
Abstract
Background
Nintedanib is a substrate for p-glycoprotein which can impact bioavailability. We investigated the effects of ketoconazole, a p-glycoprotein inhibitor, and rifampicin, a p-glycoprotein inducer, on the pharmacokinetics of nintedanib.
Methods
In the ketoconazole study, 34 healthy subjects received nintedanib 50 mg orally alone and 1 h after the last dose of ketoconazole given orally at a dose of 400 mg once daily for 3 days in 1 of 2 randomized sequences. In the rifampicin study, 26 subjects received nintedanib 150 mg orally alone and the morning after the last dose of rifampicin given orally at a dose of 600 mg once daily for 7 days. The primary objective was to determine the relative bioavailability of nintedanib administered following multiple doses of ketoconazole or rifampicin versus alone, based on AUC from time 0 extrapolated to infinity (AUC0–∞) and maximum concentration (Cmax) calculated using an analysis of variance. Geometric mean ratios and 2-sided 90% CIs were calculated.
Results
Exposure to nintedanib increased when it was administered following ketoconazole versus alone (AUC0–∞: geometric mean ratio, 160.5% [90% CI, 148.2–173.7]; Cmax: geometric mean ratio, 179.6% [90% CI, 157.6–204.8]) and decreased when it was administered following rifampicin versus alone (AUC0–∞: geometric mean ratio, 50.1% [90% CI, 47.2–53.3]; Cmax: geometric mean ratio, 59.8% [90% CI, 53.8–66.4]). The time to reach Cmax (tmax) and half-life (t½) of nintedanib were unaffected by co-administration of ketoconazole or rifampicin.
Conclusions
Exposure to nintedanib is increased by co-administration of ketoconazole and decreased by co-administration of rifampicin, likely due to effects on bioavailability of the absorbed fraction.
ClinicalTrials.govidentifiers:NCT01679613, NCT01770392.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Exposure to nintedanib, a known substrate for p-glycoprotein (P-gp), is increased by co-administration of a P-gp inhibitor and decreased by co-administration of a P-gp inducer. |
This is believed to be due to effects on the bioavailability of the absorbed fraction. |
1 Introduction
Nintedanib (formerly known as BIBF 1120) is a potent intracellular inhibitor of tyrosine kinases [1, 2]. Nintedanib has been approved for the treatment of idiopathic pulmonary fibrosis (IPF) at an oral dose of 150 mg twice daily in several countries and regions including the US and Europe [3, 4] and has been approved at an oral dose of 200 mg twice daily in Europe for the treatment of non-small cell lung cancer with adenocarcinoma histology in combination with docetaxel [5].
The pharmacokinetics of nintedanib are comparable in healthy volunteers, patients with solid tumors [6,7,8,9], and patients with IPF [10]. Following oral administration, nintedanib is rapidly absorbed and reaches maximum plasma concentration after approximately 2–4 h; steady state is reached within 7 days of dosing [3,4,5]. Nintedanib displays at least biphasic disposition kinetics, with a terminal half-life of 10–15 h [9]. Nintedanib has a high total clearance (geometric mean 1390 mL/min) and a high volume of distribution (geometric mean 1050 L) [9]. All pharmacokinetic variables show moderate to high variability (geometric coefficient of variation [gCV] > 30%). The absolute bioavailability of nintedanib 100 mg in healthy volunteers is approximately 5%, with the percentage absorbed estimated to be 23%, confirming the large amount of metabolites formed during first pass metabolism [9].
The prevalent metabolic reaction for nintedanib is hydrolytic cleavage by esterases, resulting in the free acid moiety BIBF 1202 [3,4,5]. BIBF 1202 is subsequently glucuronidated by UGT enzymes, namely UGT 1A1, UGT 1A7, UGT 1A8, and UGT 1A10, to BIBF 1202 glucuronide. Only a minor extent of the biotransformation of nintedanib involves CYP pathways, with CYP3A4 being the predominant enzyme involved. In an absorption, distribution, metabolism, and elimination study conducted in healthy volunteers, the major CYP-dependent metabolite could not be detected in plasma. In vitro, CYP-dependent metabolism accounts for about 5% and ester cleavage for about 25% [3,4,5, 11]. However, nintedanib is a substrate for p-glycoprotein (P-gp) [3,4,5], which may be one factor influencing its low bioavailability [12]. Ketoconazole is an in vivo inhibitor of P-gp and CYP3A4 and rifampicin is an in vivo inducer of P-gp and CYP3A4 [12].
We conducted two studies to investigate the effects of ketoconazole and rifampicin on the pharmacokinetics of nintedanib to elucidate the effect of P-gp on the bioavailability of nintedanib.
2 Methods
Two single-center, open-label studies were undertaken in healthy subjects, one to determine the relative bioavailability of nintedanib (OFEV®, Boehringer Ingelheim Pharma GmbH & Co. KG, Germany) administered following multiple doses of ketoconazole (Polfarmex® S.A., Kutno, Poland) versus alone (ketoconazole study) and the other to determine the relative bioavailability of nintedanib administered following multiple doses of rifampicin (Eremfat®, Riemser Arzneimittel AG, Germany) versus alone (rifampicin study). Both clinical trial protocols were approved by the local independent ethics committee (Landesärztekammer Baden-Württemberg, Stuttgart, Germany) and the German Competent Authority (Bundesinstitut für Arzneimittel und Medizinprodukte, Bonn, Germany). The studies were conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Tripartite Guideline for Good Clinical Practice. Written informed consent was provided by every subject prior to entering the study.
2.1 Subjects
In both studies, healthy male subjects aged 18–55 years with a body mass index (BMI) of 18.5–29.9 kg/m2 were eligible to participate. Exclusion criteria included any clinically relevant laboratory values or findings on medical examination; cardiovascular, hepatic, hormonal, immunologic, renal, respiratory, metabolic, and gastrointestinal disorders; diseases of the central nervous system; psychiatric disorders; neurologic disorders; chronic or relevant acute infections; any other clinically relevant disease; history of orthostatic hypotension, fainting spells, blackouts; allergy/hypersensitivity; and alcohol or drug abuse.
2.2 Study Design
The ketoconazole study was an open-label, 2-way crossover study comprising two periods: a pilot period and a main study period. In the pilot period, eight healthy subjects received nintedanib 50 mg alone and 1 h after the last dose of ketoconazole administered at a dose of 400 mg once daily for 3 days, in 1 of 2 randomized treatment sequences. Randomization was undertaken using a computer-generated random code using a validated system involving a pseudo-random number generator and a supplied seed number to ensure that the allocation of subjects to treatment sequences was reproducible and non-predictable. Each dose of nintedanib was separated by a period of ≥ 14 days. A 50 mg dose of nintedanib was administered during the pilot period to ensure that exposure was not greater than that achieved with the 150 mg dose even if inhibition of P-gp by ketoconazole resulted in a greater than twofold increase in exposure. The tolerability of a 150 mg dose of nintedanib had been established in earlier trials [6,7,8]. According to the protocol, if acceptable safety and tolerability were confirmed during the pilot period, 26 different subjects were to receive nintedanib alone and with ketoconazole (400 mg once daily for 3 days) in 1 of 2 randomized treatment sequences in the main study period. The sample size of 26 healthy volunteers was determined for a precise estimation of the relative bioavailability ratio in terms of a desired precision represented by the half-width of the 90% confidence interval of the point estimate on the logarithmic scale. Based on an intra-individual variability (gCV) of 40% as observed in a previous trial, a 90% confidence interval for the ratio of the geometric means (test/reference) for the maximum concentration of drug in plasma (Cmax) would be obtained with a probability of 90% with a desired precision of about 0.24 (on the logarithmic scale). Each dose of nintedanib was to be separated by a period of ≥ 14 days. The dose of nintedanib (50 mg or 100 mg) and whether it was given 1 h before or after the last dose of ketoconazole during the main study period was based on the safety and tolerability of nintedanib 50 mg observed during the pilot period and on an interim pharmacokinetic analysis undertaken after the pilot period. According to the protocol, if the nintedanib dose used in the pilot period and main study period was the same (i.e. 50 mg), the data from both periods could be combined for the pharmacokinetic and statistical analyses.
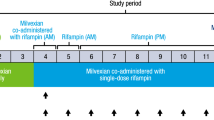
The rifampicin study was an open-label, two-period, fixed-sequence study, in which 26 healthy subjects received a single dose of nintedanib 150 mg alone and the morning after the last dose of rifampicin administered at a dose of 600 mg once daily for 7 days. Each dose of nintedanib was separated by a period of ≥ 14 days. A fixed sequence rather than a cross-over design was chosen for this study because the duration of the rifampicin effect is not known and so a suitable washout period could not be defined.
2.3 Outcomes
The primary objectives of these studies were to determine the relative bioavailability of nintedanib administered following multiple doses of ketoconazole or rifampicin versus alone based on the primary endpoints AUC from time 0 extrapolated to infinity (AUC0–∞) and Cmax. The secondary endpoint was AUC from time 0 to the last quantifiable concentration (AUC0–tz) for nintedanib. Other pharmacokinetic endpoints for nintedanib and its metabolites BIBF 1202 and BIBF 1202 glucuronide included time to achieve Cmax (tmax) and half-life (t½). The ratio of urinary concentration of 6β-hydroxycortisol to cortisol was determined in the rifampicin study. Rifampicin binds to the pregnane X receptor (PXR), which belongs to the nuclear receptor superfamily and acts as a ligand-activated transcription factor by binding to response elements in the promotor region of target genes including CYP3A and mdr1 (gene of the P-gp transporter). CYP3A is responsible for the transformation of cortisol to 6β-hydroxycortisol and hence measuring their ratio in urine provides a surrogate biomarker of the rifampicin-mediated PXR activation of mdr1 [13,14,15].
Safety and tolerability were evaluated based on adverse events (AEs), clinical laboratory tests, vital signs, 12-lead ECG, and physical examination. In the ketoconazole study, AEs occurring between the first intake of study drug and the first intake of the next study drug (including washout) or the start of the post-treatment period were assigned to the respective treatment period (nintedanib alone, ketoconazole alone, or nintedanib and ketoconazole). In the rifampicin study, AEs reported in the 72 h following administration of nintedanib alone or with rifampicin were assigned to the ‘nintedanib alone’ and ‘nintedanib and rifampicin’ periods. AEs reported more than 72 h after administration of nintedanib alone and before the first dose of rifampicin were assigned to the washout period.
2.4 Pharmacokinetic Analyses
Blood samples were collected for the quantification of plasma concentrations of nintedanib, BIBF 1202 and BIBF 1202 glucuronide. In the ketoconazole study, samples were collected pre-dose and at the following times after nintedanib administration: 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 10, 12, 15, 24, 36, 48, and 72 h. In the rifampicin study, samples were collected pre-dose and 0.5, 1, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 24, 36, 48, and 72 h after nintedanib administration.
Plasma concentrations of nintedanib, BIBF 1202 and BIBF 1202 glucuronide were determined by a validated LC MS/MS assay. All bioanalytical methods shared the same general outline. Deuterated analogs of the analytes were used as internal standards. A 50 µl plasma sample aliquot was mixed with the appropriate internal standard(s). All assays used sample clean-up by solid-phase extraction in the 96-well plate format. Linear concentration ranges, inter-assay precision, and inter-assay accuracy are summarized in Table 1. Chromatography was performed on an analytical reversed phase LC column (Phenomenex Luna (C18) 100 Å, 30 × 2.0 mm (3.0 μm particles) with gradient elution. Analytes and internal standards were detected and quantified by positive ion mode electrospray ionization tandem mass spectrometry (Sciex API 5000 LCMS/MS) in the multiple reaction monitoring mode. All samples were handled and analyzed within the time frame supported by stability experiments and within the concentration range where calibration curves were linear [9].
Cmax and tmax were determined directly from the plasma concentration–time profiles for each subject. The apparent terminal elimination rate constant λz was estimated from a regression of log(C) versus time over the terminal log-linear drug disposition portion of the concentration–time profiles. Half-life (t½) was calculated as the ratio of log(2) and λz. AUC values were calculated using the linear and log trapezoidal method for ascending and descending concentrations, respectively. AUC0–∞ was estimated as the sum of AUC to the last measured concentration, with the extrapolated area given by the quotient of the predicted last measurable concentration and λz.
For determination of the metabolic ratio of 6β-hydroxycortisol to cortisol in the rifampicin study, spot morning urine samples were collected the day before the first dose of rifampicin and before the nintedanib dose on the morning following the last dose of rifampicin. Study samples were analyzed according to a method previously validated at SGS Cephac Europe [16]. In summary, the analytical method consists of a solid phase extraction on Oasis HLB cartridges followed by reverse phase liquid chromatography with tandem mass spectrometric detection. The calibration curves of undiluted samples were linear over the range 1.00–100 ng/mL for cortisol and 10.0–3000 ng/mL for 6β-hydroxycortisol, using a urine volume of 500 μL.
2.5 Statistical Analysis
The relative bioavailability of nintedanib administered following multiple doses of ketoconazole or rifampicin (test [T]) compared with alone (reference [R]) was assessed using an analysis of variance (ANOVA) model. In the ketoconazole study, the ANOVA model included sequence, period, and treatment as fixed effects and subject within sequence as a random effect. In the rifampicin study, the ANOVA model included treatment as a fixed effect and subject as a random effect. Values for AUC0–∞, AUC0–tz and Cmax were log-transformed before fitting the ANOVA model. The difference between the expected means for log(T)–log(R) was estimated using the difference in the corresponding least squares means (point estimate), and 2-sided 90% CIs were calculated based on the t distribution. These values were back-transformed to the original scale to give the geometric mean ratio (GMR) and 2-sided 90% CIs for response under T versus R conditions. Other pharmacokinetic parameters were assessed descriptively.
3 Results
3.1 Subjects
Thirty-four subjects entered the ketoconazole study (eight in the pilot period and 26 in the main study period) (Fig. 1a). Nintedanib 50 mg 1 h after the last dose of ketoconazole was used in both the pilot and main study periods, therefore, analyses were undertaken using the combined dataset. All 34 subjects received at least one dose of study drug and were included in the treated set. Mean (SD) age of subjects was 35.9 (10.7) years, 33 (97.1%) were White, and mean (SD) BMI was 25.2 (2.4) kg/m2. During the pilot period, one subject was withdrawn due to elevated liver enzyme levels while receiving ketoconazole alone. During the main study period, two subjects were withdrawn due to elevated bilirubin levels while receiving ketoconazole alone. Thus, pharmacokinetic analyses were based on data from 31 subjects. Two further subjects were withdrawn during the main study period due to elevated bilirubin levels prior to receiving the second dose of nintedanib. These subjects had not received nintedanib + ketoconazole. The remaining 22 subjects completed the main study period. Thus, a total of 29 patients completed the pilot period or main study period.

Patient disposition in (a) the study with ketoconazole and in (b) the study with rifampicin
Twenty-six subjects entered the rifampicin study and all received at least one dose of study drug (Fig. 1b). Mean (SD) age of subjects was 37.3 (8.7) years, all were White, and mean (SD) BMI was 25.7 (2.3) kg/m2. One subject discontinued due to influenza during the washout period and did not receive nintedanib + rifampicin. The remaining 25 subjects completed the study.
3.2 Pharmacokinetics: Ketoconazole Study
Sufficient exposure to ketoconazole was achieved to inhibit CYP3A4 and P-gp after 3 days of treatment with ketoconazole 400 mg once daily (17.4 μM). Exposure to nintedanib (AUC0–∞, Cmax, AUC0–tz) was 1.6–1.7-fold higher when nintedanib was co-administered with ketoconazole than when administered alone (Fig. 2a and b; Table 2); tmax and t½ were similar (Table 2). The 90% CIs for the GMRs for AUC0–∞ and Cmax are shown in Table 3. Co-administration with ketoconazole influenced exposure to the nintedanib metabolites in a manner similar to its effects on nintedanib itself (data not shown).

gMean plasma concentration–time profiles of a single dose of nintedanib 50 mg alone and after multiple doses of ketoconazole 400 mg (a) linear scale and b semi-log scale; and a single dose of nintedanib 150 mg alone and after multiple doses of rifampicin 600 mg (c) linear scale and d semi-log scale
3.3 Pharmacokinetics: Rifampicin Study
Exposure to nintedanib (AUC0–∞, Cmax, AUC0–tz) was lower when nintedanib was co-administered with rifampicin than when it was administered alone (Fig. 2c and d; Table 4); tmax and t½ were similar (Table 4). The 90% CIs for GMRs for AUC0–∞ and Cmax are shown in Table 3. Co-administration with rifampicin influenced exposure to the nintedanib metabolites in a manner similar to its effects on nintedanib (data not shown). Rifampicin 600 mg once daily for 7 days increased the ratio of urinary concentrations of 6β-hydroxycortisol to cortisol by approximately fivefold (data not shown).
3.4 Safety
Administrations of single dose of nintedanib alone and with ketoconazole or rifampicin were well tolerated (Table 5 and 6). No serious AEs were reported. There was one severe AE (influenza during the washout period of the rifampicin study, which led to withdrawal of study treatment). There were no clinically relevant changes in laboratory measurements, ECG recordings, vital signs, or physical examination in either study.
4 Discussion
As nintedanib is a substrate for P-gp [3,4,5], two studies were undertaken to determine the effect of an inhibitor of P-gp (ketoconazole) and an inducer of P-gp (rifampicin) [12] on the pharmacokinetics of nintedanib. These studies showed that co-administration with ketoconazole increased exposure to nintedanib by about 1.6-fold based on AUC and 1.7-fold based on Cmax, while co-administration with rifampicin reduced exposure to nintedanib by about 50% based on AUC and 60% based on Cmax. These effects on nintedanib exposure are believed to be due to an increased or reduced fraction of the nintedanib dose reaching the systemic circulation when it is co-administered with ketoconazole or rifampicin, respectively. These findings in humans are consistent with observations made in rats upon pre-treatment with zosuquidar (a potent P-gp inhibitor), both with respect to the order of magnitude of the increase in exposure and translation of findings to the metabolites (data on file).
Ketoconazole is an inhibitor of CYP3A while rifampicin is an inducer of CYP3A4 in vivo [12]. Ketoconazole exposure was sufficient to inhibit CYP3A4 and P-gp [17]. Furthermore, the observed increase in the metabolic ratio of 6β-hydroxycortisol to cortisol, an established marker of human hepatic CYP3A4 induction [13], suggested adequate induction of CYP3A4 and P-gp by rifampicin. The lack of effect of ketoconazole and rifampicin on the tmax and t½ of nintedanib indicate that they did not affect the elimination kinetics of nintedanib via effects on P-gp or CYP3A. As the majority of metabolism of nintedanib is independent of CYP [11], interactions between nintedanib and CYP substrates, inhibitors, or inducers are not expected.
The increase in exposure of nintedanib when co-administered with a potent P-gp inhibitor is within the range of inter-subject variability; however, patients being treated with potent P-gp inhibitors should be monitored for tolerability of nintedanib. An a priori adjustment of the nintedanib dose is not required. Management of side effects due to higher exposure may require interruption, dose reduction, or discontinuation of nintedanib therapy [3,4,5]. On the other hand, due to the decrease in nintedanib exposure by potent P-gp inducers (e.g., rifampicin, carbamazepine, phenytoin, St. John’s Wort), co-administration of these compounds should be considered carefully [3,4,5].
5 Conclusions
Nintedanib exposure was increased when it was administered following multiple doses of the P-gp inhibitor ketoconazole and decreased following multiple doses of the P-gp inducer rifampicin. These results suggest that co-administration of P-gp inhibitors may increase exposure to nintedanib due to an increase in the bioavailability of the absorbed fraction while co-administration of P-gp inducers may decrease exposure to nintedanib. Guidance on how to manage patients who require administration of nintedanib and P-gp inhibitors/inducers is provided in the product labels.
References
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A, Rettig WJ. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–82.
Hostettler KE, Zhong J, Papakonstantinou E, Karakiulakis G, Tamm M, Seidel P, Sun Q, Mandal J, Lardinois D, Lambers C, Roth M. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15:157.
Boehringer Ingelheim Pharmaceuticals, Inc. (2017). OFEV™ (nintedanib) prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/205832s004lbl.pdf. Accessed 27 Apr 2017.
Boehringer Ingelheim (2015). OFEV (nintedanib) summary of product characteristics. February 2017. https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003821/WC500182474.pdf. Accessed 27 Apr 2017.
Boehringer Ingelheim International GmbH (2015). Vargatef (nintedanib) summary of product characteristics. March 2017. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002569/WC500179970.pdf. Accessed 27 Apr 2017.
Mross K, Stefanic M, Gmehling D, Frost A, Baas F, Unger C, Strecker R, Henning J, Gaschler-Markefski B, Stopfer P, de Rossi L, Kaiser R. Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res. 2010;16:311–9.
Okamoto I, Kaneda H, Satoh T, Okamoto W, Miyazaki M, Morinaga R, Ueda S, Terashima M, Tsuya A, Sarashina A, Konishi K, Arao T, Nishio K, Kaiser R, Nakagawa K. Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors. Mol Cancer Ther. 2010;9:2825–33.
Reck M, Kaiser R, Eschbach C, Stefanic M, Love J, Gatzemeier U, Stopfer P, von Pawel J. A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer. Ann Oncol. 2011;22:1374–81.
Dallinger C, Trommeshauser D, Marzin K, Liesener A, Kaiser R, Stopfer P. Pharmacokinetic properties of nintedanib in healthy volunteers and patients with advanced cancer. J Clin Pharmacol. 2016;56:1387–94.
Ogura T, Taniguchi H, Azuma A, Inoue Y, Kondoh Y, Hasegawa Y, Bando M, Abe S, Mochizuki Y, Chida K, Klueglich M, Fujimoto T, Okazaki K, Tadayasu Y, Sakamoto W, Sugiyama Y. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1382–92.
Stopfer P, Rathgen K, Bischoff D, Lüdtke S, Marzin K, Kaiser R, Wagner K, Ebner T. Pharmacokinetics and metabolism of BIBF 1120 after oral dosing to healthy male volunteers. Xenobiotica. 2011;41:297–311.
Food and Drug Administration (FDA). Guidance for industry: drug interaction studies–study design, data analysis, implications for dosing, and labeling recommendations. February 2011. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed 4 Mar 2016.
Tran JQ, Kovacs SJ, McIntosh TS, Davis HM, Martin DE. Morning spot and 24-hour urinary 6 beta-hydroxycortisol to cortisol ratios: intraindividual variability and correlation under basal conditions and conditions of CYP 3A4 induction. J Clin Pharmacol. 1999;39:487–94.
Lim YP, Huang JD. Interplay of pregnane X receptor with other nuclear receptors on gene regulation. Drug Metab Pharmacokinet. 2008;23:14–21.
Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14851–7.
Brault M. Development and validation of an LC-MS/MS method for the quantification of cortisol and 6-beta-hydroxycortisol in human urine. SGS Cephac Europe Analytical Report Reference No. CP095124, 01 Sept 2010.
Sekar VJ, Lefebvre E, De Pauw M, Vangeneugden T, Hoetelmans RM. Pharmacokinetics of darunavir/ritonavir and ketoconazole following co-administration in HIV-healthy volunteers. Br J Clin Pharmacol. 2008;66:215–21.
Acknowledgements
All authors were involved with drafting this article or revising it critically for important intellectual content. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Clare Ryles and Wendy Morris of FleishmanHillard Fishburn, London, UK during the preparation of this article. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
These studies were funded by Boehringer Ingelheim.
Conflict of interest
All authors are employees of Boehringer Ingelheim.
Ethical Approval
All procedures in these studies were carried out in accordance with the Helsinki declaration and its amendments and the ethical committee or institutional review board which approved these studies.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Luedtke, D., Marzin, K., Jungnik, A. et al. Effects of Ketoconazole and Rifampicin on the Pharmacokinetics of Nintedanib in Healthy Subjects. Eur J Drug Metab Pharmacokinet 43, 533–541 (2018). https://doi.org/10.1007/s13318-018-0467-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-018-0467-9