
Abstract
Danirixin (GSK1325756) is a small, high-affinity, selective and reversible CXCR2 antagonist in development for treatment of chronic obstructive pulmonary disease. The objective of the study was to evaluate the relative bioavailability, including the inter-subject variability, of a conventional immediate-release (IR) formulation and two prototype bioenhanced formulations of danirixin during gastric acid suppression in a healthy, elderly population. A single-centre, crossover study in healthy male and female volunteers aged 65–80 years was conducted. Subjects were randomised to receive danirixin 50 mg IR in the fed and fasted states and danirixin 50 mg Bioenhanced Formulation 1 and 2 in the fasted state. All subjects also received omeprazole 20 mg each morning beginning 4 days prior to the first treatment period and continuing through danirixin dosing in the final treatment period. Twenty subjects were randomised and completed the study. Bioenhanced Formulation 2 in the fasted state demonstrated the highest adjusted geometric means for AUC(0–t), AUC(0–inf), AUC(0–24) and C max. Danirixin IR demonstrated adjusted means that were higher in the fed state compared with the fasted state. For all formulations tested, there was substantial inter-subject variability (CVb >100 % for all formulations). The overall incidences of adverse events (AEs) were 10 % for danirixin IR (both in the fed and fasted states) and 15–20 % for the bioenhanced formulations. The majority of AEs were mild in intensity. There were no serious AEs. Concomitant use of omeprazole resulted in large inter-subject variability in the exposure to danirixin. Bioenhanced formulation strategies could not overcome the effect of omeprazole on exposure and variability between subjects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Danirixin (GSK1325756) is a small, non-peptide, high-affinity (IC50 for CXCL8 binding 12.5 nM), selective and reversible CXCR2 antagonist. Danirixin has demonstrated potent antagonism of CXCR2 activity, both in vitro and in vivo in preclinical studies (GSK, data on file). Its oral potency and duration of action support its potential use as an oral, anti-inflammatory agent in the treatment of disorders associated with the accumulation of neutrophils.
CXCR2, one of a family of CXC chemokine receptors, is necessary for the chemokine-mediated recruitment of neutrophils to sites of inflammation (Chapman et al. 2009). The neutrophil is thought to be an important contributor, via the release of tissue-destructive proteases and other mediators, to excess mucus production, airway stenosis, and destruction of the lung parenchyma which, in part, is responsible for the decline in lung function associated with chronic obstructive pulmonary disease or COPD (Stockley 2002; Quint and Wedzicha 2007). Levels of CXCL8 (interleukin-8) and other CXCR2 ligands (e.g. CXCL5 or ENA-78) are elevated in bronchoalveolar lavage fluid and the sputum of patients with COPD (Mukaida 2003). Several molecules with CXCR2 antagonist activity have been shown to reduce neutrophils migration to the lung in experimental medicine studies and in diseased subjects (Holz et al. 2010; Lazaar et al. 2011; Nair et al. 2012; Rennard et al. 2013). Selective antagonism of the CXCR2 receptor provides a potential strategy for reducing the underlying inflammation that contributes to the progression of COPD and other neutrophil-mediated diseases (Chapman et al. 2009).
Danirixin has its highest solubility at pH <2. The formulation strategy for the clinical development of danirixin has been focussed on optimising dissolution in the stomach to deliver a solution of the drug for absorption in the small intestine. A raised intra-gastric pH could have a significant impact on the dissolution and hence absorption of danirixin. In fact, inter-subject variability in danirixin exposure in elderly subjects, particularly in the fasted state and when given on a background of proton pump inhibitor use has been demonstrated, raising an important consideration for designing clinical trials of danirixin in patient populations (GSK 2013b).
Proton pump inhibitors and histamine H2-receptor antagonists are widely used for the management of disorders associated with excess gastric acid production (Blume et al. 2006). Because these drugs are typically used for long-term treatment, the potential for clinically significant drug interactions exists. Gastroesophageal reflux disease is a co-morbidity associated with COPD (Bor et al. 2010) and gastric acid-suppressing medications are commonly used by COPD patients. Though metabolic drug interactions (e.g. with the cytochrome P450 system) are a concern with several proton pump inhibitors, these agents may also alter the exposure to drugs for which a low gastric pH is required for optimal dissolution and subsequent absorption (Shi and Klotz 2008; Blume et al. 2006; Budha et al. 2012). Additionally, reduced production of gastric acid in some elderly subjects may also contribute to altered pharmacokinetics of some drugs in this population (Russell et al. 1993).
The aims of the current study were to evaluate the pharmacokinetics, with particular attention to inter-subject variability of several formulations of danirixin, including two bioenhanced formulations under conditions of gastric acid suppression in healthy elderly subjects. The bioenhanced formulations were developed to determine if the impact of a raised gastric pH on the pharmacokinetics of danirixin could be overcome through an alternative formulation approach.
2 Methods
2.1 Danirixin formulations
The structure of danirixin is shown in Fig. 1.

Danirixin free base
The original immediate-release (IR) tablets contained 50 mg of danirixin free base and standard excipients including mannitol, microcrystalline cellulose, HPMC, croscarmellose sodium and magnesium stearate.
Two bioenhanced, immediate-release tri-layer tablet formulations were manufactured by standard manufacturing procedures. These formulations were developed with the potential to enhance bioavailability compared to the initial immediate-release formulation under conditions of higher gastric pH. Tablets contained 25 mg of danirixin free base. The two formulations were similar to the original IR formulation with the exception of the addition of citric acid in Bioenhanced Formulation 1 (BE1) and both citric acid and poloxamer 407 in Bioenhanced Formulation 2 (BE2). In BE1, these modifications were made to enhance the solubility of danirixin in higher pH environments by reducing the localised pH around the disintegrating tablet, and in BE2 poloxamer was also added to improve wetting of the drug to aid dissolution.
2.2 In vitro solubility testing of danirixin
The determination of the pH solubility profile of danirixin was performed by adding excess danirixin to the appropriate pH Britton–Robinson buffer solutions in a vial. The vial was then placed on a roller mixer for 4 h at ambient temperature followed by filtration of a sample with centrifugation using a microcentrifuge tube with a 0.45 μm polyvinylidene difluoride membrane. The clear solution was then assayed using a gradient reversed-phase HPLC procedure to determine the danirixin content in solution. The procedure used a Waters Sunfire C18 column with a mobile phase of water and acetonitrile, containing 0.05 % trifluoroacetic acid with detection by UV at 220 nm.
2.3 Subjects
Eligible subjects were male or non-childbearing female, 65–80 years of age, healthy as assessed by medical history, physical examination, laboratory tests and cardiac monitoring, with a body weight ≥60 kg for men and ≥45 kg for women, a body mass index 19–32 kg/m2, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase and bilirubin ≤1.5 × upper limit of normal, and resting blood pressure of ≤160/95 mmHg, irrespective of anti-hypertensive medication status.
Key exclusion criteria included current or chronic history of liver disease or known hepatic or biliary abnormalities, a positive hepatitis B surface antigen or positive hepatitis C antibody within 3 months of screening or a positive test for human immunodeficiency virus antibody. Patients were also excluded if they had a pre-study positive drug/alcohol result, a history of regular alcohol consumption or previous history of smoking within 6 months of study, screening QT interval corrected using Bazett’s formula >450 ms, a PR interval outside of 120–220 ms or an electrocardiogram (ECG) not suitable for QT measurements, a history of sensitivity to any of the study treatments or its components, exposure to >4 new chemical entities within 12 months prior to first dose, or use of prescription or non-prescription drugs 7 months or five half-lives prior to first dose.
2.4 Study design and treatment
This was an open-label, five-period crossover study (http://www.clinicaltrials.gov identifier NCT01453478; GSK study code CX3115550) conducted at a single centre (Quotient Clinical Ltd., Nottingham, UK) between 12 October 2011 and 19 December 2011. The study was approved by an independent ethics committee (Capenhurst Independent Research Ethics Committee, Wirral, UK) and conducted in accordance with the Declaration of Helsinki 2008 (WMA Declaration of Helsinki 2008) and ICH Good Clinical Practice guidelines (ICH 1996). All subjects provided written informed consent prior to the start of the study.
Following screening, subjects were randomised to receive single doses of the following medications during treatment periods 1 through 4: danirixin IR 50 mg in the fed state (i.e. after a light breakfast), danirixin IR 50 mg in the fasted state (i.e. after an overnight fast), danirixin BE1 50 mg (2 × 25 mg) in the fasted state or danirixin BE2 50 mg (2 × 25 mg) in the fasted state. In each treatment period, all subjects received omeprazole 20 mg QD in the morning for 5 days (Days −4 to Day 1) for gastric acid suppression. On Day 1, the morning dose of omeprazole was administered together with the applicable formulation of danirixin 50 mg within a 5 min period. All treatment periods were separated by a minimum washout period of 3 days. Subjects would be eligible to continue to Treatment Period 5 if results of the interim analysis of the PK profile and relative bioavailability of each formulation following completion of Treatment Periods 1 through 4 met the pre-specified criteria (i.e. inter-subject variability <50 %). If these criteria were not fulfilled, the study would be terminated following the completion of Treatment Period 4.
2.5 Outcomes and assessments
The primary PK end points included determination of area-under-the-blood-concentration curve (AUC) data for danirixin IR and two prototype bioenhanced formulations of danirixin for the calculation of relative bioavailability under fed and fasted conditions. Secondary PK end points included characterisation of C max (maximum observed concentration), T max (time of occurrence of C max) and apparent terminal elimination half-life (t 1/2). Safety and tolerability were assessed by adverse events (AEs), vital signs, ECGs, changes in clinical laboratory parameters including clinical chemistry, haematology and urinalysis, and changes in clinical signs and symptoms from physical examination. Safety and tolerability data were summarised by treatment group.
2.6 Measurements
PK samples (whole blood) were collected at pre-dose and on Day 1 (0.5, 1, 2, 3, 4, 6, 8, and 12 h post-dose), Day 2 (24 and 36 h post-dose) and Day 3 (48 h post-dose). Whole blood concentrations of danirixin free base were determined using a validated analytical method based on extraction from a dried blood spot (Spooner et al. 2009), with a 3 mm disc being punched from a 0.015 mL sample on Whatman FTA™ card. Danirixin was extracted using methanol (0.1 mL) containing isotopically labelled [2H7]-danirixin (racemic version of danirixin) at a concentration of 50 ng/mL as an internal standard. The extraction tubes were shaken for 1 h at ambient temperature, prior to the supernatant being transferred to clean tubes. The supernatant was injected (5 μL) onto a high-performance liquid chromatography system utilising a Thermo Scientific Hypersil Gold C18 (5 μm packing 50 × 4.6 mm) column (supplied by Thermo Scientific, Hempstead, UK), eluted using an isocratic composition: 55:45 (v/v) of aqueous 10 mM ammonium formate containing 0.1 % formic acid (A) and acetonitrile (B). Danirixin free base has a retention time of approximately 1 min and was detected using tandem mass spectrometry on a Sciex 5000 (Applied Biosystems, Warrington, UK) using TurboIon Spray in positive polarity mode. Mass transitions monitored were 84 and 89 from precursor ions of 442 and 449 for danirixin and the internal standard. The assay had a linear dynamic range of 5–1,000 ng/mL and quantification was performed using peak area ratios with 1/x2 weighted linear regression. Assay quality control samples were 15 ng/mL, 200 ng/mL and 800 ng/mL. The average within-run precision (%CV) was 7.1, 3.2 and 3.3, respectively, for the low (15 ng/mL), mid (200 ng/mL) and high (800 ng/mL) quality controls.
Interim and final PK analyses were carried out using standard non-compartmental analysis and WinNonlin Professional v5.2. All calculations of non-compartmental parameters were based on nominal sampling times (relative to dosing) for interim analyses and actual sampling times for final analyses.
2.7 Statistical analysis
This sample size was chosen to provide acceptable precision to assess the study objectives and assuming similar variability to previous studies (GSK 2013a, b). The All Subjects population was defined as all subjects who received at least one dose of danirixin. The PK population was defined as all subjects in the All Subjects population for whom a PK sample was obtained and analysed.
Analysis was performed following completion of Treatment Period 4 and used the PK data collected during Treatment Periods 1 through 4. The analysis compared danirixin IR in the fed and fasted states, both bioenhanced formulations in the fasted state with danirixin IR in the fed and fasted states, and BE1 in the fasted state with BE2 in the fasted state. The PK parameters AUC(0–t), AUC(0–inf), AUC(0–24) and C max of danirixin underwent a natural log transformation (loge). A mixed effects model was used to analyse PK data and included an appropriate covariance structure to provide different estimates of variance for each treatment group; period, treatment and gender were used as fixed effects and subject as a random effect. Point estimates and 90 % confidence intervals (CIs) for each of the treatment comparisons were constructed using the error variance obtained from the statistical analysis and were back transformed to obtain estimates of the ratios of the adjusted geometric means. The variability was assessed by obtaining the within-subject variability (CVw) and CVb from the statistical analysis. Distributional assumptions underlying the statistical analyses were assessed by visual inspection of studentised residual plots, and normality was examined by normal probability plots (data not shown).
3 Results
3.1 In vitro solubility profile of danirixin free base
The pH solubility profile of danirixin free base is shown in Table 1. At pH values above 2, the solubility of danirixin free base was markedly reduced compared to the solubility at a pH of 2.
3.2 Study population
Twenty healthy, white, elderly subjects were randomised and all completed Treatment Periods 1 to 4 (Table 2). The population was equally split between male and female subjects; the mean age was 68.1 years (range 65–77 years).
3.3 Pharmacokinetic analysis
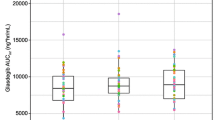
All subjects completed the four treatment periods and were included in the pharmacokinetic analysis. The criteria to progress to Treatment Period 5 were not fulfilled and the study was terminated after Treatment Period 4. Pharmacokinetic parameters derived from whole blood concentrations of danirixin and subjected to statistical analysis are shown in Table 3. Individual subject plots of whole blood concentrations of danirixin over 48 h are shown in Fig. 2. The adjusted geometric means for AUC(0–t), AUC(0-inf), AUC(0–24) and C max were significantly higher for BE2 in the fasted state compared with danirixin IR and BE1 in the fasted state. In addition, adjusted geometric means for AUC(0–24) and C max were significantly higher for BE2 in the fasted state compared to danirixin IR in the fed state. The adjusted geometric means for AUC(0–t), AUC(0–inf), AUC(0–24) and C max were similar for BE1 in the fasted state and danirixin IR in the fed state. All adjusted geometric mean parameters for BE1 in the fasted state were significantly higher than danirixin IR in the fasted state, and parameters for danirixin IR in the fed state were significantly higher than danirixin IR in the fasted state. Comparisons of AUC and C max for the various formulations are shown in Table 4.

Individual subject time–blood concentration profiles
For all formulations, there was substantial inter-subject variability in exposure to danirixin; CVb values for all AUC and C max parameters were greater than 100 %.
A statistically significant period effect was observed for all parameters (AUC(0–t), p = 0.0010; AUC(0–inf), p = 0.0011; AUC(0–24), p = 0.0005; C max, p < 0.0001), which suggests that exposures in Treatment Period 1 were higher than those observed for Treatment Periods 2 through 4.
For the IR formulation, the median T max was 3.5 h (2.0 − 4.0 h) in the fed state and 2 h (1.0 − 3.0 h) in the fasted state. The apparent terminal half-life (t 1/2) of danirixin was similar across the formulations (approximately, 7–9 h).
3.4 Safety
Nine of the 20 subjects (45 %) reported AEs during the study. There were no serious AEs and no withdrawals due to AEs. There were no differences in the incidence of reported AEs when danirixin IR was administered in the fed or fasted state (both 10 %). The incidence of AEs after administration of the BE1 and BE2 was 20 % and 15 %, respectively. The only AE reported by more than one subject was headache; 20 % of the subjects reported headaches during the study. Two incidences of headache (one after receiving danirixin IR and one after receiving BE2) were considered to be drug related. All AEs were reported to be mild in intensity, except for an AE of headache in a subject administered BE2, reported to be of moderate intensity; all AEs resolved. Haematology, clinical chemistry and vital sign abnormalities of potential clinical importance were isolated occurrences and were only slightly outside the reference range. There were no clinically significant ECG findings.
4 Discussion
In a previous study, when dosed concomitantly with the proton pump inhibitor omeprazole, an immediate-release formulation of danirixin free base showed reduced exposure and wide inter-subject variability compared to the non-gastric acid suppression state. Though the effect of omeprazole was expected based on the pH solubility profile of danirixin free base, this drug interaction might limit the use of danirixin in patient populations in which use of proton pump inhibitors and histamine H2-receptor antagonists is common. The primary goal of the present study was to determine if bioenhanced formulations of danirixin free base could reduce the wide inter-subject variability in exposure on a background of concomitant proton pump inhibitor use (GSK 2013b). The study evaluated the relative bioavailability, including the inter-subject variability of danirixin IR in the fed and fasted states and of two prototype bioenhanced formulations of danirixin in the fasted state during gastric acid suppression.
All formulations of danirixin, regardless of fed or fasted state, exhibited substantial inter-subject variability (CVb values >100 % for all formulations). This wide variability in exposure is consistent with the results of a previous study that investigated danirixin in the fed and fasted states (GSK 2013b). However, the previous study did demonstrate reduced variability in exposure when danirixin was administered with food. The lack of improvement in variability with the bioenhanced formulations of danirixin in the present study indicates that the tested formulations cannot overcome the effects of gastric acid suppression on danirixin pharmacokinetics. Regular, daily use of proton pump inhibitors such as omeprazole will increase intragastric pH levels to above 4.0 (Blum et al. 1997) and this likely accounts for the reduction in exposure and the inter- and intra-subject variability observed in the present study.
Systemic exposure to danirixin, in terms of AUC and C max, was generally highest following Bioenhanced Formulation 2 compared with the other formulations in the fasted state, but this was not associated with any reduction in inter-subject variability. Systemic exposure following the danirixin IR formulation was higher in the fed state compared with the fasted state. The estimated terminal t 1/2 values were similar for the different danirixin formulations in this study (7–9 h) and were in line with previously determined values, suggesting that the elimination of danirixin was independent of formulation dose or fed/fasted status, supporting the conclusion that dissolution and absorption in the gastrointestinal track are major determinants of systemic exposure following oral administration (GSK 2013b).
In the present study danirixin exposures in Treatment Period 1 were higher than exposures for the subsequent treatment periods. It is unclear whether this unexpected ‘period’ effect is real as the exposure and variability of danirixin are consistent with a prior study in which the omeprazole interaction was evaluated (GSK 2013b). The possibility of some variability over time in gastric pH due to omeprazole is a potential explanation, though neither gastric pH nor the PK of omeprazole was measured. Steady-state levels of omeprazole should have been achieved prior to dosing on Day 1 of Treatment Period 1. It does not appear that lack of compliance with the omeprazole dosing regimen during any of the treatment periods was a factor.
Overall, all formulations of danirixin were well tolerated in the healthy elderly population assessed in this study and no major safety concerns were identified. The safety profile was similar to that observed in prior studies (GSK 2013a, b) and was not different between danirixin IR in the fed and fasted states. The incidence of reported AEs with the bioenhanced formulations was similar, but slightly higher than for danirixin IR. In the current study, there were no reports of neutropaenia or sustained decreases in neutrophil count, which is also consistent with previous single- and repeat-dose danirixin studies (GSK 2013a, b).
This study has several strengths and potential limitations. Overall, the study results are largely consistent with those from a previous study of danirixin (GSK 2013a, b) and the collective results from both studies will help to guide selection of the most appropriate formulation/dosing regimen for future investigations. The observed (and statistically significant) treatment period effect was unexpected and remains unexplained. However, this did not interfere with the study objectives as the results with the conventional immediate-release formulation were consistent with previous studies.
In conclusion, although systemic exposures were higher for Bioenhanced Formulation 2 of danirixin in the fasted state than for any other formulation of danirixin during either fasted or fed states, the bioenhanced formulations do not confer any advantage over a conventional immediate-release formulation with regard to inter-subject variability on a background of concomitant proton pump inhibitor use. Concomitant use of gastric acid suppressing medications will need to be a consideration in the selection of appropriate subjects for further clinical development of danirixin.
References
Blum RA, Shi H, Karol MD, Greski-Rose PA, Hunt RH (1997) The comparative effects of lansoprazole, omeprazole, and ranitidine in suppressing gastric acid secretion. Clin Ther 19:1013–1023
Blume H, Donath F, Warnke A, Schug BS (2006) Pharmacokinetic drug interaction profiles of proton pump inhibitors. Drug Saf 29:769–784
Bor S, Kitapcioglu B, Solak ZA, Ertilav M, Erdinc M (2010) Prevalence of gastroesophageal reflux disease in patients with asthma and chronic obstructive pulmonary disease. J Gastroenterol Hepatol 25:309–313
Budha NR, Frymoyer A, Smelick GS, Jin JY, Yago MR, Dresser MR, Holden SN, Benet LZ, Ware JA (2012) Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther 92:203–213
Chapman RW, Phillipsa JE, Hipkina RW, Currana AK, Lundella D, Fine JS (2009) CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther 121:55–68
GSK (2013a) GSK Study CX3112483 (NCT01209052). A single-centre, double-blind, placebo-controlled study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of single, oral, ascending doses and repeat oral doses of GSK1325756 in healthy male subjects. Results summary available at: http://www.gsk-clinicalstudyregister.com. Last accessed 07 June 2013
GSK (2013b) GSK Study CX3113722 (NCT01209104). A single-dose, open, randomized, crossover investigation of age, gender, food-effect and proton-pump inhibitor interaction on the pharmacokinetics of GSK1325756 in healthy adult subjects and a single-dose, double blind, parallel-group, placebo-controlled investigation of the pharmacokinetics of GSK1325756 in healthy elderly adult subjects. Results summary available at: http://www.gsk-clinicalstudyregister.com. Last accessed 07 June 2013
Holz O, Khalilieh S, Ludwig-Sengpiel A, Watz H, Stryszak P, Soni P, Tsai M, Sadeh J, Magnussen H (2010) SCH527123, a novel CXCR2 antagonist, inhibits ozone-induced neutrophilia in healthy subjects. Eur Respir J 35:564–570
International Conference on Harmonisation Tripartite Guideline: Guidance for Good Clinical Practice E6 (R1). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf; 1996. Last accessed: April 24, 2013
Lazaar AL, Sweeney LE, MacDonald AJ, Alexis NE, Chen C, Tal-Singer R (2011) SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br J Clin Pharmacol 72:282–293
Mukaida N (2003) Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol 284:L566–L577
Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O’Byrne PM, Stryszak P, Gann L, Sadeh J, Chanez P (2012) Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy 42:1087–1103
Quint JK, Wedzicha JA (2007) The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol 119:1065–1071
Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, Watz H, Lu S, Strysak P, Rosenberg E, Staudinger H (2013) CXCR2 antagonist MK-7123—a phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med 187:A6071
Russell TL, Berardi RR, Barnett JL, Dermentzoglou LC, Jarvenpaa KM, Schmaltz SP, Dressman JB (1993) Upper gastrointestinal pH in seventy-nine healthy, elderly, North American men and women. Pharm Res 10:187–196
Shi S, Klotz U (2008) Proton pump inhibitors: an update of their clinical use and pharmacokinetics. Eur J Clin Pharmacol 64:935–951
Spooner N, Lad R, Barfied M (2009) Dried blood spots as a sample collection technique for the determination of pharmacokinetics in clinical studies: considerations for the validation of a quantitative bioanalytical method. Anal Chem 81:1557–1563
Stockley RA (2002) Neutrophils and the pathogenesis of COPD. Chest 121:151S–155S
WMA Declaration of Helsinki—ethical principles for medical research involving human subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964 and amended (latest) by the 59th WMA General Assembly, Seoul; 2008. http://www.wma.net/en/30publications/10policies/b3/index.html. Date last updated: October 2008. Last accessed: 22 April 2013
Acknowledgments
Editorial support in the form of development of a manuscript first draft in consultation with the lead author, fact checking, referencing and assembling tables was provided by Tara N Miller, PhD at Gardiner-Caldwell Communications (Lyndhurst, US), and support in the form of graphic services was provided by Laura Maguire, MChem at Gardiner-Caldwell Communications (Macclesfield, UK); editorial support was funded by GlaxoSmithKline. Study CX3115550 was funded by GlaxoSmithKline.
Conflict of interest
All authors were employees of GlaxoSmithKline (GSK) at the time the study was conducted. GSK funded study CX3115550.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Miller, B.E., Smart, K., Mistry, S. et al. The pharmacokinetics of conventional and bioenhanced tablet formulations of danirixin (GSK1325756) following oral administration in healthy, elderly, human volunteers. Eur J Drug Metab Pharmacokinet 39, 173–181 (2014). https://doi.org/10.1007/s13318-014-0179-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-014-0179-8