
Abstract
Background and Objectives
The importance of quercetin and flavonoids in the diet and as food supplements is well known, and literature studies support their potential use to treat several human diseases. Many beneficial properties have been described for quercetin, so much effort has been directed into overcoming the major drawbacks of this natural compound—its poor solubility and low oral absorption. The aims of this study were to compare a new food-grade lecithin-based formulation of quercetin, Quercetin Phytosome®, to unformulated quercetin in terms of solubility in simulated gastrointestinal fluids and oral absorption in a randomized crossover pharmacokinetic study of healthy volunteers.
Methods
The solubility of the new formulation was determined by in vitro incubation in simulated gastrointestinal fluids, and quercetin was detected by ultra performance liquid chromatography. A single-dose, randomized, six-sequence/three-period crossover clinical trial (3 × 3 × 3 crossover design) with a balanced carryover effect was conducted in healthy volunteers under fasting conditions. Twelve healthy volunteers of both sexes with an age range of 18–50 years were recruited; one dose of quercetin and two different doses of Quercetin Phytosome were administered orally as film-coated tablets. Pharmacokinetic samples were collected at twelve time points (from 0 h to 24 h) after administration, and quercetin levels were measured by HPLC/MS/MS. Data were analyzed using the Phoenix WinNonlin (v.6.4) software package, and the most significant pharmacokinetic parameters were calculated. Statistical analysis involved performing a two-way ANOVA with repeated measures followed by post hoc analysis (Tukey’s test).
Results
Significant improvements in both in vitro solubility and oral absorption (in terms of both exposure and maximum concentration achieved) by healthy volunteers in a human clinical study were obtained with the Quercetin Phytosome formulation as compared to unformulated quercetin.
Conclusions
A more soluble formulation of quercetin based on lecithin, Quercetin Phytosome, has recently been developed, and was found to facilitate the attainment of very high plasma levels of quercetin—up to 20 times more than usually obtained following a dose of quercetin—when the novel formulation was administered orally in human volunteers, and it did not have any notable side effects. These results suggest that Quercetin Phytosome allows the oral administration of quercetin in a safe and bioavailable manner, thus facilitating the effective utilization of this natural compound to treat various human diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
It is well known from the literature that flavonoids, especially quercetin, are very important biological molecules, strongly suggesting their potential use to treat several human diseases |
A new food-grade lecithin-based formulation of quercetin, Quercetin Phytosome, was developed and validated in healthy volunteers. |
Quercetin Phytosome overcomes the low bioavailability hurdle of quercetin and should help to fulfill the great health benefit potential of this flavonoid in the diet and as food supplements |
1 Introduction
Quercetin (3,3′,4′,5,7-pentahydroxyflavone) is a natural flavonoid compound widely found in vegetables, fruits, and nuts. Major dietary sources of quercetin are apple, onions, tomatoes, broccoli, lettuce, and black and green tea [1]. A great number of important biological activities of quercetin have been discovered in recent years by different groups, so interest in giving benefit to human health by administering quercetin as a food supplement or dietary component has rapidly grown. Quercetin has been reported to exhibit antioxidant, antitumoral, antinflammatory, antimicrobial, antibacterial, and antiviral properties [2, 3]; moreover, it has been found to exert anti-aging, antithrombotic, antiaggregatory, and vasodilatory effects [4,5,6].
However, the low solubility and low absorption of quercetin limits its practical use [2], so a great deal of research has been directed into overcoming those liabilities. Different delivery systems to enhance the water solubility of quercetin have been developed, including systems based on liposomes, nanoparticles, nanoemulsions, and micelles. The encouraging results of those studies, mainly performed in vitro and evaluated in vivo in rodent species, support the hypothesis that increasing the water solubility of quercetin would enhance its oral bioavailability. For this reason, in the work reported in the current paper, we focused on a new formulation of quercetin called Quercetin Phytosome, in which food-grade lecithin is utilized to deliver the quercetin. This novel formulation of quercetin was studied and tested to evaluate its potential advantages in terms of both solubility and bioavailability in healthy human volunteers over unformulated quercetin.
2 Methods
2.1 Formulation
Quercetin (batch no.: 45092, formula: C15H10O7, CAS no. 117-39-5) and its lecithin formulation Quercetin Phytosome® (QUERCEFIT™, batch no.: 06/16/PA) used in the study were prepared and provided by Indena SpA (Milan, Italy). Quercetin Phytosome consists of quercetin and sunflower lecithin in a 1:1 weight ratio along with about a fifth part of food-grade excipients that are added to improve the physical state of the product and to standardize it to a HPLC-measured total quercetin content of about 40% (patent application no. 171816341).
For the clinical study, 500 mg of quercetin were formulated by Indena SpA into film-coated tablets containing anhydrous calcium phosphate (Di-Cafos® A150, Budenheim, Germany), hydroxypropylmethylcellulose (Methocel™ E15, Dow, Germany), silicon dioxide (Syloid® 244FP, Grace GmbH, Germany), polyvinylpolypyrrolidone (Kollidon® CL, BASF, Germany), talc (Microtalc Pharma 50, Mondo Minerals BV, Netherlands), and magnesium stearate (Ligafood®, Peter Greven, Netherlands). For the clinical study, 250 mg of Quercetin Phytosome were formulated by Indena SpA into film-coated tablets containing anhydrous calcium phosphate, Isomalt (GalenIQ™ 960, Beneo GmbH, Germany) polyvinylpolypyrrolidone, silicon dioxide, talc, and magnesium stearate.
All tablets were coated with a hydroxypropylmethylcellulose-based film coating system (Opadry White, Colorcon Inc., USA). Before releasing the film-coated tablets containing quercetin (batch no. 89107) or Quercetin Phytosome (batch no. 89108), their appearance, average mass, uniformity of mass, HPLC-measured content of quercetin, disintegration time, and microbiological quality were tested.
2.2 Solubility Study
The solubility of Quercetin Phytosome was determined under saturation conditions and compared with that of unformulated quercetin under the same saturation conditions and in the following simulated gastrointestinal media: FaSSGF pH 1.6 (fasted-state simulated gastric fluid), FaSSIF pH 6.5 (fasted-state simulated intestinal fluid), and FeSSIF pH 5.0 (fed-state simulated intestinal fluid). In order to clarify the influence of the manufacturing process of Quercetin Phytosome on the solubility of quercetin, a physical mixture with the same quali/quantitative composition as Quercetin Phytosome was also prepared and submitted to the solubility study. Biological media (Biorelevant.com, London, UK) were prepared according to the manufacturer’s instructions. In order to ensure that the quercetin concentrations in all of the samples were very similar, about 20 mg of quercetin, about 50 mg of Quercetin Phytosome and about 50 mg of the physical mixture, with the same composition as Quercetin Phytosome, were added to 10 ml of each simulated biological fluid. The resulting suspensions were left for 2 h at room temperature under constant magnetic stirring. After that period, an aliquot of each suspension was filtered through a 0.2-µm polytetrafluoroethylene (PTFE) syringe disposable filter, and 1 µl of the clarified solution was injected and analyzed for its content of quercetin by ultra performance liquid chromatography (UPLC). Chromatographic separation of quercetin was achieved by a reversed-phase UPLC method, with UV detection at 371 nm, using a Waters Acquity UPLC system. Briefly, a Waters Acquity BEH C18 column (100 mm × 2.1 mm, particle size 2.7 µm) kept at 27 °C was eluted at constant flow of 0.368 ml/min with a gradient of H3PO4 0.3% in water as solvent A and acetonitrile as solvent B according to the following timetable (linear gradient): initial conditions B 15%; 1.55 min B 15%; 5.33 min B 44%; 7.59 min B 50%; 8.73 min B 55%; 9.10 min B 95%; 10.60 min B 95%; total run time (including the re-equilibration step): 12.00 min. The samples were kept at 5 °C in silanized glass vials prior to analysis.
2.3 Clinical Study
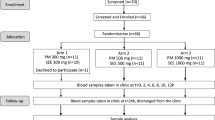
A single-dose, randomized, six-sequence/three-period crossover clinical trial (3 × 3 × 3 crossover design) with a balanced carryover effect was performed in healthy volunteers under fasting conditions to evaluate the oral absorption of the Quercetin Phytosome in comparison to that of quercetin.
2.4 Subjects
Healthy volunteers of both sexes who were within the age range of 18–50 years inclusive and had body mass index values within the range 18.5–27 participated in the study. No evidence of significant organic or psychiatric diseases (based on history, physical examination, and additional tests) was observed, and negative serology for hepatitis B (HBV) and C (HCV) viruses as well as for human immunodeficiency virus (HIV) was verified. Laboratory tests (blood count, biochemistry, and urine sediment) were performed according to the normal reference values of the laboratory of biochemistry, University Araba Hospital, Txagorritxu headquarters, Vitoria-Gasteiz. Whether variations were acceptable depended on the clinical judgment of the investigator. Vital signs (blood pressure, heart and respiratory rate, temperature) and ECG results were monitored to ensure that they were within normal limits, and pregnancy tests were performed in the women by determining plasma human β-chorionic gonadotropin (β-HCG) levels during the selection phase and in urine before each experimental period.
The study was carried out in accordance with the relevant guidelines of the Declaration of Helsinki (1964) and its amendments and the general principles of the ICH Harmonised Tripartite Guidelines for Good Clinical Practice (ICH Topic E6, CPMP/ICH/135/95). At the beginning of the study, written informed consent (reviewed by the ethics committee) was obtained from all individual participants included in the study, which was performed in accordance with the ICH-GCP, the Declaration of Helsinki, and the regulatory and legal requirements of Spain. The Spanish Research Ethics Committee of Araba University Hospital (Vitoria-Gasteiz, Alava, Spain) approved the study on 20th January 2017.
2.5 Study Design
A randomized crossover pharmacokinetic clinical study of the three different treatments of quercetin administered in a single dose to healthy volunteers under fasting conditions was performed. Patients were told not to consume quercetin-containing foods from at least 72 h prior to day 1 until the end of the study. There was no control group who used a placebo or another treatment; each individual acted as his/her own control. The volunteers were selected, and in each case an anamnesis was compiled and a physical examination, ECG, and analytical tests were performed. Participants were asked to refrain from excessive consumption of quercetin-containing foods (a list of quercetin-containing foods was provided to them) at least from 72 h prior to day 1 until the end of the study. Each experimental session required hospitalization, and participants remained in hospital under the supervision of qualified personnel for up to 12 h after the administration of the product, with subsequent monitoring at 24 h post-administration. Additional tests for substance abuse and pregnancy were performed before the administration of the product.
On each experimental day (a day when the product was given), an indwelling cannula for blood sample collection in a forearm vein was placed, the product was administered orally, and blood sampling was performed at the predefined times.
At least 1 week after completing the third experimental period, the final examination was performed. This involved a new clinical and analytical evaluation of all participants, including a new physical examination, ECG, and analytical tests that were similar to the original set of tests but with the serology (hepatitis, HIV), substance abuse test, and β-HCG determination omitted. Therefore, clinical safety (evaluation of vital signs and adverse systemic effects) and biological safety (evaluation of each subject’s blood count and blood chemistry results) were monitored to determine the tolerability of the treatments.
Each volunteer received one film-coated tablet of quercetin 500 mg (treatment A, batch no. 89107), one film-coated tablet of Quercetin Phytosome 250 mg (treatment C, batch no. 89108), and two film-coated tablets of Quercetin Phytosome 250 mg (500 mg total; treatment B, batch no. 89108) on the three experimental days, administered according to a previously randomized sequence. Treatments were known to the investigator (the clinical research products were identified as A, B, or C), but the aliquoted samples for pharmacokinetic analysis were not labeled according to the treatment applied, i.e., the analyst was blinded to the treatment associated with each sample.
A balanced carryover effect in healthy volunteers was realized, with washout periods of at least one week between the three treatment periods.
In order to determine quercetin levels, blood samples were collected at the following 12 time points: before dosing (time 0), at 15, 30, 45, and 60 min, and at 2, 3, 4, 6, 8, 12, and 24 h after the administration of the compounds.
2.6 Sample Preparation and Analysis
Blood and blank samples (8 ml each) were transferred to pre-labeled K2-EDTA Vacutainer tubes on ice. After centrifugation, 1 ml of each plasma sample was mixed with 100 µl of ascorbic acid solution 10% v/v as a preservative and stored at − 80 °C until it was analyzed. Pretreatment of the blank human plasma with ascorbic acid (10% v/v) was necessary to stabilize the quercetin before the extraction process. Samples (220 µl) were analyzed using a method previously validated by Kymos Pharma Services S.L. Briefly, the free quercetin component was determined by HPLC MS/MS (HPLC: Agilent series 1100 with a Luna C18(2) column, 5 μm 4.6 × 50 mm, Phenomenex 008-4252-EO; MS: MDS Sciex API-3200 equipped with a TurboIonSpray ion source, used in conjunction with the Analyst software package, version 1.4.2) after a liquid–liquid extraction with ethyl acetate. The same procedure was applied for total quercetin (free and conjugated), but the samples were subjected to enzymatic hydrolysis by β-glucuronidase from Helix pomatia (BBI Enzymes GH2G) before liquid extraction. Analysis was performed in the presence of the internal standard quercetin-d3. The concentration range of the method, in which a linear fitting model (1/χ2) was applied, was set from the lower limit of quantification (1 ng/mL) to 1000 ng/mL. The method proved to be selective, linear, precise, and accurate when applied to quercetin determination. The plasma samples were analyzed in four chromatographic batches for free quercetin and six chromatographic batches for total quercetin. Each batch included a set of calibration standards (concentration range: 1–1000 ng/mL), blanks (blank human plasma), a zero sample (blank spiked with internal standard), and quality control samples at three different concentrations (nominal concentrations of 3, 30, and 800 ng/mL). The chromatographic batches were accepted if they complied with the acceptance criteria defined for the calibration curve and quality control samples. Quality control samples corresponding to three concentration levels (3, 30, and 800 ng/mL) were prepared in each validation batch. In each chromatographic batch, the minimum number of quality control samples (in multiples of three) was at least 5% of the number of unknown samples or six quality control samples, whichever was greater.
Plasma concentrations were analyzed by a noncompartmental model, and the following major pharmacokinetic parameters were calculated by Phoenix WinNonlin (v.6.4; Pharsight): Cmax (maximum plasma concentration), Tmax (time to achieve Cmax), AUClast (area under the plasma concentration vs time curve), t1/2 (elimination half-life), and MRT (mean residence time).
Statistical data analysis was performed by two-way ANOVA with repeated measures followed by post hoc analysis (Tukey’s test).
3 Results
3.1 Solubility Studies
The results of the comparative solubility study of quercetin, Quercetin Phytosome, and the physical mixture with the same quali/quantitative composition as Quercetin Phytosome in simulated gastrointestinal fluids are reported in Table 1. Under strongly acidic conditions (FaSSGF pH 1.6), no detectable solubility was observed in the quercetin samples, and the quercetin in both the Quercetin Phytosome and the physical mixture was found to have very low solubility. In FeSSIF pH 5.5, the phospholipid was seen to have a positive effect on quercetin solubility, as there was a clear increase in the solubility of the quercetin in the Quercetin Phytosome as compared to that in the physical mixture. This improvement in solubility was also noted, albeit to a lesser extent, when FaSSIF pH 6.5 was utilized.
3.2 Pharmacokinetic Studies
Taking into account that solubilization in the gastrointestinal fluids is a well-known prerequisite for effective absorption, and based on the highly promising results for Quercetin Phytosome obtained in the solubility study described above, a clinical study in healthy volunteers was performed in order to verify that the enhanced solubility of the new phytosome-based formulation permits greater absorption of quercetin in humans. The aim of this study was to compare the pharmacokinetic profiles of quercetin and Quercetin Phytosome® when used as active ingredients. The influence of the dosage on this comparison was minimized because the two formulations involved in the study (both of which were film-coated tablets) were characterized by similar disintegration times. A total of 12 subjects were enrolled in the study. The mean ± SD values for the age, weight, bone mineral density, and height were 25.0 ± 7.5 years, 64.1 ± 11.0 kg, 22.5 ± 2.4 g/m2, and 168.4 ± 9.0 cm, respectively.
No withdrawal occurred in the trial.
All chromatographic batches were found to be acceptable based on the acceptance criteria defined for quality control samples (see the “Methods” section). For free quercetin, the mean precision was below or equal to 6.75%, and the accuracy of the quality control samples ranged between − 0.43% and 7.39%. The correlation coefficients were higher than 0.99 and the intercepts were close to zero.
For total quercetin, the mean accuracy ranged between 2.13% and 7.17% and the precision was below or equal to 5.07%. Figure 1 shows chromatograms of a blank sample only, a zero sample (basal) with the internal standard, and samples spiked with quercetin standards of various concentrations. The quercetin plasma concentrations, expressed in ng/mL, obtained for each treatment during the clinical study are reported in Fig. 2. The concentrations observed with both doses of Quercetin Phytosome were statistically significantly higher than those seen after quercetin administration. While the quercetin concentration was always below 10 ng/ml in the quercetin-treated group, 1 tablet (250 mg) and 2 tablets (250 mg each) of Quercetin Phytosome led to maximum quercetin concentrations of about 100 ng/mL and 170 ng/mL, respectively.

Original MS/MS chromatograms of quercetin in plasma for a a blank sample only (left) and with the internal standard (right), b a zero sample only (left) and with the internal standard (right), c a sample with the quercetin standard corresponding to the lower limit of calibration (1 ng/mL; left) and the internal standard (right), and d a sample with the quercetin standard corresponding to the upper limit of calibration (1000 ng/mL; left) and with the internal standard (right)

Pharmacokinetic profile of quercetin in the clinical study. The plasma concentrations of quercetin obtained after single oral administration of the unformulated quercetin at 500 mg/tablet and after single oral administration of its corresponding lecithin formulation, Quercetin Phytosome, at a dose of either 500 or 250 mg are shown. Data are plotted as the mean value + SD, n = 12 for each point. *P < 0.0001 in comparison to treatment A (Tukey’s test). §P < 0.005 in comparison to treatment C (Tukey’s test)
Table 2 shows the calculated pharmacokinetic parameters obtained with the three treatments. A Cmax of 223 ng/ml was achieved with Quercetin Phytosome 500 mg, i.e., about 20-fold higher than that attained with quercetin, and the AUC for Quercetin Phytosome 500 mg was about 18-fold larger than that of quercetin, demonstrating a statistically significant improvement in quercetin absorption when using the lecithin formulation rather than the unformulated quercetin at the same dose.
Administering Quercetin Phytosome 250 mg resulted in the same pharmacokinetic profile as produced by Quercetin Phytosome 500 mg but with the values of Cmax and AUC half those seen with the highest dose, implying the presence of a dose-dependent relationship.
3.3 Safety
No significant differences were observed between the three treatments in vital signs, physical examination results, or ECG results, so the study demonstrated that the new formulation is safe and well tolerated, and is as safe as unformulated quercetin.
4 Discussion
In the present work, a more soluble quercetin utilizing a delivery system based on food-grade lecithin—Quercetin Phytosome—was developed. The solubility of quercetin in that formulation was shown to be considerably higher than that of unformulated quercetin, leading to a significant improvement in quercetin bioavailability when the new formulation was administered to human volunteers in a clinical study. Dose linearity was also demonstrated for the new quercetin formulation, based on the two doses administered. It is noteworthy that the Quercetin Phytosome treatment was well tolerated and that no notable side effects were reported.
Like other similar antioxidant flavonoids, quercetin is considered to be a free-radical scavenger of highly reactive species such as peroxynitrite and the hydroxyl radical [7]; for that reason, quercetin is considered to have notable beneficial health effects. Quercetin has been reported to exhibit antimicrobial [8], anti-allergic and anti-inflammatory [9, 10], angioprotective (in heart disease) [11], anticancer [12,13,14], anti-obesity, anti-diabetic, and gastroprotective [15, 16] effects. Therefore, quercetin (together with flavonoids in general) is an important dietary supplement for relieving human disorders.
It is well known that quercetin is a compound with low water solubility (only 0.01 mg/ml [17]), low stability (which depends on the temperature, pH, presence of metal ions, and glutathione), and low bioavailability. These characteristics limit its benefits and mean that a high dosage (500–1000 mg) must be applied to achieve the desired biological effects. The poor water solubility of this compound has always made oral delivery of quercetin problematic, because the compound must ideally be in solution for it to be well absorbed from the gastrointestinal tract.
Furthermore, when it is administered orally, quercetin undergoes a series of metabolic degradation steps [2, 18] at different levels of the gastrointestinal system: interactions with salivary gland proteins to form aggregates in the mouth [19], degradation to phenolic acids (e.g., protocatechuic acid) with limited absorption in the stomach at low pH, as well as extensive glucuronidation, sulfation, and methylation at the intestinal and hepatic levels [20]; and reconversion of the resulting glucuronide derivatives to quercetin by enzymes (β-glucosidase) present in microbiota [21]. A large number of metabolites are therefore produced, including quercetin-3-O-β-d-glucuronide (Q3GA), which is the major metabolite of quercetin that circulates in the bloodstream.
Several attempts have been made to improve quercetin bioavailability by reformulating the quercetin. Quercetin-loaded nanoparticles [22], where the hydrophobic quercetin fits well into the lipid structure of a nanocarrier, have been utilized to deliver quercetin to cancer cells [23]. Self-assembling lecithin-based mixed polymeric micelles (LMPM) containing quercetin [24] have also been described. Good results were obtained with these LMPM after oral exposure of male Sprague–Dawley rats, including an approximately threefold increase in AUC, a longer half-life, and reduced clearance when compared to unformulated quercetin. However, no data are currently available for quercetin-LMPM in humans. The same profile in terms of improved values of pharmacokinetic parameters in rat species was achieved with a quercetin-containing self-nanoemulsifying drug delivery system (Q-SNEDDS), a formulation described in 2014 [25]. These nanotechniques show great promise and have enabled researchers to identify new drug delivery systems; however, they also require highly specialized laboratories and facilities.
A number of recent studies utilizing less sophisticated but still useful and complex techniques have shown the beneficial effects of phospholipid complexation for improving the solubility, oral absorption, and biological effects of active substances [26]. A solubility study demonstrated that phospholipid complexation increased the water solubility of quercetin approximately 13-fold; when the resulting formulation was administered orally to rats, three- to fourfold increases in the AUC and Cmax were observed with respect to the corresponding values of free quercetin.
Liposomes can also improve the water solubility, reduce the toxic effects of, and control the release of substances entrapped in them. The liposomes protect the encapsulated compound from external stimuli, such as light, enzymes, extreme temperatures, and pH variations [27, 28]. Polymeric micelles have also been found to be very useful for encapsulating many drugs, including quercetin [29].
In the present work, a new formulation of quercetin based on a phytosome delivery system created, developed, and industrialized by Indena was studied. A phytosome is a solid dispersion of natural substances including lecithin. Therefore, phytosomes can be used to naturally promote the solubilities of poorly bioavailable active natural ingredients and their abilities to cross biological barriers, leading to significant improvements in bioavailability and thus biological activity [30]. Water-soluble phytoconstituents such as quercetin are incorporated into the phospholipids in a phytosome, in contrast to a liposome, which is an aggregate of phospholipid molecules, including unbound active molecules. The hydrophilic quercetin is released from the phytosome into the lipid environment of an enterocyte membrane, allowing it to penetrate into the bloodstream. Data in the present work showed that incorporation into phytosomes led to increased quercetin solubility, and consequently to a significant improvement in its bioavailability in humans. Results obtained from pharmacokinetic human studies showed substantial increases in absorption, AUC, and Cmax when Quercetin Phytosome was administered rather than quercetin alone. Dose linearity was demonstrated for Quercetin Phytosome, based on the two doses administered. In addition, the Quercetin Phytosome treatment was well tolerated and no notable side effects were reported.
5 Conclusions
In conclusion, a new safe formulation of quercetin, Quercetin Phytosome, was successfully developed and validated in human healthy volunteers. The application of this new quercetin formulation overcomes the low bioavailability hurdle of quercetin.
References
Rothwell JA, Perez-Jimenez J, Neveu V, Medina-Remón A, M’hiri N, García-Lobato P, et al. Phenol-Explorer 3.0: a major update of the Phenol-Explorer database to incorporate data on the effects of food processing on polyphenol content. Database (Oxford). 2013;2013:bat070.
Wang W, Sun C, Mao L, Ma P, Liu F, Yang J, et al. The biological activities, chemical stability, metabolism and delivery systems of quercetin: a review. Trends Food Sci Technol. 2016;56:21–38.
D’Andrea G. Quercetin: a flavonol with multifaceted therapeutic applications? Fitoterapia. 2015;106:256–71.
Chondrogianni N, Kapeta S, Chinou I, Vassilatou K, Papassideri I, Gonos ES. Anti-ageing and rejuvenating effects of quercetin. Exp Gerontol. 2010;45(10):763–71.
Chopra M, Fitzsimons PE, Strain JJ, Thurnham DI, Howard AN. Nonalcoholic red wine extract and quercetin inhibit LDL oxidation without affecting plasma antioxidant vitamin and carotenoid concentrations. Clin Chem. 2000;46(8 Pt 1):1162–70.
Erlund I, Kosonen T, Alfthan G, Mäenpää J, Perttunen K, Kenraali J, et al. Pharmacokinetics of quercetin from quercetin aglycone and rutin in healthy volunteers. Eur J Clin Pharmacol. 2000;56(8):545–53.
Amić A, Lučić B, Stepanić V, Marković Z, Marković S, Dimitrić Marković JM, et al. Free radical scavenging potency of quercetin catecholic colonic metabolites: thermodynamics of 2H+/2e− processes. Food Chem. 2017;218:144–51.
Ganesan S, Faris AN, Comstock AT, Wang Q, Nanua S, Hershenson MB, et al. Quercetin inhibits rhinovirus replication in vitro and in vivo. Antiviral Res. 2012;94(3):258–71.
Shaik YB, Castellani ML, Perrella A, Conti F, Salini V, Tete S, et al. Role of quercetin (a natural herbal compound) in allergy and inflammation. J Biol Regul Homeost Agents. 2006;20(3–4):47–52.
Chirumbolo S. The role of quercetin, flavonols and flavones in modulating inflammatory cell function. Inflamm Allergy Drug Targets. 2010;9(4):263–85.
Larson AJ, Symons JD, Jalili T. Therapeutic potential of quercetin to decrease blood pressure: review of efficacy and mechanisms. Adv Nutr. 2012;3(1):39–46.
Russo GL, Russo M, Spagnuolo C, Tedesco I, Bilotto S, Iannitti R, et al. Quercetin: a pleiotropic kinase inhibitor against cancer. Cancer Treat Res. 2014;159:185–205.
Sak K. Site-specific anticancer effects of dietary flavonoid quercetin. Nutr Cancer. 2014;66(2):177–93.
Li S, Yuan S, Zhao Q, Wang B, Wang X, Li K. Quercetin enhances chemotherapeutic effect of doxorubicin against human breast cancer cells while reducing toxic side effects of it. Biomed Pharmacother. 2018;100:441–7.
Zhao Y, Chen B, Shen J, Wan L, Zhu Y, Yi T, et al. The beneficial effects of quercetin, curcumin, and resveratrol in obesity. Oxid Med Cell Longev. 2017;2017:1459497.
Kawabata K, Mukai R, Ishisaka A. Quercetin and related polyphenols: new insights and implications for their bioactivity and bioavailability. Food Funct. 2015;6(5):1399–417.
Gao L, Liu G, Wang X, Liu F, Xu Y, Ma J. Preparation of a chemically stable quercetin formulation using nanosuspension technology. Int J Pharm. 2011;404(1–2):231–7.
Mullen W, Edwards CA, Crozier A. Absorption, excretion and metabolite profiling of methyl-, glucuronyl-, glucosyl- and sulpho-conjugates of quercetin in human plasma and urine after ingestion of onions. Br J Nutr. 2006;96(1):107–16.
Manach C, Scalbert A, Morand C, Rémésy C, Jiménez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr. 2004;79(5):727–47.
Rich GT, Buchweitz M, Winterbone MS, Kroon PA, Wilde PJ. Towards an understanding of the low bioavailability of quercetin: a study of its interaction with intestinal lipids. Nutrients. 2017;9(2):111.
Németh K, Plumb GW, Berrin JG, Juge N, Jacob R, Naim HY, et al. Deglycosylation by small intestinal epithelial cell beta-glucosidases is a critical step in the absorption and metabolism of dietary flavonoid glycosides in humans. Eur J Nutr. 2003;42(1):29–42.
Bonferoni MC, Rossi S, Sandri G, Ferrari F. Nanoparticle formulations to enhance tumor targeting of poorly soluble polyphenols with potential anticancer properties. Semin Cancer Biol. 2017;46:205–14.
Sarkar A, Ghosh S, Chowdhury S, Pandey B, Sil PC. Targeted delivery of quercetin loaded mesoporous silica nanoparticles to the breast cancer cells. Biochim Biophys Acta. 2016;1860(10):2065–75.
Chen LC, Chen YC, Su CY, Hong CS, Ho HO, Sheu MT. Development and characterization of self-assembling lecithin-based mixed polymeric micelles containing quercetin in cancer treatment and an in vivo pharmacokinetic study. Int J Nanomed. 2016;11:1557–66.
Tran TH, Guo Y, Song D, Bruno RS, Lu X. Quercetin-containing self-nanoemulsifying drug delivery system for improving oral bioavailability. J Pharm Sci. 2014;103(3):840–52.
Chen ZP, Sun J, Chen HX, Xiao YY, Liu D, Chen J, et al. Comparative pharmacokinetics and bioavailability studies of quercetin, kaempferol and isorhamnetin after oral administration of Ginkgo biloba extracts, Ginkgo biloba extract phospholipid complexes and Ginkgo biloba extract solid dispersions in rats. Fitoterapia. 2010;81(8):1045–52.
Gang W, Jie WJ, Ping ZL, Ming DS, Ying LJ, Lei W, et al. Liposomal quercetin: evaluating drug delivery in vitro and biodistribution in vivo. Expert Opin Drug Deliv. 2012;9(6):599–613.
Kuo YC, Tsao CW. Neuroprotection against apoptosis of SK-N-MC cells using RMP-7- and lactoferrin-grafted liposomes carrying quercetin. Int J Nanomed. 2017;12:2857–69.
Lv L, Liu C, Li Z, Song F, Li G, Huang X. Pharmacokinetics of quercetin-loaded methoxy poly(ethylene glycol)-b-poly(l-lactic acid) micelle after oral administration in rats. Biomed Res Int. 2017;2017:1750895.
Semalty A, Semalty M, Rawat MS, Franceschi F. Supramolecular phospholipids–polyphenolics interactions: the PHYTOSOME strategy to improve the bioavailability of phytochemicals. Fitoterapia. 2010;81(5):306–14. https://doi.org/10.1016/j.fitote.2009.11.001.
Acknowledgements
We would like to express our gratitude to Dr. Paola Misiano for her valuable editorial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors Antonella Riva, Massimo Ronchi, Giovanna Petrangolini, Stefania Bosisio, and Pietro Allegrini are employees of Indena SpA.
Funding
We did not receive any specific grant for this research from funding agencies in the public or not-for-profit sectors. The study was funded by Indena SpA.
Ethical approval
All procedures in this study are in accordance with the 1964 Helsinki Declaration (and its amendments) and the criteria of the ethics committee that approved the study.
Informed consent
Written informed consent, as reviewed by the ethics committee, was obtained individually from all subjects who participated in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Riva, A., Ronchi, M., Petrangolini, G. et al. Improved Oral Absorption of Quercetin from Quercetin Phytosome®, a New Delivery System Based on Food Grade Lecithin. Eur J Drug Metab Pharmacokinet 44, 169–177 (2019). https://doi.org/10.1007/s13318-018-0517-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-018-0517-3