentration values > 100 ng/ml were diluted up to tenfold with blank plasma and re-assayed. Elacestrant plasma concentrations below the lower limit of quantification (LLOQ, < 0.05 ng/ml) were set equal to zero for calculations of summary statistics.
2.3 Statistical Analyses
2.3.1 Pharmacokinetic Analyses
All subjects who received treatment and for whom the pharmacokinetic data were interpretable were included in the pharmacokinetic analyses. Concentration-time data were presented using descriptive statistics. Missing pharmacokinetic parameter data were not imputed. To assess the bioavailability of elacestrant, pharmacokinetic parameters were dose-normalized, logarithmically transformed and evaluated with an analysis of variance (ANOVA, with treatment as the fixed factor and subject as a random factor). Based upon the residual variation from the ANOVA, the ratio of least-square means and corresponding 90% confidence intervals (CIs) were calculated for subjects in the absolute bioavailability assessment (Study 001 Cohort 4 using area under the curve [AUC]) and for subjects in the relative bioavailability assessment (food effect, Cohort 3, using AUC and Cmax).
To explore the dose proportionality, the geometric means of Cmax and AUC were examined against dose. Dose proportionality was assessed using a power model (i.e., Log (AUC) = α + β × log (dose) + error). An ideal proportional model corresponds to β = 1. Linear dose proportionality was present if the 90% CI for β included 1.
2.3.2 Safety Analyses
All subjects who received at least one dose of study drug (elacestrant or placebo) were included in the safety analyses. Adverse events were coded using the Medical Dictionary for Regulatory Activities Terminology. Descriptive analyses were used. In Study 001, AEs were recorded as “mild,” “moderate” or “severe,” and their relationship with the study drug was coded as “none,” “remote,” “possible,” “probable” or “definite.” In Study 004, severity of AEs was graded using the Common Terminology Criteria for Adverse Events (CTCAE) 5-point scale.
2.3.3 Sample Size
Both Studies 001 and 004 were dose-finding; hence, no prospective calculations of statistical power were made. The sample sizes were selected to provide information on pharmacokinetics, pharmacodynamics, safety, tolerability and MTD of elacestrant.
3 Results
3.1 Subject Disposition and Demographics
Study 001 was conducted between February 13, 2008, and August 20, 2008. Study 004 was conducted between June 3, 2014, and April 20, 2015. Subject dispositions for Study 001 and Study 004 are shown in Figs. 1 and 2, respectively. In total, 80 subjects received at least one dose of study drug (62 received elacestrant and 18 received placebo). All Study 001 subjects completed the study except for one participant in Cohort 4 of the SAD part who withdrew consent after the first dosing period. In Study 004, 47 subjects were randomized and received at least one dose of study drug in Cohorts 1 through 4 (39 received elacestrant and 8 received placebo), while 13 subjects in Cohort 6 received elacestrant. Among Study 004 subjects, 43 receiving elacestrant and all subjects receiving placebo completed the study; 9 subjects treated with elacestrant at doses ≥ 500 mg daily withdrew because of AEs, as shown in Fig. 2. The demographics of the study populations in Studies 001 and 004 are summarized in Table 1. Subjects in both studies were primarily non-Hispanic or of white ethnicity and were similar in age and BMI.
3.2 Pharmacokinetic Profile
3.2.1 Study 001
The pharmacokinetic parameters for elacestrant in Study 001 are summarized in Table 2. After a single oral dose under fasted conditions, elacestrant was absorbed rapidly with mean tmax ranging from 1.6 to 3.3 h (Table 2 and Fig. 3a). Based on geometric mean plasma concentrations, for doses ≥ 10 mg, secondary peaks were observed at 4.5–5.0 h post-dose. The mean t1/2 with single oral doses up to 200 mg ranged from 27.4 to 32.5 h.

Geometric mean elacestrant plasma concentration values plotted on a logarithmic scale against time since last dose. Individual plots were derived from subjects in: a Study 001, SAD part, following a single dose of elacestrant. b Study 001, MAD part, following the last dose (day 7) of elacestrant. c Study 004, following the last dose (day 7) of elacestrant. aThe elacestrant plasma concentrations for this group were below LLOQ (< 0.05 ng/ml) and therefore these data could not be presented. SAD single ascending dose, MAD multiple ascending dose
Plasma concentration-time curves plotted after the last dose of elacestrant in the MAD part of Study 001 are shown in Fig. 3b. The mean plasma concentrations of elacestrant were notably higher after the last dose than the first dose, a pattern consistent with accumulation associated with once-daily administration. The geometric means of trough plasma concentrations plateaued around day 6, suggesting a steady state was achieved after approximately six doses. Steady-state t1/2s with multiple dosing (31.1– 47.3 h) were higher than those seen with single doses (27.4–32.5 h), possibly related to the longer observation phase following the last dose of the multiple-dose regimen (72 h) compared to the observation phase following single doses (48 h).
Urine excretion of elacestrant was low after single and multiple dosing, with a maximum of 0.04% of a given dose recovered in the urine and a renal clearance of ≤ 2.3 ml/min.
Oral elacestrant pharmacokinetic exposure increased slightly more than linear dose proportionality. Comparing the pharmacokinetic parameters across the dose range of 1–200 mg in the SAD part, the slopes (β) of the power model equation [log(AUC) = α + β·log(dose)] for AUC0–last, AUC0–∞ and Cmax were 1.34 (90% confidence interval [CI] 1.21–1.47), 1.40, (90% CI 1.30–1.50) and 1.21 (90% CI 1.13–1.28), respectively. Similarly, in the MAD part, comparing the steady-state pharmacokinetic parameters across the dose range of 10–200 mg on Day 7, the slopes (β) of the power model equation for AUC0–τ and Cmax were 1.38 (90% CI 1.29–1.46) and 1.35 (90% CI 1.27–1.43). The values of β were all slightly > 1, suggesting slightly nonlinear dose-proportional increases in elacestrant Cmax and AUC.
3.2.2 Absolute Bioavailability and Food Effect
The absolute bioavailability of elacestrant was calculated in Study 001 by comparing pharmacokinetic parameters following oral (100 mg) vs. intravenous (1 mg) administration of a single dose in the same cohort of subjects receiving both treatments in the fasted state (Cohort 4, Fig. 1 and Table 2). An exploratory analysis estimated that the absolute bioavailability of the oral dose of elacestrant using logarithmically transformed ratios of AUC0–last and AUC0–∞ was 0.10 (90% CI 0.08–0.13) and 0.11 (90% CI 0.08–0.14), respectively (Supplemental Table S1).
Elacestrant plasma concentrations increased more slowly when administered in the fed state (FDA recommended high-fat, high-calorie meal) compared to the fasted stated (Fig. 1), increasing the mean tmax by about 2 h, from 1.9 h in fasted subjects to 4.2 h in fed subjects (Table 2). The high-fat, high-calorie meal also increased relative bioavailability. Comparing the 50 mg single oral dose in fed vs. fasted conditions, the ratio of geometric means for Cmax was 2.06 (90% CI 1.62–2.62), and the ratios for AUC0–last and AUC0–∞ were 1.57 (90% CI 1.38–1.80) and 1.57 (90% CI 1.39–1.78), respectively (Supplemental Table S1).
3.2.3 Study 004
The geometric mean elacestrant plasma concentrations after 7 days of oral dosing up to 1000 mg daily are plotted in Fig. 3c. The mean tmax ranged between 3.3 and 4.5 h and was independent of dose. Trough plasma concentrations, which were assessed from day 5 onward, showed that steady state was already achieved by day 5 (Supplemental Figure S1). Steady-state t1/2s ranged from 37.5 to 41.6 h, similar to values observed with multiple oral dosing of 10–200 mg daily in the MAD part of Study 001, and appeared similar regardless of dose.
3.3 Pharmacodynamic Results and BBB Penetration
In Cohort 6 of Study 004, ERα occupancy in the brain, pituitary, and uterus was assessed using static and/or dynamic PET scanning. The Vt and SUV in the pituitary could not be corrected for background signal because no suitable non-target reference region was available. Hence, the Vt and SUV in the pituitary without background correction consist partly of receptor-mediated signal and partly of non-specific uptake, leading to an underestimation of receptor occupancy. In contrast to pituitary, individual brain regions showed only very low levels of (non-specific) tracer uptake, both on baseline and post-dose PET scans. As a result, ERα occupancy in the brain could not be assessed. Results from static PET scans of the uterus showed 83% ERα occupancy in subjects receiving 200 mg elacestrant daily and 92% in subjects receiving 500 mg daily (Table 3 and Fig. 4). ERα occupancy in the pituitary (not corrected for background) ranged from 33 to 42%.

Representative ERα occupancy based on FES-PET imaging of the uterus at baseline and after 6 days of oral elacestrant administration at: a 200 mg and b 500 mg. 18F-FES-PET scans 4 h post-dose on day 6 in Cohort 6 of Study 004 in a subject 3 from subgroup 1 (elacestrant 200 mg daily) and b subject 7 from subgroup 2 (elacestrant 500 mg daily)
The CSF concentrations and the ratio of CSF to plasma concentrations of elacestrant were also assessed in Cohort 6 of Study 004. The median CSF concentrations were 0.0966 and 0.155 ng/ml for subjects receiving 200 mg and 500 mg, respectively, and the corresponding median CSF-to-plasma concentration ratios were 0.205% and 0.126%, respectively (Supplemental Table S2). A plot of elacestrant concentrations in CSF compared to plasma in individual subjects is shown in Supplemental Figure S2.
3.4 Safety and Tolerability
The safety analysis of Study 001 included all subjects who received at least one dose of elacestrant (n = 62) or placebo (n = 18; Fig. 1). Because each subject participated in two study periods, with different doses of elacestrant or fasted vs. fed conditions in each period, for the purpose of calculating the proportion of subjects with treatment-related TEAEs in elacestrant and placebo groups, each subject was counted for the number of periods they had taken at least one dose of study drug (n = 85 for elacestrant treatment periods and n = 26 placebo treatment periods; Tables 4, 5). Among all cohorts in Study 001, 33 of 85 (39%) elacestrant-treated subjects and 7 of 26 (27%) placebo-treated subjects experienced at least one treatment-related TEAE. All were of mild intensity, and none led to study discontinuation. In addition, there were no clinically significant changes in laboratory tests, vital signs, physical examination findings or ECGs, and there were no deaths or other serious AEs. The most common treatment-related TEAEs were in the system organ classes of gastrointestinal disorders (mainly nausea and dyspepsia) and nervous system disorders (mainly headache). In Study 001 SAD part (single-dose elacestrant), among all oral doses tested from 1 to 200 mg, and intravenous dose of 1 mg, the most common treatment-related TEAEs were dry mouth, esophageal pain, nausea and headache (each 4%) (Table 4). In Study 001 MAD part, with multiple oral dosing up to 200 mg daily, the most common treatment-related TEAEs reported by ≥ 10% of subjects included dyspepsia and headache (each 18%), nausea (16%) and dizziness (11%) (Table 5).
The safety analysis of Study 004 included all subjects who received at least one dose of elacestrant (n = 44) or placebo (n = 8; Fig. 2). Among treatment-related TEAEs reported by ≥ 10% of subjects, the most common were in the system organ class of gastrointestinal disorders (89% in elacestrant-treated subjects vs. 38% in those taking placebo; Table 6). Common gastrointestinal treatment-related TEAEs in elacestrant-treated subjects included nausea (43%), dyspepsia (36%), vomiting (32%), abdominal pain and esophageal pain (each 23%), salivary hypersecretion (18%) and diarrhea and dysphagia (each 16%). Most of these treatment-related gastrointestinal TEAEs were grade 1 or 2 in severity. Treatment-related TEAEs of Grade 3 intensity in elacestrant-treated subjects included two events of esophageal spasm (750 mg and 1000 mg dose groups), one event of vomiting (500 mg dose group), one event of esophageal pain (1000 mg dose group) and one event of syncope (1000 mg dose group). There were no treatment-related TEAEs of Grade 4 severity. Treatment-related TEAEs in the system organ class of nervous system disorders were reported in 41% of subjects taking elacestrant vs. 25% in those taking placebo, with headache being the most common (23%). Sensation of foreign body (16% vs. 0%), hot flush (25% vs. 13%) and hiccups (20% vs. 0%) were also more common in elacestrant-treated subjects. All non-gastrointestinal treatment-related TEAEs occurring in ≥ 10% of subjects were Grade 1 or 2 in severity.
Most gastrointestinal TEAEs (including nausea, dyspepsia, vomiting and esophageal pain) occurred within 30 min of dosing, except for diarrhea and abdominal discomfort, which occurred from 1 to 5 h after dosing. Likewise, the time of onset for sensation of foreign body was generally within 15 min after dosing. Most nervous system TEAEs (including headache) were reported 5 h after dosing.
There were no deaths or other serious AEs. Nine subjects, all of whom were in elacestrant groups at oral daily doses of ≥ 500 mg, were withdrawn from treatment because of 1 or more TEAEs (Table 6), including 3 of 14 subjects in the 500 mg dose group, 2 of 8 subjects in the 750 mg dose group and 4 of 7 subjects in the 1000 mg dose group. Only one of the TEAEs (Grade 1 fatigue), reported by a subject who withdrew, was considered unrelated to treatment, and this subject also had treatment-related gastrointestinal TEAEs of Grade 2 and 3 severity. The majority of treatment-related TEAEs leading to withdrawal from the study were gastrointestinal TEAEs (Grade 3 for 3 subjects, Grade 2 for 5 subjects and Grade 1 for 1 subject). All TEAEs that led to treatment withdrawal were transient and resolved within 1–5 days.
An exploratory analysis was performed to evaluate the relationship between elacestrant plasma concentrations and values of corrected QT intervals (QTcF) (Supplemental Figure S3A) and QTcF change from baseline (Supplemental Figure S3B) from study 004 (200–1000 mg daily for 7 days). The analysis included only data when both elacestrant plasma concentration and QTcF measurements were obtained at the same time point. Slight trends toward longer QTcF and toward decreased QTcF change from baseline were observed with increasing elacestrant plasma concentrations. Across all elacestrant doses, the maximum QTcF measured was 467 ms, and the maximum QTcF change from baseline was 24 ms, suggesting that oral elacestrant doses of up to 1000 mg daily (2.5 × the anticipated therapeutic dose of 400 mg) do not adversely affect cardiac repolarization.
Given that the majority of treatment-related TEAEs were gastrointestinal AEs, mostly Grade 1–2 in severity, and that there were no clinically significant changes in laboratory tests, vital signs, physical examination findings or ECGs, deaths or other serious AEs, the investigators determined that the MTD for elacestrant was not reached.
4 Discussion
Elacestrant is a novel, nonsteroidal SERD that is currently being evaluated as a potential therapy for ER+ advanced or metastatic breast cancer [10, 21]. The aims of the two first-in-human phase 1 studies in healthy postmenopausal women presented here were to (1) characterize the pharmacokinetic profile of elacestrant (including bioavailability and food effect) following single and repeated oral dosing ranging from 1 to 1000 mg daily; (2) assess the pharmacodynamic profile of elacestrant via ERα occupancy in the uterus, pituitary and brain based on 18F-FES-PET imaging; (3) assess the BBB penetration of elacestrant; (4) evaluate the safety and tolerability across the dose ranges tested; (5) establish the MTD of elacestrant in healthy postmenopausal women.
Elacestrant exhibits rapid oral absorption. Under the fasted condition, mean tmax ranged from 1.0 to 3.9 h after single and repeated dosing across the 10–200 mg dose range evaluated in Study 001. Plasma concentration profiles of elacestrant exhibited secondary peaks at 4.5–5.0 h after administration, suggesting enterohepatic circulation of elacestrant and/or metabolites.
For IV administration, the mean systemic clearance (36.8 l/h [Table 2]) represents approximately 2/3 of the typical liver plasma flow rate (approximately 55 l/h, corresponding to liver blood flow rate of 90 l/h). This suggests that the high plasma protein binding of elacestrant (> 99% bound) does not limit the systemic clearance. Similarly, the large mean volume of distribution (1730 liters, [Table 2], approximately 25 l/kg) indicates that elacestrant is extensively distributed out of the bloodstream where it binds to the body’s tissues. This also suggests that the high plasma protein binding of elacestrant does not limit its distribution. The mean t1/2 for IV administration was 33.4 h (Table 2).
A food effect on elacestrant pharmacokinetics was observed. A high-fat breakfast 1 h before the intake of an oral elacestrant 50 mg capsule delayed the peak plasma concentration by approximately 2 h and increased the overall Cmax by approximately twofold and AUC by 1.6-fold compared to the fasted state, possibly because of delayed gastric emptying or increased solubility in the fed state. Although the effect of a light meal was not directly evaluated, comparing across studies, the 200 mg dose group in Study 004 (in which an oral elacestrant capsule was administered after a light meal) exhibited only slightly higher Cmax (51.6 vs. 43.5 ng/ml) and AUC values (695 vs. 627 ng·h/ml) on day 7 compared to the 200 mg group in Study 001 at day 7 (fasting administration). For this same comparison of 200 mg between Study 001 (fasted administration) and Study 004 (light meal), the mean tmax was 3.3 and 3.4 h, respectively, which also indicates a smaller food effect for the light meal than for the high-fat, high-calorie meal.
The t1/2 of elacestrant ranged from 27.4 to 47.3 h in both studies. Consistent with the long t1/2 of elacestrant, steady-state was achieved after 5 or 6 days of once-daily dosing. Elacestrant was absorbed after oral dosing with an absolute bioavailability of 10%. The bioavailability of elacestrant was likely limited by the low, pH-dependent solubility (≥ 5 mg/ml at pH 4.5 and 0.0174 mg/ml at pH 6.8) and low permeability of elacestrant, but it is sufficient, along with the other pharmacokinetic characteristics, to allow elacestrant to be administered orally once a day. In comparison, the much lower bioavailability and pre-systemic metabolism of fulvestrant, the only SERD currently marketed, require that a long-acting formulation be administered as two 5-ml intramuscular injections, one in each buttock, at an outpatient clinic every month [4, 22].
Systemic exposure (AUC) and Cmax increased with dose in a slightly more than dose-proportional manner over the dose range of 25–200 mg in Study 001 and across the 200–1000 mg dose range in Study 004. For Study 001 and Study 004, the power model equation produced slope parameters (β) ranging from 1.21 to 1.47, suggesting slightly nonlinear dose proportionality. To put these slopes into context, these equations predict that a doubling (i.e., a twofold increase) in dose would be expected to increase the Cmax or AUC by 2.31-fold to 2.77-fold (i.e., 21.21-fold to 21.47-fold), which are only slightly higher than the expected 2.00-fold increase with linear dose proportionality. This represents a small deviation from linear dose proportionality that may not be important for dose titration in individual patients. Correspondingly, a reduction in dose from 400 to 300 mg, which may occur during treatment with elacestrant, would be expected to decrease the Cmax or AUC to 0.706-fold to 0.655-fold of the original exposure (i.e., 0.751.21-fold to 0.751.47-fold).
The PET imaging results showed that uterine ERα occupancy was robust, with > 75% occupancy in the 200 mg dose group and > 90% in the 500 mg dose group. Thus, higher doses are likely unnecessary to achieve maximum activity. The high ERα occupancy of elacestrant compares favorably with previous data that suggest incomplete ER inhibition in 6 out of 16 (38%) patients treated with fulvestrant at the current standard dose of 500 mg [23]. The robust ERα occupancy may at least in part explain the preliminary antitumor activity demonstrated in preclinical models and observed in a phase 1 study in postmenopausal women with ER+ breast cancer [11,12,13, 17].
Elacestrant penetrates the BBB, but concentrations in the CSF were low. Although fulvestrant has been shown to penetrate the brain and hypothalamic tissue in a rat model [24], its ability to cross the BBB has not been reported in humans. In an intracranial MCF-7 MBC mouse model, elacestrant-treated animals survived longer than those treated with either fulvestrant or control [12]. In that study, elacestrant concentrations in plasma and in the intracranial tumor were 738 ± 471 ng/ml and 462 ± 105 ng/ml, respectively, suggesting that elacestrant effectively crossed the BBB. In contrast, our clinical study showed very low CSF concentrations of elacestrant. However, given that our attempt to evaluate ERα occupancy in the brain was unsuccessful, coupled with low CSF concentrations, the potential of elacestrant in the treatment of ER+ MBC that has metastasized to the brain remains unclear. Nevertheless, BBB penetration may be improved in patients with brain metastases where the BBB may be disrupted, and these are the patients most likely to benefit from the treatment.
Elacestrant was well tolerated by most participants with oral doses up to 500 mg daily. At doses > 500 mg daily, gastrointestinal events were poorly tolerated, with some subjects discontinuing treatment because of the events. The most frequently reported TEAEs were nausea, dyspepsia, vomiting and headache. Gastrointestinal TEAEs appear to be dose-related. Some of the upper gastrointestinal TEAEs, such as esophageal spasms and pain, may be related to the rapid release of drug from the capsule formulation and the high number of capsules (up to 7) that had to be taken by the participants. Indeed, gastrointestinal toxicity appeared to be reduced in postmenopausal breast cancer subjects treated with the 400 mg tablet formulation compared to those treated with the 400 mg capsule formulation in Study RAD1901-005 [17]. Importantly, there were no deaths, other serious AEs or safety signals based on laboratory, ECG or physical findings. Based on these findings, the MTD was not reached. Although there were more subjects with TEAEs leading to study discontinuation in the 1000 mg dose group, this dose was 2.5-fold higher than the dose currently being studied (400 mg QD) in patients with ER+/HER2− advanced or MBC.
Limitations of both studies (001 and 004) include: (1) these were first-in-human dose-finding studies with small sample size designed to provide information on the pharmacokinetics, pharmacodynamics, safety, tolerability and MTD of elacestrant; (2) studies were carried out in healthy subjects with intact BBB; (3) both studies were conducted with the capsule formulation of elacestrant, which has since been replaced with a tablet formulation.
5 Conclusions
Data from 2 phase 1 studies in healthy postmenopausal subjects demonstrate that elacestrant is safe and well tolerated at oral doses up to 500 mg per day. Robust ERα occupancy (75–90%) was observed at doses of 200–500 mg daily. The bioavailability and long t1/2 of elacestrant support a once-daily oral dosing strategy. Collectively, the pharmacokinetic profile of elacestrant, ERα occupancy and safety data in the dose range tested support the 400 mg daily oral dosing strategy selected for further evaluation in an ongoing phase 3 clinical trial (NCT03778931) in postmenopausal women and men with ER+/HER2− advanced or metastatic breast cancer [10, 21].
Change history
30 August 2020
Authors would like to correct the errors in table 2.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. https://doi.org/10.3322/caac.21551.
Huang HJ, Neven P, Drijkoningen M, Paridaens R, Wildiers H, Van Limbergen E, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol. 2005;58(6):611–6. https://doi.org/10.1136/jcp.2004.022772.
National Comprehensive Cancer N. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Breast Cancer Version 2.2020-Feb 9, 2020.
FASLODEX [prescribing information]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2019.
Robertson JFR, Bondarenko IM, Trishkina E, Dvorkin M, Panasci L, Manikhas A, et al. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. Lancet. 2016;388:2997–3005. https://doi.org/10.1016/S0140-6736(16)32389-3.
Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425–39. https://doi.org/10.1016/S1470-2045(15)00613-0.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018;36(24):2465–72. https://doi.org/10.1200/JCO.2018.78.9909.
Sledge GW, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2-advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875–84. https://doi.org/10.1200/JCO.2017.73.7585.
Turner NC, Slamon DJ, Ro J, Bondarenko I, Im S-A, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379(20):1926–36. https://doi.org/10.1056/NEJMoa1810527.
Bardia A, Aftimos P, Bihani T, Anderson-Villaluz AT, Jung J, Conlan MG, et al. EMERALD: phase III trial of elacestrant (RAD1901) vs endocrine therapy for previously treated ER+ advanced breast cancer. Future Oncol. 2019;15(28):3209–18. https://doi.org/10.2217/fon-2019-0370.
Bihani T, Patel HK, Arlt H, Tao N, Jiang H, Brown JL, et al. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor acvity in multiple ER(+) breast cancer patient-derived xenograft models. Clin Cancer Res. 2017;23(16):4793–804. https://doi.org/10.1158/1078-0432.CCR-16-2561.
Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs. 2015;26(9):948–56. https://doi.org/10.1097/cad.0000000000000271.
Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer. 2015;22(5):713–24. https://doi.org/10.1530/erc-15-0287.
Arlt H, Garner F, Bihani T. Elacestrant (RAD1901) demonstrates anti-tumor activity in a fulvestrant-resistant PDX model [abstract]. In: Proceedings of 2017 San Antonio breast cancer symposium; 5–9 Dec 2017; San Antonio, TX. Cancer Res. 2018;78(4 Suppl):P4-04-17.
Patel H, Tao N, Arlt H, Bihani T. Anti-tumor activity of elacestrant (RAD1901) in models harboring ESR1 mutations resistant to standard of care therapies [abstract]. In: Proceedings of the 2018 San Antonio breast cancer symposium; 4–8 Dec 2018; San Antonio, TX. Cancer Res. 2019;74 (4 Suppl):P6-20-08.
Patel HK, Tao N, Lee KM, et al. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res. 2019;21(1):146. https://doi.org/10.1186/s13058-019-1230-0.
Kaklamani V, Bardia A, Wilks S, Weise A, Richards D, Harb W, et al. Final analysis of phase 1 study of elacestrant (RAD1901), a novel selective estrogen receptor degrader (SERD), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2−) advanced breast cancer. Cancer Res. 2020;80(4 Suppl):PD7-07.
FDA. Guidance for industry: food-effect bioavailability and fed bioequivalence studies. Rockville: Food and Drug Administration; 2002.
Römer J, Steinbach J, Kasch H. Studies on the synthesis of 16-alpha-[18F]fluoroestradiol. Appl Radiat Isot. 1996;47:395–9.
Khayum MA, de Vries EFJ, Glaudemans AWJM, Dierckx RAJO, Doorduin J. In vivo imaging of brain estrogen receptors in rats: a 16α-18F-fluoro-17β-estradiol PET study. J Nucl Med. 2014;55:481–7.
ClinicalTrials.gov. Phase 3 trial of elacestrant vs. standard of care for the treatment of patients with ER+/HER2− advanced breast cancer (EMERALD). https://clinicaltrials.gov/ct2/show/NCT03778931. Accessed 12 Sept 2019.
Robertson JF, Harrison M. Fulvestrant: pharmacokinetics and pharmacology. Br J Cancer. 2004;90(Suppl 1):S7–10. https://doi.org/10.1038/sj.bjc.6601630.
van Kruchten M, de Vries EG, Glaudemans AW, van Lanschot MC, van Faassen M, Kema IP, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 2015;5(1):72–81. https://doi.org/10.1158/2159-8290.Cd-14-0697.
Alfinito PD, Chen X, Atherton J, Cosmi S, Deecher DC. ICI 182,780 penetrates brain and hypothalamic tissue and has functional effects in the brain after systemic dosing. Endocrinology. 2008;149(10):5219–26. https://doi.org/10.1210/en.2008-0532.
Acknowledgements
The authors thank Phillips Gilmore Oncology Communications, Inc., for professional assistance with manuscript preparation, which was funded by Radius Health, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by Radius Health, Inc., on behalf of its wholly owned subsidiary, Radius Pharmaceuticals, Inc.
Competing interests
EFJDV: received payment from Radius Health, Inc., for the execution and analysis of the PET scans. AWJMG: no competing interest. YW, ST and MGC are employees and stockholders of Radius Health, Inc.
Ethics approval
The clinical study protocols and informed consent forms were reviewed and approved by an independent ethics committee, and all subjects provided written informed consent. Both studies were conducted in accordance with the principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonisation (ICH) E6 Guideline for Good Clinical Practice (GCP) (Committee for Proprietary Medicinal Products (CPMP) guideline CPMP/ICH/135/95), and with the EU CTD: Directive 2001/20/EC.
Informed consent
All subjects were informed verbally and in writing of the objectives, procedures and risks of study participation including possible side effects and potential interactions. All participants signed the informed consent form before starting the studies.
Availability of data and materials
Data that underlie the results reported in a published article may be requested for further research 6 months after completion of FDA or EMA regulatory review of a marketing application or 18 months after trial completion (whichever is latest and/or if not associated with a marketing application). Radius will review requests individually to determine whether (1) the requests are legitimate and relevant and meet sound scientific research principles and (2) are within the scope of the participants’ informed consent. Prior to making data available, requestors will be required to agree in writing to certain obligations, including without limitation, compliance with applicable privacy and other laws and regulations. Proposals should be directed to info@radiuspharm.com.
Additional information
The original version of this article was revised: Errors in table 2 corrected.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Conlan, M.G., de Vries, E.F.J., Glaudemans, A. et al. Pharmacokinetic and Pharmacodynamic Studies of Elacestrant, A Novel Oral Selective Estrogen Receptor Degrader, in Healthy Post-Menopausal Women. Eur J Drug Metab Pharmacokinet 45, 675–689 (2020). https://doi.org/10.1007/s13318-020-00635-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-020-00635-3