
Abstract
Fusarium brachygibbosum Padwick is a phytopathogen with a widespread distribution, infecting various host plants. In South Africa, there is a limited number of studies on the genetic diversity of fusaria, particularly in undisturbed soils. In the current study, the genetic diversity of F. brachygibbosum was investigated using inter-simple sequence repeat (ISSR) and mating type genes. The F. brachygibbosum isolates were collected from four different geographic regions within the grassland biome of South Africa. A total of ten ISSR primers produced 64 distinct and reproducible amplicons, with 100% polymorphism. The ISSR markers revealed high levels of intraspecific variation, which is depicted by various genetic diversity indexes. In the analysis of mating type genes, the MAT1-1-2 gene was found among the four populations, but the MAT1-2-1 gene was only present in the population from the Groenkloof Nature Reserve. The data from this study will contribute substantially to knowledge of grassland fusaria of South Africa and their population structure in the grassland biome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fusarium brachygibbosum is a plant pathogen that was initially described by Padwick (1945) based on morphological characteristics. Molecular identification of F. brachygibbosum have been performed using the internal transcribed spacer (ITS) of the rDNA (Al-Sadi et al. 2012; Al-Mahmooli et al. 2013; Mirhosseini et al. 2014; Trabelsi et al. 2018; Ali et al. 2020), the translation elongation factor 1α (TEF-1α) (Al-Mahmooli et al. 2013; Trabelsi et al. 2018; Laraba et al. 2021) RNA polymerase II largest subunit (RPB1) and RNA polymerase second largest subunit (RPB2) gene regions. This phytopathogen is geographically widely distributed, and can be found in a wide range of host plants such as oleander (Nerium oleander L.) in Iran (Mirhosseini et al. 2014), sugar beet (Beta vulgaris L.) in China (Cao et al. 2018), marijuana (Cannabis sativa L.) in California, USA (Punja et al. 2018), and olive trees (Olea europaea L.) in Tunisia (Trabelsi et al. 2018). The life cycle of F. brachygibbosum has not been reported in detail (Ali et al. 2020), which makes it challenging to determine the exact disease cycle of this pathogen in host plants.
Trabelsi et al. (2018) identified F. brachygibbosum as a causative agent of dieback and vascular wilt of olive trees based on morphological and microscopic characteristics that coincided with those reported by Al-Mahmooli et al. (2013). Tan et al. (2011) associated F. brachygibbosum with the production of mycotoxins in legume pastures, namely Medicago polymorpha L. (annual medic), Trifolium subterraneum L. (subterranean clover), and M. sativa L. (lucerne) in Australia. According to the results, F. brachygibbosum produced type A trichothecene derivatives and fusarenon-X (type B trichothecene).
Increased reports of F. brachygibbosum occurring as a phytopathogen on a variety of hosts raises the concern that it has the potential to develop into a significant future pathogen. The current study was conducted to reveal the genetic diversity of F. brachygibbosum, which F. brachygibbosum was only recently reported from South Africa in wheat grains (Beukes et al. 2017) and has been detected as one of the most predominant Fusarium species isolated from the South African grassland biome (Mojela 2017; Jacobs et al. 2018).
The aim of the current study was to determine the population diversity of F. brachygibbosum from undisturbed soils of four different geographic regions within the grassland biome of South Africa (Mojela 2017; Jacobs et al. 2018; Mavhunga et al. 2021). The first objective of the study was to identify inter-simple sequence repeat (ISSR) loci, which are highly polymorphic genetic markers that can reveal intraspecific genetic variation (Nayaka et al. 2011). Secondly, the molecular markers were used to analyze F. brachygibbosum isolates from the above-mentioned localities in order to test for the presence of two closely related species. Lastly, the mating types were investigated to further understand the potential sexual behaviour of F. brachygibbosum, since sexual recombination contributes to higher genetic variability.
Materials and methods
Fungal isolates and DNA extraction
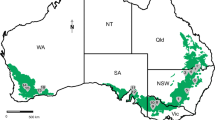
This study was conducted on 104 isolates of F. brachygibbosum obtained from the National Collection of Fungi (NCF), Agricultural Research Council-Plant Health and Protection (ARC-PHP). The isolates were used in previous studies (Jacobs et al. 2018; Mavhunga 2021) on fusaria biodiversity from undisturbed soils in four different geographical regions Groenkloof (GK, 30 isolates) Nature Reserve (25.7934°S 28.2039°E), Willem Pretorius (WP, 33 isolates) Nature Reserve (28.306°S 27.236°E), Rietvlei (RV, 21 isolates) Nature Reserve (25°53′49″S 28°17′38″E), and Melvillekoppies (MK, 20 isolates) Nature Reserve (26.1674986°S 28.0020311°E) within the grassland biome of South Africa (Fig. 1). The isolates were grown on full strength potato dextrose agar (PDA) (Biolab) in the dark at 28 °C for seven days. Mycelium was scraped from the surface of the growth medium and ground to a fine powder using liquid nitrogen. Total genomic DNA was extracted using a modification of the CTAB (cetyl trimethylammonium bromide) extraction method (Leslie and Summerell 2006). The extracted DNA was stored at -20 °C.

The F. brachygibbosum isolates were sampled from the soils of the four geographic regions (black dots) within the grassland biome (gray area) of South Africa. These sampling regions are depicted by the closed circles
PCR and gel electrophoresis
The first four ISSR primers in Table 1 (i.e., Primer 1, Primer 2, Primer 3, and Primer 4) were obtained from Mishra et al. (2003). Amplification reactions for these primers were performed in 20 μl reaction volumes containing 1x DreamTaq reaction buffer, 0.2 mM dNTPs, 0.5 μM of each ISSR primer, 1 U of DreamTaq polymerase, and 20 ng/μl of gDNA. The PCR amplifications were performed under the following conditions: initial denaturation was for 2 min at 95 °C, followed by 40 cycles of 1 min at 94 °C, 2 min at 55 °C (primer 1 and 2), or 48 °C (primer 3 and 4) and 30 s at 72 °C, with a final extension of 5 min at 72 °C. The PCR products were separated using 6% denaturing polyacrylamide gel electrophoresis (PAGE). The gel was electrophoresed in a 1x TBE (Tris-borate-EDTA) buffer (89 mM Tris, 89 mM boric acid, and 2 mM EDTA) at 5 V/cm. The gel was stained with ethidium bromide and visualized using a UV light transilluminator.
The software program Genome-wide Microsatellite Analysing Tool (GMATo) (Wang et al. 2013) was used to mine seven additional ISSR primers. The full genome of the closely related species F. sambucinum strain F-4 was obtained from NCBI and uploaded into GMATo to obtain the ISSR loci. The PCR was optimized using the Taguchi methods as described in Cobb and Clarkson (1994). The PCR reactions for primers FS-03, FS-05, FS-06, FS-12 and FS-13 contained 1x DreamTaq reaction buffer, 0.38 mM dNTPs, 2.5 mM MgCl2, 0.5 μM of each primer, 1 U of DreamTaq polymerase and 20 ng/μl of gDNA. The PCR reactions for primers FS-08 and FS-11 contained 1x reaction buffer, 0.31 mM dNTPs, 3.13 mM MgCl2, 0.5 μM of each ISSR primer, 1 U DreamTaq polymerase and 20 ng/μl gDNA. The PCR reactions were performed in 20 μl volume under the following conditions: initial denaturation was for 2 min at 95 °C, followed by 40 cycles of 1 min at 94 °C, 2 min for annealing temperature, and 30 s at 72 °C, with a final extension of 5 min at 72 °C. The PCR products were electrophoresed on 6% PAGE/Urea. The MAT gene primers were obtained from Montoya-Martínez et al. (2019).
The PCR amplifications for the gene MAT1-1-2 from the MAT1-1 idiomorph were performed using the primer sets M112f1 (forward) and M112r1 (reverse) and should yield a fragment of 465 bp. The PCR amplifications for the gene MAT1-2-1 from the MAT1-2 idiomorph were performed using the primer sets M121f2a (forward) and M121r2 (reverse) with an expected band size of 330 bp. The PCR for the MAT1-1-2 and MAT1-2-1 genes were conducted in a 30 μl volume containing 1x DreamTaq reaction buffer, 0.2 mM of dNTPs, 0.25 μM of each primer sets, 1 U of DreamTaq polymerase, and 20 ng/μl of gDNA. The PCR was conducted under the following conditions: initial denaturation was 90 s at 94 °C, followed by 40 cycles of 30 s at 94 °C, 40 s at 55 °C (MAT1-1-2 gene), and 61 °C (MAT1-2-1 gene) and 60 s at 68 °C, with a final extension of 5 min s at 68 °C. The PCR products were electrophoresed on a 2% agarose gel to confirm the presence of amplicons.
Population genetics analyses
The bands for each locus were scored manually as either present “1” or absent “0”, and the bands that could not be distinguished were denoted as missing data “n”. The generated binary data matrix was used to calculate genetic diversity, number of polymorphic loci, and the percentages of polymorphic loci within each population and within the combined populations were calculated using the software program POPGENE version 1.32 (Yeh et al. 1999). This software was also used to calculate Nei’s genetic diversity index (h), and Shannon’s Information index (I) was calculated according to the formula I=-\({\sum }_{}^{}Pilog2Pi\) (Lewontin 1972), in which Pi represents the frequency of the presence of bands. Nei’s (1973) gene diversity was calculated according to the formula H = 1–Σxk2 (xk is the frequency of the kth allele). The other genetic diversity parameters were also calculated: Ht represented total genetic diversity within the populations, Hs is the diversity within each population and Gst is the coefficient of allelic differentiation. These parameters were calculated using the software GenAlEx 6.501 (Peakall and Smouse 2006, 2012). The gene flow estimates (Nm) were calculated according to the formula Nm = 0.5(1 – Gst)/Gst (McDermott and McDonald 1993), where Nm is the number of allelic migrants per generation. The software program MultiLocus version 1.2.2 (Agapow and Burt 2001) was used to calculate the θ-values of population differentiation between the populations. The θ-values were calculated using Weir and Cockerham’s (1984) modification of Wright’s (1949) FST for haploid data, which is given by the formula θ = Q2-Q3/1-Q3, where Q2 is the probability that two alleles within the same population are identical, and the probability that two alleles from different populations are identical is given by Q3. Moreover, the θ-values were used to calculate the theoretical number of migrants per generation (Slatkin 1995), which is given by the formula \(\stackrel{\prime }{M}\)=\(\frac{1}{2}\left(\frac{1}{\theta }-1\right)\), where \(\stackrel{\prime }{M}\) is the number of migrants per generation. The additional parameters were number of alleles (Na), effective number of alleles (Ne) (Kimura and Crow 1964), Shannon’s Information index (I), Nei’s diversity (h), and unbiased diversity (uh). The distribution of genetic variation within and between the populations were determined by analysis of molecular variance (AMOVA) (Excoffier et al. 1992) using the software GenAlEx 6.501. A two-sample t-test assuming unequal variances was conducted using Microsoft Excel Ver. 16.0 to compare differences in genetic diversity using Nei’s diversity (h) between populations that are encroached by urban development (GK and MK) and populations located in agriculturally intensive area (WP and RV). Dendrograms were constructed using distance matrices generated using Nei and Li’s coefficient (Nei and Li 1979). The software program PAUP 4.0* (phylogenetic analysis using parsimony; Swofford 2002) was used to construct UPGMA (un-weighted pair group method using arithmetic averages) dendrograms. The dendrograms were constructed using the heuristic search protocol and bootstrap analysis with 1 000 replicates was done using a full heuristic search protocol.
Results
Genetic diversity
In the analysis of the first four ISSR primers (Table 1), three of the four tested markers resulted in positive amplifications, while Primer 2 did not produce amplicons. The seven mined ISSR primers (Fig. 2; Table 1) all resulted in positive amplifications with the results from primer FS-08 on selected isolates from Groenkloof nature reserve shown in Fig. 2. In total, the ten ISSR primers produced 64 distinct and reproducible bands, with 100% polymorphism. The mean percentage of polymorphic loci was 91.02% with a range from 68.75% (WP) to 100% (MK). The values of Shannon’s information index (I) and Nei’s genetic diversity (h) both range from 0 to 1, where the values closer to 1 indicate high genetic diversity. In the analysis of Shannon’s information index (I), the mean I for GK was I = 0.459, for WP it was I = 0.273, for RV it was I = 0.518, and for MK it was I = 0. 537.The overall mean Shannon information index (I) within the four populations was I = 0.447. In the analysis of Nei’s genetic diversity (h), the mean h for GK was h = 0.299, for WP it was h = 0. 171, for RV it was h = 0. 346, and lastly, for MK it was h = 0. 360. The mean Nei’s genetic diversity for the combined populations was h = 0.294. Shannon’s information index (I) and Nei’s genetic diversity (h) revealed high genetic diversity within the four populations. The other genetic diversity indexes (Tables 2 and 3) also indicated high levels of genetic diversity within the four populations.

ISSR fingerprint of F. brachygibbosum isolates from Groenkloof nature reserve visualized on 6% PAGE/Urea. Primer FS-08 was used to produce the profiles in both gels. Lane M: 2 kb ladder, Lane 1: PPRI 16,109, Lane 2: PPRI 16,116, Lane 3: PPRI 16,126, Lane 4: PPRI 16,137, Lane 5: PPRI 16,139, Lane 6: PPRI 16,603, Lane 7: PPRI 16,621, Lane 8: PPRI 16,124, Lane 9: PPRI 16,633, Lane 10: PPRI 17,527, Lane 11: PPRI 17,909, Lane 12: PPRI 17,916, Lane 13: PPRI 17,921, Lane 14: PPRI 17,926, and Lane 15: Non target control
The genetic identity between the populations was calculated to determine the degree of identical alleles between the four populations. In the analysis of Nei’s Genetic Identity, the highest identity (0.9339) was observed between populations RV and MK, while populations GK and WP had the lowest genetic identity (0.8657). Genetic distance measures the genetic divergence between the populations. The highest genetic distance (0.1443) was observed between populations WP and GK, while the lowest genetic distance (0.0684) occurred between the populations MK and RV. In the analysis of the coefficient of gene differentiation (Gst) among the populations, the mean Gst was 0.1779, indicating the presence of 17.79% of genetic variation among the four populations (with an estimated 82.21% variation within the populations). AMOVA was done for the four populations combined, and according to the results, most of the variation was distributed within the populations (80%) rather than among the two populations (20%).
Population differentiation
The number of allelic migrants per generation was Nm = 2.3102, which suggested a significant migration rate among the four populations. Since Nm used allelic frequencies to determine the migration rate, the population differentiation (θ) was calculated to determine the true value of migrants per generation (\(\stackrel{\prime }{M}\)) using genotypic frequencies. The population differentiation (θ) for the four populations was θ = 0.2135. The θ-value was used to calculate the number of migrants per generation (\(\stackrel{\prime }{M}\)) among the four populations. The number of migrants per generation was \(\stackrel{\prime }{M}\) = 1.8419. The value for \(\stackrel{\prime }{M}\) indicated that the estimated migration rate among the four populations was low.
Two-sample t-tests
The two-sample t-tests assuming unequal variances were calculated on the mean Nei’s diversity (h) to determine the significance of differences in genetic diversity. The null hypothesis stated that there will be no significant difference in the mean Nei’s diversity (h) between populations that are encroached by urban development (GK and MK) and populations located in agriculturally intensive area (WP and RV). Therefore, t-values of P\(\le =\) 0.05 indicated significant difference in mean Nei’s diversity. According to the results, the significant difference in the mean Nei’s diversity (h) between the two stated groups was P = 0.0004981. Therefore, the null hypothesis was rejected since there was a significant difference in Nei’s diversity.
Clustering analysis
Cluster analysis was done using UPGMA based on Nei and Li’s coefficients of similarity (Nei and Li 1979) from 64 loci for the ten ISSR markers. Overall, the clustering analysis produced two major clades comprising of isolates from the four nature reserves (Fig. 3). Therefore, this indicated that the geographic origins of the isolates did not have a major impact on the clustering of the isolates. The two major clades for the dendrogram wase significantly supported with a bootstrap value of 100%. The isolate PPRI 14,375 from the GK nature reserve formed an outgroup (OG) in the dendrograms. The tree length was 889, the consistency index (CI) = 0.0719, and the retention index (RI) = 0.4691.

UPGMA based on Nei and Li’s coefficients of similarity for the F. brachygibbosum isolates from this study. Bootstrap support values are indicated by the numbers on the branches. Tree statistics include length = 889, the consistency index (CI) = 0.0719, and the retention index (RI) = 0.4691
Mating type genes
Both the MAT1-1-2 and the MAT1-2-1 genes were amplified for the four nature reserves (Fig. 4). In the population from the GK nature reserve, both genes were present with only one isolate (PPRI16109) amplifying the MAT1-2-1 allele. In the populations from the WP, RV and MK nature reserves, only the MAT1-1-2 genes amplified, and the MAT1-2-1 gene did not produce amplicons.

PCR products for the primer sets M112f1 and M112r1 (MAT1-1-2 gene) for F. brachygibbosum isolates from Willem Pretorius nature reserve visualized on a 2% agarose gel., Lane 1: PPRI 19,118, Lane 2: PPRI 19,125, Lane 3: PPRI 19,133, Lane 4: PPRI 19,139, Lane 5: PPRI 19,150, Lane 6: PPRI 19,166, Lane 7: PPRI 19,169, Lane 8: PPRI 19,170, Lane 9: PPRI 19,175, Lane 10: PPRI 19,232, Lane 11: PPRI 19,236, Lane 12: PPRI 20,527, Lane 13: PPRI 20,720, Lane 14: PPRI 20,729, Lane 15: PPRI 20,734, Lane 16: PPRI 20,745, Lane 17: NTC, Lane 18: PPRI 20,755, Lane 18: PPRI 21,041, Lane 19: PPRI 21,042, Lane 20: PPRI 21,049, Lane 21: PPRI 21,050, Lane 22: PPRI 21,050, Lane 23: PPRI 21,052, Lane 24: PPRI 21,058, Lane 25: PPRI 21,061, Lane 26: PPRI 21,063, Lane 27: PPRI 21,082, Lane 28: PPRI 21,084, Lane 29: PPRI 21,086, Lane 30: PPRI 21,087, Lane 31: PPRI 21,088, Lane 32: PPRI 21,093, Lane 33: 21,094, Lane 34: PPRI 21,100, and Lane 35: Non template control
Discussion
In the current study, the application of ten ISSR markers have demonstrated a high level of genetic diversity among isolates from four populations of F. brachygibbosum. The ISSR markers showed that isolates from the Rietvlei and Melvillekoppies nature reserves have a higher genetic diversity compared to those from the Groenkloof and Willem Pretorius nature reserves. This is interesting since the Melvillekoppies nature reserve is encroached by urban development, while the Willem Pretorius nature reserve is located in open grasslands, surrounded by farms. Van Rensburg (2010) conducted a survey on the biodiversity and distribution of microorganisms collected from soils in the grasslands encroached by urban development and concluded that the biodiversity of fungal populations was higher in rural areas. This suggests that increased human activity in urban areas have an influence on the biodiversity of fungal populations. Recently, Laraba et al. (2021) performed a phylogenetic analysis of the Fusarium sambucinum species complex (FSamSC). Included in this analysis was 50 of the 171 isolates designated as closely related to F. brachygibbosum. The phylogenetic analyses revealed that within the F. brachygibbosum clade, there were two described species (i.e., F. brachygibbosum and F. transvaalense) and 11 novel phylogenetically distinct species. This high level of closely related potential novel species within this complex sheds light on the observation of higher population diversity levels in this study. Thus, our analysis supports the finding that multiple novel species may be found within the F. brachygibbosum clade based on the diversity that was observed. The population analysis further supports the observation that these species are closely related and may be difficult to resolve. The high level of genetic diversity observed from the ISSR markers is attributed to the high discriminatory power of these markers to differentiate and reveal intra- and interspecific variations in various Fusarium species (Mishra et al. 2003, 2004; Bayraktar and Dolar 2011). The analysis of additional genetic diversity parameters demonstrated a high number of allelic migrants per generation (Nm), but the true migration rate among the four populations was given by \(\stackrel{\prime }{M}\), which demonstrated lower migration rates among the four populations. The lower migration rates coincide with the significant geographic distance among the four populations. Additionally, the high value for allelic migrants can be ascribed to size homoplasy of the markers.
Nei’s genetic distance among the four populations was substantially high, which corroborates the occurrence of recombination. Mating type genes were investigated to further explain the presence of high genetic variation among the four populations. From this analysis, both MAT1 idiomorphs (i.e., MAT1-1 and MAT1-2) were found in the isolates among the four populations. However, both MAT1-1-2 and MAT1-2-1 genes were only found in the population from the GK nature reserve, while only the MAT1-1-2 gene was found in the isolates from the other three nature reserves. Zeller et al. (2003) used an integrated approach to characterize F. anthophilum isolates recovered from the uncultivated grasslands in Kansas. From this study, the putative isolates of F. anthophilum presented two closely related species. Therefore, a new species, F. konzum was described using morphological characteristics, mating compatibility, AFLP markers, and β-tubulin DNA sequencing. This demonstrates the power of population studies to discern between closely related species that are morphologically similar and occur together. The clustering of the F. brachygibbosum and the detection of both mating type genes will facilitate the delimitation of several species that are reported for the F. brachygibbosum clade within the FSamSC.
References
Agapow PM, Burt A (2001) Indices of multilocus linkage disequilibrium. Mol Ecol Notes 1:101–102
Al-Mahmooli IH, Al-Bahri YS, Al-Sadi AM, Deadman ML (2013) First report of Euphorbia larica dieback caused by Fusarium brachygibbosum in Oman. Plant Dis 97:687–687
Al-Sadi AM, Al-Jabri AH, Al-Mazroui SS, Al-Mahmooli IH (2012) Characterization and pathogenicity of fungi and oomycetes associated with root diseases of date palms in Oman. Crop Prot 37:1–6
Ali HZ, Hameed MS, Abdulrahman AA, Saood HM (2020) First Report on Fusarium brachygibbosum isolate FIR 16_ITS isolated from iraqi wheat plant. J Ecol Eng 21:81–86
Bayraktar H, Dolar FS (2011) Molecular identification and genetic diversity of Fusarium species associated with onion fields in Turkey. J Phytopathol 159:28–34
Beukes I, Rose LJ, Shephard GS, Flett BC, Viljoen A (2017) Mycotoxigenic fusarium species associated with grain crops in South Africa-A review. South Afr J Sci 113(3–4):1–12
Cao S, Yang N, Zhao C, Liu J, Han C, Wu X (2018) Diversity of Fusarium species associated with root rot of sugar beet in China. J Gen Plant Pathol 84:321–329
Cobb BD, CIarkson JM (1994) A simple procedure for optimising the polymerase chain reaction (PCR) using modified Taguchi methods. Nucleic Acids Res 22:3801–3805
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131(2):479–491
Jacobs A, Mojela L, Summerell B, Venter E (2018) Characterisation of members of the Fusarium incarnatum-equiseti species complex from undisturbed soils in South Africa. Antonie Van Leeuwenhoek 111:1999–2008
Kimura M, Crow JF (1964) The number of alleles that can be maintained in a finite population. Genetics 49:725–738
Laraba I, McCormick SP, Vaughan MM, Geiser DM, O’Donnell K (2021) Phylogenetic diversity, trichothecene potential, and pathogenicity within Fusarium sambucinum species complex. PLoS ONE 16:1–30
Leslie JF, Summerell BA (2006) The Fusarium laboratory manual. Blackwell Publishing Professional, Ames
Lewontin RC (1972) The apportionment of human diversity. Evol Biol 6:381–398
Mavhunga M, Kwinda GT, Summerell BA, Venter BA, Jacobs A (2021) Genetic diversity of Fusarium oxysporum complex isolated from the grassland biome of South Africa. Phytopathol 111:1459–1469
McDermott JM, McDonald BA (1993) Gene flow in plant pathosystems. Ann Rev Phytopathol 31:353–373
Mirhosseini HA, Babaeizad V, Hashemi L (2014) First report of Fusarium brachygibbosum causing leaf spot on oleander in Iran. J Plant Pathol 96:431
Mishra PK, Fox RT, Culham A (2003) Inter-simple sequence repeat and aggressiveness analyses revealed high genetic diversity, recombination and long‐range dispersal in Fusarium culmorum. Ann Appl Biol 143:291–301
Mishra PK, Tewari JP, Clear RM, Turkington TK (2004) Molecular genetic variation and geographical structuring in Fusarium graminearum. Ann Appl Biol 145:299–307
Mojela ML (2017) The diversity of the genus Fusarium in soil from the Willem Pretorius Nature Reserve, South Africa (Masters thesis, University of Johannesburg (South Africa))
Montoya-Martínez AC, Rodríguez-Alvarado G, Fernánadez-Pavía SP, Proctor RH (2019) Design and validation of a robust multiplex polymerase chain reaction assay for MAT idiomorph within the Fusarium fujikuroi species complex. Mycologia 111:772–781
Nayaka SC, Wulff EG, Udayashankar AC, Nandini BP, Niranjana SR, Mortensen CN, Prakash HS (2011) Prospects of molecular markers in Fusarium species diversity. Appl Microbiol Biotechnnol 90:1625–1639
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci 70:3321–3323
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci 76:5269–5273
Padwick GW (1945) Notes on indian fungi. HI Mycological Papers 12:1–15
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28:2537–2539
Punja ZK, Scott C, Chen S (2018) Root and crown rot pathogens causing wilt symptoms on field-grown marijuana (Cannabis sativa L.) plants. Can J Plant Pathol 40:528–541
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Swofford DL (2002) PAUP: phylogenetic analysis using parsimony (and other methods). Version 4.0. Sinauer Associates, Sunderland, Massachusetts
Tan DC, Flematti GR, Ghisalberti EL, Sivasithamparam K, Chakraborty S, Obanor F, Barbetti MJ (2011) Mycotoxins produced by Fusarium species associated with annual legume pastures and ‘sheep feed refusal disorders’ in western Australia. Mycotoxin Res 27:123–135
Trabelsi R, Gdoura R, Triki MA (2018) Fusarium brachygibbosum and Fusarium Chlamydosporum causing wilt and die-back of olive in Tunisia. In: Kallel A, Ksibi M, Dhia HB, Khélifi N (eds) Recent advances in Environmental Science from the Euro-Mediterranean and surrounding regions. EMCEI 2017. Advances in Science, Technology & Innovation. IEREK Interdisciplinary Series for Sustainable Development. Springer, Cham, pp 581–582
Van Rensburg JPJ (2010) Investigation of the microbial diversity and functionality of soil in fragmented South African grasslands along an urbanization gradient (Masters thesis, North-West University (South Africa))
Wang X, Lu P, Luo Z (2013) GMATo: a novel tool for the identification and analysis of microsatellites in large genomes. Bioinformation 9:541–544
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wright’s (1949) The genetical structure of populations. Ann Eugen 15:323–354
Yeh FC, Yang RC, Boyle T (1999) POPGENE. Microsoft Windows-Based Freeware for Population Genetic Analysis: release 1.31. University of Alberta, Edmonton
Zeller KA, Summerell BA, Bullock S, Leslie JF (2003) Gibberella konza (Fusarium konzum) sp. nov. from prairie grasses, a new species in the Gibberella fujikuroi species complex. Mycologia 95:943–954
Acknowledgements
The project was funded by NRF Thutuka (TTK13070120147, TTK160530166659), NRF FBIP (FBIS150520118174) and the University of Johannesburg URC/FRC grants.
Funding
Open access funding provided by University of Johannesburg.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare that are relevant to this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dhlamini, L., Jacobs, A., van der Merwe, N.A. et al. The population structure of the Fusarium brachygibbosum species complex in the grassland biome of South Africa. Australasian Plant Pathol. 53, 31–39 (2024). https://doi.org/10.1007/s13313-023-00943-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13313-023-00943-7