
Abstract
Background and Objective
Puquitinib mesylate (XC-302) is a new multiple-target anticancer inhibitor, which directly suppresses the activity of phosphatidylinositol 3-kinase (PI3K). This study was aimed to develop a sensitive and specific liquid chromatography electrospray ionization tandem mass spectrometry (HPLC–ESI MS/MS) method for the quantification and pharmacokinetic investigation of plasma puquitinib in cancer patients.
Methods
The analytes of human plasma were prepared by liquid–liquid extraction using methyl-t-butyl ether (MTBE). The plasma analytes were separated by HPLC on Thermo ODS Hypersil column (2.1 × 150 mm; 3 μm) at 25 °C with 5 mmol/L ammonium acetate (A)-acetonitrile (B) (30:70, v/v) as the mobile phase.
Results
The total run time was 3.5 min and the elution of puquitinib was at 1.38 min. The detection were analyzed by multiple reaction monitoring (MRM) mode with positive-ion electrospray ionization (ESI) interface using the respective [M + H]+ ions: m/z 318.2 → 261.1 for puquitinib and m/z 258.2 → 121.0 for the internal standard (etofesalamide). The optimized method provided a good linear relation over the concentration range of 1.00-500.00 ng/mL (r = 0.9944) for puquitinib. The intra-day and inter-day precision (relative standard deviation [RSD%]) were within 9.83%, and the intra-day and inter-day accuracy ranged from 91.05 to 103.26%. The lower limit of quantitation (LLOQ) was 1.00 ng/mL. The absolute extraction recovery was on an average of 50.43% for puquitinib and 49.3% for internal standard. In addition, the maximum plasma concentration (Cmax) of puquitinib in dosage from 50 to 800 mg/m2 in the human study showed an increased linearly (57.1–1289.2 ng/mL), which displayed that the concentrations had reached effective levels.
Conclusions
The optimized method was successfully applied to the pharmacokinetic profile study in human cancer patient plasma after the oral administration of puquitinib.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Established a sensitive, specific and precise HPLC–ESI MS/MS method for the determination of puquitinib levels in human plasma. |
HPLC–ESI MS/MS method is very suitable and valuable for the quantification and pharmacokinetic investigation of plasma puquitinib in cancer patients. |
1 Introduction
The signaling pathway of phosphatidylinositol 3-kinase (PI3K) is an essential component of malignant cellular processes [1, 2], which is very critical for the development, growth progression, proliferation and invasion of the malignant tumors [1,2,3,4]. The small molecule inhibitors targeting for the proteins of PI3K-pathway have been investigated as new anticancer [5,6,7,8,9]. Currently, several small molecule inhibitors of PI3K-pathway proteins are in early phase clinical trials [6, 8, 10]. Puquitinib Mesylate (XC-302) is a new multiple-target anticancer inhibitor, which directly suppresses the activity of PI3K. XC-302 has been developed independently by Xinchang Pharmacy Corp. (Zhejiang Medicine Co., Ltd. China) [9]. Previous studies have reported that XC-302 could inhibits the cellular proliferation of several cancer cell lines, such as lung, breast, ovarian, colon, prostate cancer, leukemia, lymphoma and osteosarcoma with the half-maximal inhibitory concentration (IC50) ranged between 0.5 and 2.0 μ [9, 10]. In addition, the preliminary studies have showed that XC-302 has a clear antitumor efficacy in vivo xenograft mice model [11, 12]. Although there have many pharmacological researches about XC-302, there is no any standard analytical method for XC-302 in human blood distribution, so it is necessary to develop analytical methods for the effective determination of XC-302 in biological fluids.
At present, many analytical methods have been described in the literature for the determination of small molecule drugs in biological or other matrices, such as high-performance liquid chromatography with ultraviolet detection (HPLC–UV), high-performance liquid chromatography with evaporative light scattering, gas chromatography coupled with mass (GC–MS), or liquid chromatography with mass spectrometry (LC–MS) [13, 14]. However, the above analytical methods have a lower limit of detection (LOD), which is not enough for pharmacokinetic and bioavailability studies. As we know, high-performance liquid chromatography-tandem mass spectrometry (HPLC–MS/MS) method could increase the throughput, detection sensitivity and chromatographic peak resolution [15, 16]. Until now there is no HPLC–MS/MS method exists for the determination of puquitinib to characterize pharmacokinetic properties. Thus, this study was mainly aimed to develop a sensitive and specific liquid chromatography electrospray ionization mass spectrometry method for the quantification and pharmacokinetic investigation of plasma puquitinib in cancer patients.
2 Methods
2.1 Chemicals and Reagents
Puquitinib mesylate was supplied by Xinchang Pharmaceutical Factory Zhejiang Medicine Co., Ltd. (Zhejiang, China, 99.4%); Etofesalamide (internal standard) was purchased from Guangzhou Institute for Drug Control (SFDA, 99.9%).
HPLC-grade reagents (methanol, acetonitrile and methyl-t-butyl ether) were obtained from Tedia Company Inc. (Fairfield, U.S.A.). Other HPLC-grade reagents were obtained from the Chemical Reagent Factory of Guangdong (Guangdong, China). Ultrapure water (Guangdong Shenzhou Pharmaceutical Co., Ltd.) was prepared by Milli-Q system. Blank human blood was collected from healthy drug-free volunteers. Plasma was obtained by centrifugation of blood treated with the anticoagulant sodium heparin. The patient samples were stored at − 80 °C in ultra cold freezers up to the time of analysis.
2.2 Instrumentation and Chromatographic ESI MS/MS Detection Conditions
An Agilent 1200 HPLC system (Agilent, U.S.A.) equipped with a vacuum degasser and an auto sampler was used in the study. The mass spectrometer was an ABI 2000 mass spectrometer from ABI (ABI, U.S.A.), and the chromatographic data system was Analyst version 1.4.2 (Agilent, U.S.A.).
The analytics were separated on a Thermo ODS Hypersil column (150 × 2.1 mm; 3 μm) with column temperature 25 °C. The mobile phase consisted of 5 mmol/L ammonium acetate and acetonitrile (30:70), and filtered with 0.45 μm filters in a Millipore solvent filtration apparatus. The flow-rate was 300 μL/min.
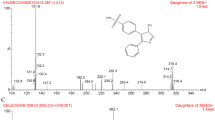
The compounds were ionized using electrospray ionization (ESI) ion source in the positive ion mode of the mass spectrometer. The scan mode was multiple reaction monitoring using the precursor ion at m/z (M + 1)+ (m/z: 318.2 for puquitinib, 258.2 for IS, Fig. 1) and the product ions (m/z: 261.1 for puquitinib, 121.0 for IS, Fig. 1) were used for quantification of puquitinib mesylate and IS, respectively. The detection conditions were as follows: CurtainGas (CUR): 35 eV; Ionspray Voltage (IS): 4500 eV; Temperature (TEM): 450 °C; Ion Source 1 (GAS 1): 0 Pa; Ion Source 2 (GAS 2): 65 Pa; Declustering Potential (DP): 35 eV; Focusing Potential (FP): 400 eV; Entrance Potential (EP): 10 eV; Collision Energy (CE): 40 eV.

Mass spectrum and the structure of puquitinib and internal standard (etofesalamide)
2.3 Standard and Quality Control
A stock solution containing 100 μg/mL of puquitinib was prepared by dissolving the appropriate amount of pure substance in methanol. Working solutions were obtained by diluting the stock solutions with methanol. All of the standard solutions were stored at − 20 °C.
Routine daily calibration curves were prepared in drug-free plasma. Appropriate volumes of working solution and drug-free human plasma were added to each test tube. The final concentrations were 1.00, 2.00, 10.00, 25.00, 100.00, 250.00 and 500.00 ng/mL. Quality control (QC) samples that were run in each assay were prepared in the same way, and the final concentrations were 3.00, 250.00, 400.00 ng/mL.
2.4 Sample Preparation
A volume of 200 μL plasma was added to a disposable Eppendorf tube, followed by spiking with 300 μL of internal standard (IS) solution (100 ng/mL) and 200 μL of 0.1 mol/L sodium hydroxide with vortex-mixed for 20 s, then added 3 mL methyl-t-butyl ether. The samples were vortex-mixed for approximately 2 min and centrifuged at 3000 rpm, 2 min. The upper layer of the organic phase was transferred to another set of clean glass tubes and evaporated to dryness under a stream of nitrogen at 40 °C. The residue was reconstituted in 200 μL mobile phase and vortex-mixed for 60 s; 20 μL solution was injected for analysis through the autosampling injector.
2.5 Validation of the Methods
Method validation assays were carried out according to the currently accepted FDA guidelines [17]. Validation runs were conducted on three consecutive days. The peak area ratios of puquitinib to IS of QC samples were interpolated from the calibration curve on the same day to give the concentration of puquitinib. Triplicate results from QC samples were used to evaluate the precision and accuracy of the method developed.
The selectivity was investigated by comparing chromatograms of six different batches of blank plasma from six subjects to those of corresponding standard plasma samples spiked with puquitinib and IS (Etofesalamide) and plasma sample after oral doses of puquitinib tablets.
2.6 Linearity
Each calibration curve consisted of seven calibration points covering the range from 1.00 to 500.00 ng/mL for puquitinib in plasma. The peak area ratios for puquitinib and IS were measured and a standard curve without the zero concentration was constructed. Calibration curves of seven concentrations of puquitinib from 1.00 to 500.00 ng/mL were extracted and assayed with weighted(1/x2)least squares linear regression.
2.7 Lower Limit of Quantitation (LLOQ)
For the LLOQ detection, a series of appropriate concentrations (0.50–500.00 ng/mL) of the blank plasma and blank plasma added with IS were analyzed to confirm the absence of interferences but were not used to construct the calibration, the aliquot of the diluted solutions was injected into HPLC for analysis. The LLOQ is defined as the lowest amount of an analyte in a sample such that the ratio of signal to noise was more than 10 with acceptable precision and accuracy.
2.8 Precision and Accuracy
The precision of the assay was determined from the QC plasma samples by replicate analyses of three concentration levels of Puquitinib (3.00, 250.00, 400.00 ng/mL). Within-batch precision and accuracy were determined by repeated analyses on three consecutive days (n = 6 series per day). The precision was calculated using the relative standard deviation (RSD) with RSD% = (standard deviation of the mean/mean) × 100%. Accuracy was calculated as the relative error (RE) with RE% = (measured concentration-nominal concentration)/nominal concentration × 100%. The concentration of each sample was determined using the calibration curve prepared and was analyzed on the same batch.
2.9 Extraction Recovery and Matrix Effect
The absolute recovery of extraction was calculated by comparing the results from the analysis of validation samples (heparinized) at analyzed concentrations of 3.00, 250.00, and 400.00 ng/mL with direct injection of solutions of puquitinib (in extracted blank plasma) at concentrations representing 100% recovery. The internal standard peak from 9 injections of validation samples was compared to that from 9 direct injections of internal standard (in extracted blank blood), representing 100% recovery.
Matrix effect was determined by comparing the areas from the analysis of puquitinib in extracted blank heparinized plasma at concentration of 250.00 ng/mL. Blank heparinized plasma samples from 6 different volunteers were analyzed for possible chromatographic interference. Puquitinib was also added to the 6 different heparinized plasma samples, and the matrix effect among plasma from different sources was analyzed.
2.10 Stability
For short-term stability, QC extracted plasma samples at three concentration levels were kept at room temperature for a period that exceeded the routine preparation time of the samples (approximately 72 h). For freeze and thaw stability, validation samples containing nominal puquitinib concentrations of 3.00, 250.00, and 400.00 ng/mL were frozen at − 80 °C and were then thawed to room temperature. This action was repeated 3 times a month prior to analysis in triplicate. For long-term stability, QC plasma samples at three concentration levels were kept at a low temperature (− 80 °C) and were studied for a period of 1 month.
2.11 Application to the Pharmacokinetic Study
The method was applied to evaluate the pharmacokinetics of puquitinib mesylate tablets in cancer patients. The study was approved by the Human Ethics Committee of Sun Yat-sen University Cancer Center. Informed consent was obtained from all subjects. The starting dose was 50 mg per m2 per patient, which was escalated to 100, 200, 400, and 800 mg per m2 per patient, all the patients were fasting and water or any other food restrictions for 8 h before the first blood harvested. Puquitinib Mesylate (XC302, Xinchang Pharmaceutical Factory Zhejiang Medicine Co., Ltd) was administered orally on day 1. Sixteen patients were enrolled in this trial, with a mean age of 53.69 years. Their mean height, weight and the body surface area were 163.97 cm, 60.91 kg and 1.66 m2, respectively (details shown in Table 1). A 4 mL blood sample was collected before dosing and 0.5, 1.0, 2.0, 3.0, 4.0, 5.0, 9.0, 12.0, 24.0, 48.0 and 72.0 h post-dosing. The patients were all taken care of for 24 h, i.e. during the entire experiment, in order to ensure their safety. Samples were centrifuged at 3000 rpm for 10 min to separate the plasma fractions. The collected plasma samples were stored at − 80 °C before analysis.
Plasma concentration was determined by HPLC–MS/MS. Data for puquitinib were analyzed using WinNonlin (Version 5.0). Peak concentrations (Cmax) were determined by visual inspection. The terminal elimination rate constants (λz) were determined by linear regression analysis of the terminal log-linear part of the concentration–time curve. The total area under the observed plasma concentration–time curve (AUC) and the area from time zero to the last measured concentration were determined using the linear-log trapezoidal rule. AUC values were extrapolated from the last observed time point to infinity by dividing the last measured concentration by λz [18].
3 Results
3.1 Methods Validation
3.1.1 Linearity
Calibration curves followed the regression equation y = a + bx over the concentration range from 1.00 to 500.00 ng/mL (regression equation: y = 6.14x-0.0038, r = 0.9944). A linear least-squares regression with a weighting index of 1/x2 was carried out on the peak area ratios of puquitinib and IS versus puquitinib concentrations of the six human plasma standards (in duplicate) to generate a calibration curve. The data of the linearity parameters of the method during the method validation are given in Table 2.
According to the data in vitro, the IC50 of puquitinib ranged from 0.5 to 2.0 μM; the LLOQ validated was 1.00 ng/mL (S/N > 10), which was defined as lowest concentration (RSD 7.76%). This satisfied the requirements of the pharmacokinetic assay.
3.1.2 Accuracy, Precision and Sensitivity
The mean extraction recoveries (mean ± SD), methodology recoveries (means ± SD), intra-day and inter-day precision for the analyte are shown in Table 3. The average extraction recovery for puquitinib was between 44.6 and 53.9%. The average methodology recovery for internal standard was 49.3%. The intra-day and inter-day RSDs were less than 9.83%.
3.1.3 Specificity
LLE chromatograms produced clean extracts with no interference from endogenous compounds with the retention of puquitinib or IS. Figure 2 shows the representative chromatograms of blank human plasma, as well as the LLOQ of puquitinib and IS in normal plasma and plasma from cancer patients. Typical retention time was sharp and symmetrical. The total run time was about 3.5 min.

HPLC–MS/MS chromatograph and mass spectrometric conditions for puquitinib. a Blank plasma; b blank plasma with internal standard; c: blank plasma with puquitinib and internal standard
3.1.4 Stability
Table 4 summarized the result of the short-term stability, freeze and thaw stability, and long-term stability of puquitinib. All of the results showed good stability during these tests; there were no stability-related problems during routine analysis of samples for pharmacokinetics studies.
3.1.5 Extraction Recovery and Matrix Effect
The extract recoveries of puquitinib from human plasma were 44.6, 53.9 and 52.8% at concentrations of 3.00, 250.00, and 400.00 ng/mL respectively. The mean extraction recovery of IS was 49.3%. Thus, the consistency in recoveries of puquitinib and IS supported the extraction procedure for its application to routine sample analysis.
The matrix effect is due to co-elution of some components present in biological samples. These components may not give a signal in MRM of target analyte but can certainly decrease or increase the analyte response dramatically to affect the sensitivity, accuracy and precision of the method. Thus, the evaluation of matrix effect due to the influence of co-eluting components on analyte ionization is necessary for an HPLC MS/MS method. All of the ratios defined in Sect. 2 were between 85 and 115%. No significant matrix effect for puquitinib was observed, indicating that no co-eluting substance influenced ionization of the analytes and IS.
3.2 Pharmacokinetic Study
The present method was successfully applied to the pharmacokinetics studies after oral administration of 50-800 mg puquitinib tablets to 16 cancer patients. Mean concentration–time profiles of puquitinib are displayed in Fig. 3. The parameters were calculated by a non-compartmental model. Pharmacokinetics parameters of puquitinib in dosage from 50 to 800 mg/m2 are shown in Table 5. As the dose increased, Cmax and AUC increased linearly. The results showed that absorption has reached the peak and the outcomes are consistent with those obtained from animal experiments: the concentrations had reached effective levels [19, 20].

Mean (SD) concentration–time curve of puquitinib in dosage from 50 to 800 mg/m2
4 Discussion
HPLC–ESI MS/MS operation parameters were carefully optimized for the determination of puquitinib. A standard solution (1000.00 ng/mL) of puquitinib and etofesalamide in methanol was directly infused into the mass spectrometer, with ESI as the ionization source. The mass spectrometer was tuned in both positive and negative ionization modes for puquitinib, which contains a secondary amino group. The response observed in positive ionization mode was higher than that observed in the negative ionization mode. In the precursor ion full-scan spectra, the most abundant ions were protonated molecules [M + H]+ at m/z 318.2 and 258.2. Parameters such as desolvation temperature, ESI source temperature, capillary and cone voltage, flow rate of desolvation gas and cone gas were optimized to obtain the highest intensity of protonated puquitinib molecules. The product ion scan spectra showed high abundance fragment ions at m/z 261.1 and 121.0 for puquitinib and IS, respectively. The collision gas pressure and collision energy of collision-induced decomposition (CID) were optimized for maximum response of the fragmentation of m/z 261.1 for puquitinib. The ion transitions of m/z 318.2 → 261.1 for puquitinib and m/z 258.2 → 121.0 for IS were selected for MRM (Fig. 1).
Chromatographic conditions were optimized to obtain high sensitivity and sample throughput. The mobile-phase systems of acetonitrile–water and methanol–water were tested at various proportions. The signal-to-noise (S/N) ratio of puquitinib was obviously higher with acetonitrile–water as the mobile phase than with methanol–water. Subsequent experiments utilized acetonitrile proportion from 60 to 90% in the mobile phase. In view of the response of puquitinib, retention times and peak shapes of both puquitinib and IS, 70% acetonitrile was optimal. The ionization of puquitinib and etofesalamide was increased by adding the additive in the mobile phase. Therefore, formic acid and ammonium acetate were added to the mobile phase to improve the response. The response of puquitinib was distinctly increased by adding ammonium acetate. The effect of ammonium acetate at 2 mmol/L, 5 mmol/L and 10 mmol/L in aqueous phase on the response and peak shape of puquitinib was investigated; 5 mmol/L ammonium acetate was found to be the best. Finally, acetonitrile–water containing 5 mmol/L ammonium acetate (70:30, v/v) was adopted as the mobile phase. The use of a C18 short column facilitated retention of the analyte and separation from endogenous interference. It provided a run time as short as 1.38 min per sample, which was shorter than values reported by others. Although the chromatography was performed on an HPLC column, the small dead volume of the Acuity system improved the separation efficiency and run time. Two channels were used for recording the response: channel 1 for puquitinib at a retention time of 1.38 min and channel 2 for the IS at a retention time of 2.20 min. No interference was observed for either puquitinib or IS.
In addition, as puquitinib is a lipophilic compound, liquid–liquid extraction (LLE) was chosen as the sample preparation method. The aim for sample preparation was to remove interferences from the biological sample. It should also be reproducible, with high recovery involving a minimum number of working steps. LLE gave both high recovery and good chromatography. Sodium hydroxide (1%) was added to the plasma to minimize protein binding. LLE was advantageous because this technique not only extracted the analyte and IS with sufficient efficiency and specificity but also minimized experimental cost. Ethyl acetate, diethyl ether and methyl-t-butyl ether were all tested as extraction solvents, and methyl-t-butyl ether was finally adopted because of its high extraction efficiency.
Furthermore, according to FDA guidelines [17], the internal standard for analysis of biological samples can be a structurally similar analog of the analyte or a stable labeled compound. Deuterated standard would be a preferred internal standard in the HPLC–MS assay; however, it is not always commercially available. Therefore, a compound that has structure, extraction recovery, and chromatographic and mass spectrometric behavior similar to the analyte may be considered. In our case, puquitinib and etofesalamide, which belong to different classes of compounds but exhibit structural similarity to the analyte, were tested as the internal standard.
5 Conclusion
The study described a sensitive, specific, accurate and precise HPLC–ESI MS/MS method for the determination of puquitinib levels in human plasma. Good linearity was observed in the range from 1.00 to 500.00 ng/mL. The LLOQ (1.00 ng/mL) makes this method of real practical use for pharmacokinetics, bioavailability and bioequivalence studies. The method was successfully applied to a pharmacokinetics study of puquitinib given in tablet form to cancer patients. As far as we know, this is the first study to investigate the validity of the HPLC MS/MS method for determination of puquitinib concentrations in vivo.
References
Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–62.
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19.
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44.
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62.
Hou MM, Liu X, Wheler J, Naing A, Hong D, Coleman RL, Tsimberidou A, Janku F, Zinner R, Lu K, Kurzrock R, Fu S. Targeted PI3K/AKT/mTOR therapy for metastatic carcinomas of the cervix: a phase I clinical experience. Oncotarget. 2014;5:11168–79.
Subbiah V, Brown RE, McGuire MF, Buryanek J, Janku F, Younes A, Hong D. A novel immunomodulatory molecularly targeted strategy for refractory Hodgkin’s lymphoma. Oncotarget. 2014;5:95–102.
Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–83.
Wang KF, Yang H, Jiang WQ, Li S, Cai YC. Puquitinib mesylate (XC-302) induces autophagy via inhibiting the PI3K/AKT/mTOR signaling pathway in nasopharyngeal cancer cells. Int J Mol Med. 2015;36:1556–62.
Yang H, Wang Y, Zhan J, Xia Y, Sun P, Bi XW, Liu PP, Li ZM, Li S, Zou BY, Jiang WQ. Puquitinib mesylate, an inhibitor of phosphatidylinositol 3-kinase p110δ, for treating relapsed or refractory non-Hodgkin’s lymphoma. Oncotarget. 2015;6(41):44049–56.
Thorpe PE, Chaplin DJ, Blakey DC. The first international conference on vascular targeting: meeting overview[J]. Cancer Res. 2003;63:1144–7.
Stephanic G, Jacqueline B, John C. Clinical trials in oncology. 3rd ed. Chapman & Hall/CRC; 2012. p. 30–34
Wang SH, Wu HY, Huang XL, Geng PW, Wen CC, Ma JS, Zhou YF, Wang XQ. Determination of N-methylcytosine in rat plasma by UPLC–MS/MS and its application to pharmacokinetic study. J Chromatogr B. 2015;990:118–24.
Wen CC, Wang SH, Huang XL, Liu ZZ, Lin YY, Yang SP, Ma JS, Zhou YF, Wang XQ. Determination and validation of hupehenine in rat plasma by UPLC–MS/MS and its application to pharmacokinetic study. Biomed Chromatogr. 2015;29(12):1805–10.
Ng SW, Ching CK, Chan AY, Mak TW. Simultaneous detection of 22 toxic plant alkaloids (aconitum alkaloids, solanaceous tropane alkaloids, sophora alkaloids, strychnos alkaloids and colchicine) in human urine and herbal samples using liquid chromatography-tandem mass spectrometry. J Chromatogr B. 2013;942–943:63–9.
Churchwell MI, Twaddle NC, Meeker LR, Doerge DR. Improving LC-MS sensitivity through increases in chromatographic performance: comparisons of UPLC-ES/MS/MS to HPLC–ES/MS/MS. J Chromatogr B. 2005;825:134–43.
United States Food and Drug Administration. Guidelines for mass spectrometry for confirmation of the identity of animal drug residues. Washington, DC, United States Government Printing Office, Docket No. 01D-0224:118–127.2001.
Eisenhauer EA, O’Dwyer PJ, Christian M, Humphrey JS. Phase I clinical trial design in cancer drug development. J Clin Oncol. 2000;18:684–92.
Parulekar Wendy R, Eisenhauer Elizabeth A. Phase I trial design for solid tumor studies of targeted, non-cytotoxic agents: theory and practice. J Natl Cancer Inst. 2004;96(13):990–7.
Gelmon KA, Eisenhauer EA, Harris AL, Ratain MF, Workman P. Anticancer agents targeting signaling molecules and cancer cell environment: challenges for drug development[J]. J Natl Cancer Inst. 1999;91:1281–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors are very grateful to Xinchang Pharmaceutical Factory Zhejiang Medicine Co., Ltd. (Zhejiang, China), for generously providing the puquitinib reference standard and tablet formulations.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethics Approval
All procedures in this study were in accordance with the 1964 Helsinki declaration (and its amendments), and the details of the Ethics Committee or institutional review board which approved the study.
Informed Consent
Informed consent was obtained from all subjects.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhan, J., Ding, Y., Zou, B. et al. Determination of Puquitinib in Human Plasma by HPLC–ESI MS/MS: Application to Pharmacokinetic Study. Eur J Drug Metab Pharmacokinet 43, 555–564 (2018). https://doi.org/10.1007/s13318-018-0468-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-018-0468-8