
Abstract
The study aimed to examine the effect of sunitinib on the plasma exposure of intravenous paracetamol and its major metabolite, paracetamol glucuronide. Both drugs share metabolic pathways in the liver, and the drug interactions between sunitinib and paracetamol administered in higher doses were reported. These interactions resulted in hepatotoxicity. The adult New Zealand male rabbits were divided into three groups (6 animals each): rabbits receiving sunitinib and paracetamol (SUN + PC), rabbits receiving sunitinib (SUN), and a control group receiving paracetamol (PC). Sunitinib was administered orally (25 mg) and paracetamol was administrated intravenously (35 mg/kg). Blood samples for sunitinib and SU12662 assays were collected up to 96 h after drug administration and for paracetamol and paracetamol glucuronide up to 300 min after drug administration. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), and bilirubin were analysed before and after drug administration. A number of pharmacokinetic parameters were analysed. There were no differences in the levels of AST, ALT, and bilirubin among the groups at either time point. Significantly higher values of AUC0–t , AUC0–∞ , and C max and lower clearance and volume of distribution of paracetamol were observed in group PC vs. group SUN + PC (p < 0.01). The maximum plasma concentration of paracetamol glucuronide tended to be higher in group PC 213.27 μg/mL (90 % CI 1.06, 1.25; p = 0.0267). Statistically significant differences were revealed for paracetamol glucuronide mean residence time (MRT); MRT was higher in group SUN + PC than in group PC (p = 0.0375). The mean t max of paracetamol glucuronide was similar in both groups: SUN + PC and group PC (15 and 20 min, respectively). The mean t max of sunitinib was different in groups SUN + PC and SUN (10.0 and 7.0, respectively; p = 0.0134). At the studied doses, neither of the drugs, whether administered alone or together, had hepatotoxic effects. The present study was not able to confirm that sunitinib, administered at low doses in conjunction with paracetamol, displays a hepatoprotective effect. Significant differences were observed in some pharmacokinetic parameters of paracetamol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Intravenous acetaminophen is an analgesic and antipyretic agent, recommended worldwide as a first-line agent for the treatment of pain and fever in adults and children (Bienert et al. 2012). At therapeutic doses, it is usually safe and well tolerated (Klotz 2012). Therapeutic concentrations for paracetamol range from 5 to 20 μg/mL (Bertolini et al. 2006). Paracetamol is metabolized primarily in the liver by first-order kinetics via three main pathways. The two major pathways that metabolize approximately 90 % of a given dose are conjugation with glucuronide (approximately 40–67 %) and conjugation with sulphate (20–46 %). The resulting metabolites are non-toxic and are eliminated in urine (Bertolini et al. 2006). The remaining amount of administered paracetamol is metabolized by the hepatic cytochrome P450 enzyme system, mainly by CYP2E1 isoenzyme and to a lesser extent by CYP1A2, CYP3A4 and CYP2A6 isoenzymes (Bertolini et al. 2006). From this pathway, a highly reactive intermediate is formed, N-acetyl-p-benzoquinoneimine (NAPQI; hepatocellular protein) which is subsequently quickly inactivated by glutathione to non-toxic cysteine or mercaptate conjugates and is eliminated in the urine (Bertolini et al. 2006). Several P450 isoforms including CYP3A1, 2E1, 1A2 and 2D6 are implicated in the activation of paracetamol to NAPQI in both humans and rodents (Weise et al. 2009).
Sunitinib is an oral multitargeted tyrosine kinase inhibitor that selectively inhibits class III, V, and XII split-kinase domain receptor tyrosine kinases, including vascular endothelial growth factor receptors (VEGFR-1, -2, and -3), platelet-derived growth factor receptors (PDGFR-α and -β), stem cell factor receptor (KIT), and Fms-like tyrosine kinase-3 receptor (FLT3) (Bello et al. 2010; Kawashima et al. 2012; Zhou 2012). Sunitinib is approved for advanced renal cell cancer (RCC) in adults (Kim et al. 2009a; Castellano et al. 2013) and gastrointestinal stromal tumour (GIST) after disease progression on or intolerance to imatinib mesylate (Kim et al. 2009a; Demetri et al. 2009). The maximum plasma concentration generally occurs between 6 and 12 h post-dose (Bello et al. 2009; Vázquez et al. 2012). Given the prolonged terminal half-lives of sunitinib and SU012662 of ~40–60 and 80–110 h (Bello et al. 2009, 2010), respectively, sunitinib and SU012662 accumulate 3- to 4-fold and 7- to 10-fold with repeated daily administration. Steady-state concentrations are achieved within 10–14 days, and by day 14, combined plasma concentrations of sunitinib and its active metabolite (total drug) range from 62.9 to 101 ng/mL. Preclinical studies have indicated that concentrations 50–100 ng/mL are required to inhibit receptor phosphorylation and result in antitumour activity (Bello et al. 2009, Mendel et al. 2003). Sunitinib is primarily metabolized by cytochrome P450 3A4 to an active N-desethyl metabolite (SU12662). The active metabolite is also metabolized by cytochrome P450 3A4 (Bello et al. 2009; Houk et al. 2009).
The present study was designed to examine the effect of sunitinib on the plasma exposure of intravenous paracetamol and its major metabolite: paracetamol glucuronide. Due to the fact that paracetamol is one of the most common analgesics and antipyretics, it is very likely that both drugs may interact as they are metabolised in the liver. Additionally, there were reports on hepatotoxicity and drug interactions between sunitinib and paracetamol administered at higher doses (Weise et al. 2009). We performed a National Library of Medicine’s bibliographic database (MEDLINE®) search and found no evidence in the literature regarding the effects of sunitinib on the pharmacokinetics of paracetamol.
2 Materials and methods
2.1 Reagents
Sunitinib and SU12662 were purchased from LGC Standards (Lomianki, Poland), HPLC grade acetonitrile, paracetamol and paracetamol glucuronide, perchloric acid, theophyllinum, from Sigma-Aldrich (Poland), 85 % orthophosphoric acid, 2 M sodium hydroxide and methanol from Merck (Poland), sodium sulphate anhydrous from Fluka (Poland). Water used in the mobile phase was deionized, distilled and filtered through a Millipore system before use. Sutent® was purchased (batch number P177H) from Pfizer Trading Polska Sp. z o.o., Warsaw, Poland. Perfalgan® was purchased (batch number 1K67394) from Bristol-Myers Squibb Polska Sp. z o.o., Warsaw, Poland.
2.2 Animals
The study was conducted on rabbits due to the need to collect numerous blood samples to present detailed profiles c = f(t). Adult New Zealand male rabbits, weighing 2.7–5.4 kg (mean ± SD = 3.17 ± 0.21), were used for experiments. All rabbits were kept in individual metal cages located in the animal laboratory of University of Medical Sciences, Department and Unit of Clinical Pharmacy and Biopharmacy. They were acclimatized for 2 weeks prior to the experiments and were maintained under standard conditions of temperature (23 ± 2 °C) and humidity (56–60 %) with an alternating 12 h light/dark cycles. New Zealand Rabbits were provided with 100 g of commercial pelleted diet (Labofeed KB®: 9.8 MJ/kg metabolic energy, 16.00 % total protein, 0.65 % vitamin P, 15,000 IU vitamin A, 1,500 JU vitamin D3, and 65 mg vitamin E) and tap water ad libitum. All experimental procedures related to this study were approved by the local ethics committee of the Medical University of Poznan.
2.3 Evaluation of sunitinib and SU12662, paracetamol and paracetamol glucuronide pharmacokinetics
The rabbits were divided into three groups (6 animals each): the rabbits receiving sunitinib and paracetamol (SUN + PC), the rabbits receiving sunitinib (SUN) and the control group receiving paracetamol (PC). Just before the administration of paracetamol and sunitinib and at the end of the experiment, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and bilirubin were measured in all the rabbits with an Architect CI8200 chemistry analyser (Abbott Laboratories Sp. z o.o., Poland). Sunitinib was administered p.o. at the single dose of 25 mg (suspended in 10 mL of normal saline) to group SUN + PC and SUN animals. Blood samples (2.5 mL) for sunitinib and SU12662 assays were collected via the central auricular artery (22 G catheter) before and 0.50, 1, 2, 4, 6, 7, 8, 9, 10, 11, 12, 24, 48, 72, 96 h following drug administration. Paracetamol was administrated via the ear vein in the dose of 35 mg per kg b.w. Bello et al. 2010 to the group SUN + PC and PC. Blood samples (approximately 0.5 ml) were collected before and 5, 15, 30, 60, 90, 120, 180, 240 and 300 min after drug administration. The blood samples were transferred into heparinised tubes and they were centrifuged at 4,000 rpm for 10 min at 4 °C. Next, the plasma was transferred to propylene tubes and stored at −20 °C until analysis.
The measurement of sunitinib concentration in the blood plasma was made by means of the HPLC (high-performance liquid chromatography) method with UV detection, which was a modification of the method developed by Faivre et al. (2011). Separation was achieved by isocratic elution of the mobile phase, ammonium acetate 20 mM pH 3.4 (adjusted with acetic acid)—acetonitrile (60:40, v/v), at a flow rate of 1.0 mL/min through a Symmetry® C8 column (250 mm × 4.6 mm, 5.0 μm particle size) (Waters®). The column temperature was maintained at 40 °C, the UV–Vis detection wavelength was set at 431 nm, and the injection volume was 50 μL. The total analysis time for each run was 6 min. The lower limit of quantification (LLOQ) and limit of detection (LOD) for sunitinib and SU12662 were 1.0 and 0.5 ng/mL. Intra- and inter-day precision and accuracy of the LLOQ, low-quality control (2.5 ng/mL), medium-quality control (25.0, 125.0 ng/mL), and high-quality control (45.0, 200.0 ng/mL) were well within the acceptable limit of 10 % coefficient of variation (CV %) for SU12662 and sunitinib, respectively. The calibration for sunitinib was linear in the range 1.0–250.0 ng/mL (r = 0.999), and for SU12662 in the range 1.0–50.0 ng/mL (r = 0.998).
The concentration of paracetamol and paracetamol glucuronide was assayed using HPLC method with UV detection (Brunner and Bai 1999). Separation was achieved by isocratic elution of the mobile phase, natrium sulphate 0.05 M pH 2.2 (adjusted with 85 % orthophosphoric acid)—acetonitrile (93:7, v/v), at a flow rate of 1.5 mL/min through an ODS Hypersil® C18 column (150 mm × 4.6 mm, 5.0 μm particle size) (Thermo Electron Corporation®). The column temperature was maintained at 25 °C, the UV–Vis detection wavelength was set at 261 nm, and the injection volume was 50 μL. The total analysis time for each run was 5 min. The lower limit of quantification (LLOQ) and limit of detection (LOD) for paracetamol and paracetamol glucuronide were 0.25 μg/mL and 0.1 μg/mL. Intra- and inter-day precision and accuracy of the LLOQ, low-quality control (0.5 μg/mL), medium-quality control (10.0 μg/mL), and high-quality control (150.0 μg/mL) were well within the acceptable limit of 10 % coefficient of variation (CV %) for paracetamol and paracetamol glucuronide. The calibration for paracetamol was linear in the range 0.5–100 μg/mL (r = 0.999), and for paracetamol glucuronide in the range 0.5–150 μg/mL (r = 0.997).
2.4 Pharmacokinetics analysis
Pharmacokinetic parameters were estimated by non-compartmental methods using validated software (WinNonlin® Professional Version 5.3; Pharsight® Corp., USA). The following pharmacokinetic parameters were calculated for sunitinib: absorption rate constant (k a), elimination rate constant (k el), area under the plasma concentration–time curve from time zero to infinity (AUC0–inf), area under the plasma concentration–time curve from zero to the time of last measurable concentration (AUC0–t ), maximum observed plasma concentration (C max), time to first occurrence of C max (t max), half-life in elimination phase (\(t_{{ 1/ 2k_{\text{el}} }}\)), clearance (CL), volume of distribution (V d), area under the first moment curve (AUMC0–t ), and mean residence time (MRT). The pharmacokinetic endpoints for SU12662 were AUC0–inf, AUC0–t , C max, t max, and \(t_{{ 1/ 2k_{\text{el}} }} .\) The following pharmacokinetic parameters were calculated for paracetamol: k el, AUC0–inf, AUC0–t , C max, t max, \(t_{{ 1/ 2k_{\text{el}} }}\), CL, V d, AUMC0–t , MRT. The pharmacokinetic endpoints for paracetamol glucuronide were k el, AUC0–inf, AUC0–t, C max, t max, \(t_{{ 1/ 2k_{\text{el}} }}\), AUMC0–t, MRT.
2.5 Statistical analysis
The effect of drug formulation was tested by one-way analysis of variance in PROC GLM of the SAS package (SAS Institute Inc. 2002–2003. The SAS System for Windows version 9.1. Cary, NC 27513-2414 USA). The 90 % confidence intervals for the ratio of geometric means were constructed, except for t max for which the confidence intervals were based on the difference of medians.
3 Results
Just before the administration of paracetamol and sunitinib and at the end of the experiment, the three analysed groups did not significantly different in the levels of AST (41.58 ± 12.27, 42.92 ± 6.24 U/l), ALT (52.07 ± 16.79, 38.38 ± 21.75 U/l) and bilirubin (0.28 ± 0.21, 0.24 ± 0.06 mg/dL) (p = 0.8310, p = 0.2682, p = 0.7013, respectively).
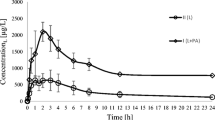
The plasma concentration–time profiles for sunitinib and SU12662, and paracetamol and its metabolite are shown in Figs. 1 and 2, respectively.

A log scale plot of arithmetic mean sunitinib (SUN) and metabolite (SU12662) plasma concentration versus time following oral administration of a single 25 mg dose of sunitinib in rabbits

A log scale plot of arithmetic mean paracetamol (PC) and paracetamol glucuronide (GLUC) plasma concentration versus time following i.v. administration of a single 35 mg per kg b.w. dose of paracetamol in rabbits
The pharmacokinetic parameters and a summary of the statistical analyses for sunitinib and SU12662, and paracetamol and its metabolite are shown in Tables 1 and 2, respectively.
Significantly upper values of AUC0–t , AUC0–∞ and C max, lower clearance and volume of distribution of paracetamol were noted in the paracetamol group (p = <0.0001, p = <0.0001, p = <0.0001, p = 0.0001, p = 0.0046, respectively).
The mean t max of paracetamol was similar in the presence and absence of sunitinib (5 min).
The maximum plasma concentration of paracetamol glucuronide tended to be higher in the paracetamol group 213.27 mg/L (90 % CI 1.06, 1.25). There were significant differences between the analysed groups (p = 0.0267). Statistically significant differences were revealed for MRT (p = 0.0375). The mean t max of paracetamol glucuronide was similar for both the paracetamol and paracetamol + sunitinib groups (15 and 20 min, respectively).
There were no significant differences among the analysed groups for the following pharmacokinetic parameters of paracetamol glucuronide: AUC0–t (p = 0.4734), AUC0–∞ (p = 0.4823), k el (p = 0.6603), AUMC0–t (p = 0.7396) and \(t_{{ 1/ 2k_{\text{el}} }}\) (p = 0.71).
The mean t max of sunitinib was not similar for sunitinib + paracetamol group and sunitinib group (10.0 and 7.0, respectively; Table 1). The comparison of the t max for the groups gave a ratio of 4 (90 % CI 1.94, 6.06). There were significant differences among the analysed groups (p = 0.0134).
The mean C max was comparable in the evaluated rabbit groups (p = 0.7959). There were no significant differences among the analysed groups for the following pharmacokinetic parameters of sunitinib: AUC0–t (p = 0.7078), AUC0–∞ (p = 0.8762), k el (p = 0.2375), AUMC0–t (p = 0.8875) and \(t_{{ 1/ 2k_{\text{el}} }}\) (p = 0.6765), Cl (p = 0.8591), V d (p = 0.9372), MRT (p = 0.3141).
There were no significant differences among the analysed groups for the following pharmacokinetic parameters of SU12662: AUC0–t (p = 0.7902), AUC0–∞ (p = 0.7122), C max (p = 0.7308) and \(t_{{ 1/ 2k_{\text{el}} }}\) (p = 0.3139).
4 Discussion
The aim of this study was to examine the effect of sunitinib on the plasma exposure of intravenous paracetamol and its major metabolite, paracetamol glucuronide, in rabbits. Rabbits are frequently applied practical experimental models in studies on the pharmacokinetics of drugs. Therefore, the authors decided to use them for the assessment of the pharmacokinetic interaction between the aforementioned tyrosine kinase inhibitor and paracetamol. It also resulted from the need to collect numerous blood samples to present detailed profiles c = f(t). Apart from that, rabbits were frequently applied experimental models in studies on sunitinib (Fallon et al. 2012a, b; Patyna et al. 2009; Meisel et al. 2011).
To the best of the authors’ knowledge, there are no data in the literature regarding the effects of sunitinib on the pharmacokinetics of paracetamol. One of the most clinically significant complications related to the use of pharmacotherapy is the potential for drug–drug interactions (DDIs) (Doligalski et al. 2012). DDIs may result in adverse clinical events by decreasing the therapeutic effect of a drug or by enhancing drug toxicity (Lim et al. 2010). Severe DDIs have been observed between anti-cancer and pain management drugs (Lim et al. 2010; Weise et al. 2009), and also between acetaminophen and other TKIs (Liu et al. 2011; Nassar et al. 2010), resulting in hepatotoxicity due to inhibition of paracetamol glucuronidation and the shared pathway of transport to the liver. The guidelines of SIGN (Scottish Intercollegiate Guidelines Network 2008) and ESMO (European Society of Medical Oncology) (Ripamonti et al. 2011) recommend acetaminophen in cancer pain management, but the FDA (Food and Drug Administration) has expressed concerns about possible toxicity due to its common use (Klotz 2012).
The importance of interactions with paracetamol is relevant to sunitinib. Weise et al. (2009) reported a patient with relapsed metastatic GIST, who, after being treated with sunitinib, paracetamol and levothyroxine, developed acute liver failure with a fatal outcome. This might suggest that administering sunitinib has an influence on the metabolism of paracetamol towards inactive paracetamol glucuronide.
However, there is a possibility that sunitinib influences the biotransformation of paracetamol towards inactive sulphate or active and toxic NAPQI (Bertolini et al. 2006; Manyike et al. 2000), which share the same isoform of P450 CYP 3A4 with metabolism of sunitinib and SU12662 (Bello et al. 2009; Houk et al. 2009; Lim et al. 2010; Mazer and Perrone 2008). When a low dose (25–80 mg/kg) of sunitinib is coadministered with acetaminophen, the formation of NAPQI is decreased and its toxicity is lowered (Lim et al. 2010). Sunitinib is a competitive inhibitor and the substrate of glutathione (GSH)-conjugate transporter (Singhal et al. 2010; Galal et al. 2012; Jaeschke et al. 2011, 2012), similar to the metabolism of cisplatin (Shimeda et al. 2005). Therefore, in higher doses (100–140 mg/kg), sunitinib (Lim et al. 2010) or SU12662 binds to GSH or reduces its availability, resulting in diminished protection of hepatocytes from accumulated NAPQI, even if the formation of NAPQI is low (Franco and Cidlowski 2009). The authors applied an oral dose of sunitinib 25 mg, which is well tolerated by animals (Meisel et al. 2011) and which is the dose applied to humans. Additionally, the dose of the drug enabled the authors to obtain the concentrations of sunitinib in the animals’ blood, which correspond to the concentrations in humans. The dose of paracetamol (35 mg/kg) resulted from the authors’ earlier experiments (Bienert et al. 2012).
Taking into account the levels of AST and bilirubin, the pre- and post-experiment concentrations of these two hepatotoxicity markers did not differ significantly between the analysed groups. This shows that, at the studied doses, neither of the drugs, whether administered alone or together, displays hepatotoxic effects.
Sunitinib does not alter the t max of paracetamol, and thus does not accelerate or decelerate the time for the analgesic effect to develop, but decreases drug exposure (lower AUC0–t , AUC0–∞ in presence of sunitinib) and dose absorption of paracetamol (lower C max in presence of sunitinib). With decreased clearance and volume distribution, it may suggest lower overall bioavailability of acetaminophen and decreased tissue concentration resulting in diminished toxicity when administered with sunitinib. However, when assessing the t max of sunitinib in the presence of paracetamol, we have noticed that paracetamol decreases the time necessary for sunitinib to reach maximum plasma concentration, but does not affect its elimination. There was no effect of acetaminophen on the pharmacokinetics of SU12662, which suggests that paracetamol does not alter the cytochrome P450 pathway of sunitinib, but may enhance bioavailability by increasing the free fraction of sunitinib, as suggested with coadministration of imatinib and acetaminophen (Nassar et al. 2009).
Taking into account paracetamol glucuronide, its MRT was noted to be higher for the group receiving both paracetamol and sunitinib. Thus, we may conclude that sunitinib elongates the time necessary to excrete paracetamol glucuronide. Sunitinib lowers maximum plasma concentrations of paracetamol glucuronide. Referring to the obtained pharmacokinetics of paracetamol, it is possible that overall bioavailability of acetaminophen is decreased in the presence of sunitinib, contrary to results of Nassar et al. (2009, 2010), suggesting different mechanisms of metabolism of acetaminophen coadministered with sunitinib than with imatinib.
Taking the above-mentioned facts into consideration, this study was not able to confirm that sunitinib, administered at low doses in conjunction with paracetamol, displays a hepatoprotective effect, as previously suggested (Weise et al. 2009; Lim et al. 2010). However, we have to remember that in the present study, the administered doses of paracetamol and sunitinib were relatively low in comparison to doses in the previously mentioned publications (sunitinib 25–140 mg/kg and paracetamol 500 mg/kg (Lim et al. 2010; Kim et al. 2009b), paracetamol 700 mg/kg (Nassar et al. 2009, 2010) and 1,000 mg/kg (Kim et al. 2011); the route of paracetamol administration was also different (Lim et al. 2010; Nassar et al. 2009, 2010; Kim et al. 2011). In higher doses, these drugs may be hepatotoxic (Weise et al. 2009; Yapar et al. 2007).
In the Bienert et al. (2012) study, rabbits received 35 mg i.v. (intravenously) of paracetamol and a high increase in ALT and AST levels was observed in the group of rabbits with diabetes in comparison to healthy rabbits receiving placebo or only acetaminophen. However, the increase of ALT and AST enzymes was not observed in rats receiving 500 mg/kg acetaminophen, but a hepatotoxic effect was seen when paracetamol was administrated 18 h earlier (Kim et al. 2009b). Also, in the study of Toyoshiba et al. (2006), lower doses (50 mg/kg) of paracetamol administrated to rats showed that genes related to the oxidative stress signalling pathway did not interact with apoptosis-related genes. Additionally, Donahower et al. (2006) examined the role of VEGF in acetaminophen hepatotoxicity in mice and observed that the VEGF receptor inhibitor SU5416 (25 mg/kg) had no effect on the toxicity of paracetamol. Furthermore, Parra et al. (2007) observed DDI interactions between warfarin, which is metabolized by cytochrome P450, and acetaminophen (200–400 mg daily) in patients, but DDI interactions with the use of lower doses of acetaminophen require further study.
In the future, a dose-escalating study of paracetamol and sunitinib should be conducted due to the lack of data concerning different doses of paracetamol coadministered with sunitinib. Taking into consideration the common use of acetaminophen, coadministration of low doses of paracetamol used for extended period of time (Kim et al. 2009b; Maciejewska-Paszek et al. 2007; Haznedar et al. 2009) should be examined.
Fasting before administration of acetaminophen (Weise et al. 2009; Lim et al. 2010; Nassar et al. 2009, 2010) could enhance the effects of low doses of acetaminophen in the present study by reducing hepatic glutathione levels (Jaeschke et al. 2011; Fernando and Ariyananda 2009).
One of the possible limitations of this study is also the fact that the analysed groups were fairly small.
5 Conclusions
We observed significant differences in the pharmacokinetic parameters of paracetamol coadministered with sunitinib.
References
Bello CL, Mulay M, Huang X et al (2009) Electrocardiographic characterization of the QTc interval in patients with advanced solid tumors: pharmacokinetic–pharmacodynamic evaluation of sunitinib. Clin Cancer Res 15(22):7045–7052
Bello CL, Garrett M, Sherman L et al (2010) Pharmacokinetics of sunitinib malate in subjects with hepatic impairment. Cancer Chemother Pharmacol 66(4):699–707
Bertolini A, Ferrari A, Ottani A et al (2006) Paracetamol—new vistas of an old drug. CNS Drug Rev 12(3–4):250–275
Bienert A, Kamińska A, Olszewski J et al (2012) Pharmacokinetics and ocular disposition of paracetamol and paracetamol glucuronide in rabbits with diabetes mellitus induced by alloxan. Pharmacol Rep 64(2):421–427
Brunner LJ, Bai S (1999) Simple and rapid assay for acetaminophen and conjugated metabolites in low-volume serum samples. J Chromatogr B Biomed Sci Appl 732(2):323–329
Castellano D, Ravaud A, Schmidinger M et al (2013) Therapy management with sunitinib in patients with metastatic renal cell carcinoma: key concepts and the impact of clinical biomarkers. Cancer Treat Rev 39(3):230–240
Demetri GD, Heinrich MC, Fletcher JA et al (2009) Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res 15(18):5902–5909
Doligalski CT, Tong Logan A, Silverman A (2012) Drug interactions: a primer for the gastroenterologist. Gastroenterol Hepatol (N Y) 8(6):376–383
Donahower B, McCullough SS, Kurten R et al (2006) Vascular endothelial growth factor and hepatocyte regeneration in acetaminophen toxicity. Am J Physiol Gastrointest Liver Physiol 291(1):102–109
Faivre L, Gomo C, Mir O et al (2011) A simple HPLC-UV method for the simultaneous quantification of gefitinib and erlotinib in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 879(23):2345–2350
Fallon EM, Nehra D, Carlson SJ et al (2012a) Sunitinib reduces recurrent pelvic adhesions in a rabbit model. J Surg Res 178(2):860–865
Fallon EM, Nehra D, Le HD et al (2012b) Effect of sunitinib on functional reproductive outcome in a rabbit model. Fertil Steril 98(2):496–502
Fernando WKBK, Ariyananda PL (2009) Paracetamol poisoning below toxic level causing liver damage in a fasting adult. Ceylon Med J 54(1):16–17
Franco R, Cidlowski JA (2009) Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ 16(10):1303–1314
Galal RM, Zaki HF, Seif El-Nasr MM et al (2012) Potential protective effect of honey against paracetamol-induced hepatotoxicity. Arch Iran Med 15(11):674–680
Haznedar JO, Patyna S, Bello CL et al (2009) Single- and multiple-dose disposition kinetics of sunitinib malate, a multitargeted receptor tyrosine kinase inhibitor: comparative plasma kinetics in non-clinical species. Cancer Chemother Pharmacol 64(4):691–706
Houk BE, Bello CL, Kang D et al (2009) A population pharmacokinetic meta-analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin Cancer Res 15(7):2497–2506
Jaeschke H, McGill MR, Williams CD et al (2011) Current issues with acetaminophen hepatotoxicity—a clinically relevant model to test the efficacy of natural products. Life Sci 88(17–18):737–745
Jaeschke H, McGill MR, Ramachandran A (2012) Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44(1):88–106
Kawashima A, Tsujimura A, Takayama H et al (2012) Importance of continuing therapy and maintaining one-month relative dose intensity in sunitinib therapy for metastatic renal cell carcinoma. Med Oncol 29(5):3298–3305
Kim A, Balis FM, Widemann BC (2009a) Sorafenib and sunitinib. Oncologist 14(8):800–805
Kim SJ, Lee MY, Kwon DY et al (2009b) Alteration in metabolism and toxicity of acetaminophen upon repeated administration in rats. J Pharmacol Sci 111(2):175–181
Kim D-W, Tan EY, Jin Y et al (2011) Effects of imatinib mesylate on the pharmacokinetics of paracetamol (acetaminophen) in Korean patients with chronic myelogenous leukaemia. Br J Clin Pharmacol 71(2):199–206
Klotz U (2012) Paracetamol (acetaminophen)—a popular and widely used nonopioid analgesic. Arzneimittelforschung 62(8):355–359
Lim AY, Segarra I, Chakravarthi S et al (2010) Histopathology and biochemistry analysis of the interaction between sunitinib and paracetamol in mice. BMC Pharmacol. doi:10.1186/1471-2210-10-14
Liu Y, Ramírez J, Ratain MJ (2011) Inhibition of paracetamol glucuronidation by tyrosine kinase inhibitors. Br J Clin Pharmacol 71(6):917–920
Maciejewska-Paszek I, Pawłowska-Góral K, Kostrzewski M (2007) The influence of small doses of paracetamol on rabbit liver. Exp Toxicol Pathol 59(2):139–141
Manyike PT, Kharasch ED, Kalhorn TF et al (2000) Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin Pharmacol Ther 67(3):275–382
Mazer M, Perrone J (2008) Acetaminophen-induced nephrotoxicity: pathophysiology, clinical manifestations, and management. J Med Toxicol 4(1):2–6
Meisel JA, Fallon EM, Le HD et al (2011) Sunitinib inhibits postoperative adhesions in a rabbit model. Surgery 150(1):32–38
Mendel DB, Laird AD, Xin X et al (2003) In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9(1):327–337
Nassar I, Pasupati T, Judson JP et al (2009) Reduced exposure of imatinib after coadministration with acetaminophen in mice. Indian J Pharmacol 41(4):167–172
Nassar I, Pasupati T, Judson JP et al (2010) Histopathological study of the hepatic and renal toxicity associated with the co-administration of imatinib and acetaminophen in a preclinical mouse model. Malays J Pathol 32(1):1–11
Parra D, Beckey NP, Stevens GR (2007) The effect of acetaminophen on the international normalized ratio in patients stabilized on warfarin therapy. Pharmacotherapy 27(5):675–683
Patyna S, Haznedar J, Morris D et al (2009) Evaluation of the safety and pharmacokinetics of the multi-targeted receptor tyrosine kinase inhibitor sunitinib during embryo-fetal development in rats and rabbits. Birth Defects Res B Dev Reprod Toxicol 86(3):204–213
Ripamonti CI, Bandieri E, Roila F (2011) Management of cancer pain: ESMO Clinical Practice Guidelines. Ann Oncol 22(6):vi69–vi77
Scottish Intercollegiate Guidelines Network (2008) Control of pain in adults with cancer. A national clinical guideline
Shimeda Y, Hirotani Y, Akimoto Y et al (2005) Protective effects of capsaicin against cisplatin-induced nephrotoxicity in rats. Biol Pharm Bull 28(9):1635–1638
Singhal SS, Sehrawat A, Sahu M et al (2010) Rlip76 transports sunitinib and sorafenib and mediates drug resistance in kidney cancer. Int J Cancer 126(6):1327–1338
Toyoshiba H, Sone H, Yamanaka T et al (2006) Gene interaction network analysis suggests differences between high and low doses of acetaminophen. Toxicol Appl Pharmacol 215(3):306–316
Vázquez S, León L, Fernández O et al (2012) Sunitinib: the first to arrive at first-line metastatic renal cell carcinoma. Adv Ther 29(3):202–217
Weise AM, Liu CY, Shields AF (2009) Fatal liver failure in a patient on acetaminophen treated with sunitinib malate and levothyroxine. Ann Pharmacother 43(4):761–766
Yapar K, Kart A, Karapehlivan M et al (2007) Hepatoprotective effect of l-carnitine against acute acetaminophen toxicity in mice. Exp Toxicol Pathol 59:121–128
Zhou A (2012) Management of sunitinib adverse events in renal cell carcinoma patients: the Asian experience. Asia Pac J Clin Oncol 8(2):132–144
Conflict of interest
The authors declare that they have no conflict of interests.
Author contribution
All authors have participated sufficiently in the work to take public responsibility for the entire content of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Karbownik, A., Szałek, E., Sobańska, K. et al. The effect of sunitinib on the plasma exposure of intravenous paracetamol and its major metabolite: paracetamol glucuronide. Eur J Drug Metab Pharmacokinet 40, 163–170 (2015). https://doi.org/10.1007/s13318-014-0191-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-014-0191-z