
Abstract
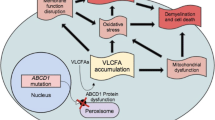
X-linked adrenoleukodystrophy (ALD) is a genetic disorder that presents neurologically as either a rapid and fatal cerebral demyelinating disease in childhood (childhood cerebral adrenoleukodystrophy; ccALD) or slow degeneration of the spinal cord in adulthood (adrenomyeloneuropathy; AMN). All forms of ALD result from mutations in the ATP Binding Cassette Subfamily D Member (ABCD) 1 gene, encoding a peroxisomal transporter responsible for the import of very long chain fatty acids (VLCFA) and results mechanistically in a complex array of dysfunction, including endoplasmic reticulum stress, oxidative stress, mitochondrial dysfunction, and inflammation. Few therapeutic options exist for these patients; however, an additional peroxisomal transport protein (ABCD2) has been successfully targeted previously for compensation of dysfunctional ABCD1. 4-Phenylbutyrate (4PBA), a potent activator of the ABCD1 homolog ABCD2, is FDA approved, but use for ALD has been stymied by a short half-life and thus a need for unfeasibly high doses. We conjugated 4PBA to hydroxyl polyamidoamine (PAMAM) dendrimers (D-4PBA) to a create a long-lasting and intracellularly targeted approach which crosses the blood–brain barrier to upregulate Abcd2 and its downstream pathways. Across two studies, Abcd1 knockout mice administered D-4PBA long term showed neurobehavioral improvement and increased Abcd2 expression. Furthermore, when the conjugate was administered early, significant reduction of VLCFA and improved survival of spinal cord neurons was observed. Taken together, these data show improved efficacy of D-4PBA compared to previous studies of free 4PBA alone, and promise for D-4PBA in the treatment of complex and chronic neurodegenerative diseases using a dendrimer delivery platform that has shown successes in recent clinical trials. While recovery in our studies was partial, combined therapies on the dendrimer platform may offer a safe and complete strategy for treatment of ALD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adrenoleukodystrophy (ALD) is an X-linked neurodegenerative disorder arising from mutations in the ATP Binding Cassette Subfamily D Member (ABCD) 1 gene encoding the peroxisomal half-transporter, adrenoleukodystrophy protein (ALDP), which imports very long chain fatty acids (VLCFA) into the peroxisome for fatty acid β-oxidation. The molecular signature of ALD is the accumulation of VLCFA within all tissues, increased oxidative stress, and peripheral axonopathy. In a third of affected individuals with ALD, and commonly young boys, the disease rapidly progresses into childhood cerebral ALD (ccALD) with neuroinflammatory cerebral demyelination resulting in severe disability and death within 2 years. The other 60–65% of male patients present during adulthood with a slowly progressive dying back axonopathy of long tracts in the spinal cord, a condition termed adrenomyeloneuropathy (AMN). AMN affects both adult males and heterozygote females although disease course is typically still more severe in males. In addition, men remain at risk for adult-onset cerebral ALD (acALD), which can be rapidly progressive similar to the phenotype observed in boys. There are over 1000 identified pathogenic variants in ABCD1 determined by elevated VLCFA, yet there remains no identified genotype–phenotype correlation, predictive biomarkers, or therapeutic options upon diagnosis. Hematopoietic stem cell transplantation, currently the only available treatment strategy, is only suitable for a subset of ccALD patients during a short window of disease, where progression may be halted without expected recovery [1,2,3]. Still, no effective therapies exist for AMN or acALD.
Fatty acids longer than 22 carbons (VLCFA) are exclusively processed in the peroxisome for β-oxidation, as opposed to the mitochondria where shorter chain fatty acids are broken down [4]. Three transporters of the ATP-binding cassette (ABC) superfamily D exist in the peroxisomal membrane which mediates a host of sometimes unique metabolic functions [4]. ABCD1, the transporter responsible for ALD, is the main transporter of saturated and monosaturated VLCFAs across the peroxisomal membrane. ABCD2 shares 80% homology with ABCD1 and thus also shares substrate specificity for VLCFAs, although not as strong an affinity for saturated VLCFA as ABCD1, and also processes long chain fatty acids [4, 5]. While ABCD3 shows some functional overlap with ABCD1, it transports the widest array of fatty acids including bile acid intermediates [6]. Due to the high homology of ABCD2 with ABCD1, ABCD2 induction has frequently been explored as a therapeutic avenue for ALD. Several pharmacologic approaches to overexpress ABCD2, such as thyroid mimetics, have had varying degrees of success in reducing VLCFAs, with the vast majority not moving beyond mouse [7]. 4-Phenylbutyrate (4PBA), the prodrug of phenylacetate, is a potent pharmacological inducer of ABCD2 and serves as an alternative peroxisomal Acyl-CoA transporter for VLCFA [5]. 4PBA is an FDA-approved compound, which normalizes CNS VLCFA levels in Abcd1 KO mice when given at very high doses [8, 9]. In contrast, in a small trial of AMN adults, 20 g of 4PBA was taken orally per day (3 × per day, or 40 pills) for approximately 3 months, and although drug was detected in CSF and brain white matter by magnetic resonance within 30 min, VLCFA remained unchanged by trial’s end (unpublished data discussed within Moser et al.) [10]. Preclinical studies and the pharmacodynamics of 4PBA point to favorable outcomes of drug use; however, complex pharmacokinetics severely limit its utility in practice and were likely the culprit of the failed clinical trial.
Advancements in nanoparticle therapeutics may offer new life to previously explored drug compounds that were deemed unsuitable due to drug dynamics. The hydroxyl polyamidoamine (PAMAM) dendrimer platform has shown significant promise for targeted intracellular delivery to disease-affected cells in the brain, from systemic administration, in many preclinical studies across six species, and in early clinical studies. These dendrimers deliver drugs across the impaired blood–brain barrier, providing efficacy at lower doses, and minimizing off-target systemic toxicity, which is especially critical for 4PBA. Dendrimer-drug conjugates improve targeted delivery across the blood–brain barrier in models of neurological conditions from traumatic brain injury to glioblastoma in rodent, rabbit, and canine models [11,12,13,14]. Here, dendrimer-conjugated 4PBA (D-4PBA) was formulated as a slow-release compound that releases drug over 1 week, thereby reducing the burden of frequent administration while enabling delivery of a steady level of drug at the site of the action.
Importantly, patients affected by AMN often do not present with any symptoms until mid-adulthood. It is, therefore, critical to determine whether any therapeutic approach can benefit individuals in the pre-symptomatic stage serving as a prophylactic to avoid future symptoms, or whether treatment later in life can still ameliorate the disease course. In this paper we describe two separate studies in which we aimed to establish and optimize parameters of D-4PBA administration in both young (7 months) and older (1 year) Abcd1− mice. The first study began early in disease pathology, representative of the pre-symptomatic patients early in disease course, and showed promise in mitigating behavioral phenotype, improving survival of spinal cord neurons, and reducing spinal cord VLCFA later in disease. The follow-up study began later in disease course with greater pathological advancement, as 1-year-old mice have been shown to have an increased accumulation of markers of oxidative stress, advancing their disease course [15, 16]. In this subset, improvements were noted in behavior at the same long-term time point; however, treatment failed to correct both histopathological outcomes and VLCFA.
Methods
D-4PBA Synthesis and Characterization
Reagents and Materials
4-Phenyl butyric acid (compound 1), tert-butyl 3-hydroxypropanoate (compound 2), N,N′-dicyclohexylcarbodiimide (DCC), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide (EDC), 4(dimethylamino)pyridine (DMAP), and trifluoroacetic acid were purchased from Sigma-Aldrich USA. All solvents (ACS grade and anhydrous) were purchased from Sigma and were used as received. Hydroxyl PAMAM dendrimer with ethylenediamine core (generation 4, 64 hydroxyl terminal groups, Pharma grade, compound 5) was purchased from Dendritech in the form of methanolic solution and the MeOH was evaporated before use. The 1000-kDa dialysis membranes for purification were purchased from Spectra/Por.
Instrumentation
The structure of intermediates and the final conjugates were characterized using proton nuclear resonance (1H NMR) spectroscopy on a 500-MHz Bruker NMR spectrometer. The chemical shifts are reported in parts per million (ppm; δ). The purity of the conjugate is analyzed using high-performance liquid chromatography (HPLC).
Synthesis Procedures
Synthesis of Compound 3
To a stirring solution of compound 1 (0.96 g, 5.84 mmol) in 15.0 mL anhydrous dichloromethane (DCM) under inert atmosphere, DMAP (285.6 mg, 2.34 mmol) was added, followed by the addition of DCC (1.327 g, 6.43 mmol). After 30 min of activation, tert-butyl 3-hydroxypropanoate (2) (1.296 mL, 8.772 mmol) diluted in 5.0 mL of anhydrous DCM was added and the reaction mixture was stirred for 12 h at room temperature. Then, the solvent was evaporated, and the crude product was purified by column chromatography using prepacked silica column and a mixture of ethyl acetate/hexane (10:90) as eluent. The pure fractions were dried under vacuo and the product was obtained as a white solid (3) (1.12 g, 81% yield).
1H NMR (500 MHz, CDCl3) δ 7.20 (dd, J = 12.3, 4.6 Hz, 2H), 7.11 (dd, J = 11.5, 7.3 Hz, 3H), 4.24 (t, J = 6.4 Hz, 2H), 2.57 (t, J = 7.6 Hz, 2H), 2.48 (t, J = 6.4 Hz, 2H), 2.25 (t, J = 7.5 Hz, 2H), 1.93–1.83 (m, 2H), 1.37 (s, 9H).
Synthesis of Compound 4
To a stirring solution of compound 3 (1.12 g) in 10.0 mL anhydrous DCM, trifluoroacetic acid (3 mL) was added at 0 °C. The reaction was stirred at 0 °C for 3 h or until completion by thin layer chromatography. Upon completion, the solvent was evaporated, and the crude was purified by column chromatography using a mixture of ethyl acetate/hexane (30:70) as eluent. The product was dried under vacuo and obtained as a white solid (4) (0.72 g, 89% yield).
1H NMR (500 MHz, CDCl3) δ 7.36–7.25 (m, 2H), 7.21 (dd, J = 13.8, 7.2 Hz, 3H), 4.37 (t, J = 6.2 Hz, 2H), 2.69 (dt, J = 15.1, 6.9 Hz, 4H), 2.35 (t, J = 7.4 Hz, 2H), 1.97 (p, J = 7.5 Hz, 2H).
Synthesis of Compound 6
To a stirring solution of compound 4 (694.68 mg, 2.94 mmol) in anhydrous dimethylformamide (DMF) (15 mL), DMAP (358.7 mg, 2.94 mmol) and EDC (842.3 mg, 4.41 mmol) were added. After stirring the reaction mixture at 0 °C for 30 min, G4-OH PAMAM dendrimer (2.1 g, 0.147 mmol) dissolved in 10.0 mL anhydrous DMF was added, and the reaction was left to stir for 48 h at room temperature. Then the reaction mixture was diluted with DMF (20 mL) and dialyzed against DMF to remove by-products and excess reactants, followed by H2O dialysis to remove DMF. Finally, the purified product was lyophilized and obtained as a white powder (2.5 g).
1H NMR (500 MHz, dimethyl sulfoxide (DMSO)) δ 8.51–7.62 (m, D-internal amide H), 7.22 (d, 4PBA H), 7.02 (m, 4PBA H), 4.76 (bs, D-OH), 4.42 (m, linker H), 4.21 (m, linker H), 4.01 (m, linker H), 3.52–2.24 (m, dendrimer − CH2 and 4PBA H), and 1.80 (m, linker H).
HPLC: purity: 98.5%; retention time: 25.6 min.
In Vitro Drug Release
In vitro drug release was carried out at intracellular conditions using citrate buffer (pH 5.5) and esterase. The conjugate was dissolved in the buffer at 1 mg/mL and 10 µL esterase was added. The experiment was done in duplicate. The tubes were incubated at physiological temperature with constant shaking. At various time points, the aliquots were taken from each tube, quenched with methanol, and stored at − 80 °C until analysis. The analysis was carried out using HPLC. A standard curve for free drug was made for various concentrations using HPLC and the amount of free drug in the samples was analyzed using standard curve by converting the area under the curve to the amount of drug.
Animals
Breeding and Care
Wild-type (WT; Abcd1+) and knockout (KO; Abcd1−) mice were bred in house with ad libitum access to food and water. Because ALD is an X-linked disorder primarily affecting males, only male mice were used in the described studies. All animal procedures were approved by the Institutional Animal Care and Use Committee of Johns Hopkins University and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Biodistribution
Localization of dendrimer within WT and KO spinal cord was determined using dendrimer conjugated to Cy5 (D-Cy5; 50 mg/kg) delivered via intraperitoneal (i.p.) injection in 8-month-old male mice, 24 h prior to tissue harvest as described below.
Dose Response
Dose efficacy of D-4PBA was determined by C26:0 lysophosphatidylcholine (LPC) levels after 8 weeks of treatment. As VLCFA are elevated at birth in Abcd1− mice, mice between the ages of 6–8 months were randomly assigned to dose response treatment groups. Once per week, mice were i.p. injected with vehicle (2.5% DMSO in phosphate-buffered saline (PBS)), 50, 100, or 200 mg/kg D-4PBA in 200 μL. One day following the final administration mice were saline perfused, and spinal cords were removed and sectioned for VLCFA LPC content as described below (the “Analysis of VLCFA” section).
Study 1 (7-Month-Old Mice)
Mice were randomly assigned to treatment groups at 7 months of age with n = 7 WT receiving vehicle, n = 6 Abcd1 KO receiving vehicle (Abcd1 KO Veh), and n = 6 Abcd1 KO receiving D-4PBA (Abcd1 KO D-4PBA). Mice were injected i.p. with vehicle or 50 mg/kg D-4PBA in 200 μL vehicle solution every other week until 16 months of age. At 16 months old and without behavioral improvement, the dose and frequency were both doubled, and mice were given 100 mg/kg D-4PBA weekly for 3 months until perfusion at 19 months of age.
Study 2 (1-Year-Old Mice)
Mice were randomly assigned to treatment groups at 12 months of age with n = 9 WT receiving vehicle, n = 7 KO Abcd1 Veh, and n = 7 Abcd1 KO D-4PBA. Mice were injected i.p. with vehicle or 100 mg/kg D-4PBA in 200 μL once weekly for 7 months beginning at 12 months of age and continuing until 19 months of age.
Behavior
For Studies 1 and 2, motor ability was assayed by accelerating Rotor Rod (San Diego Instruments) testing at baseline, then monthly for the duration of treatment. Mice ran three trials separated by at least 10 min in which latency to fall and distance travelled were averaged across trials. In case of fall prior to 20 s, the trial was excluded, and mice were given up to two additional trials. Open-field (San Diego Instruments; 45 × 45 cm) activity over a 10-min period was also assessed monthly and was analyzed using the ANY-maze system. After animals were randomly assigned to groups, drug dosing was initiated immediately following baseline testing. For weeks in which behavior coincided with dosing, behavior preceded drug administration.
Histology and Imaging
Study 1 and 2 mice were euthanized at 19 months of age with chloral hydrate and transcardially perfused with saline. Immediately following full perfusion, the spinal cord was extracted and segmented into cervical, thoracic, and lumbar regions. Each region was halved, and one half of each region was frozen fresh and the other post-fixed for 24 h in ice-cold 10% formalin. Brains were halved sagittally and similarly fixed or frozen stored.
Formalin fixed tissues were cryoprotected with a 30% sucrose gradient before embedding in OCT (Fisher Scientific) and stored at −80 °C prior to cryostat sectioning at 20 μm in a 10-series. For immunostaining, sectioned tissue was blocked (10% normal serum, 0.3% Triton-X in PBS) for 1 h at room temperature prior to overnight incubation with Iba1 (1:1000 in PBS; DAKO, Agilent, Santa Clara, CA), Cd68-AlexaFluor 647 (1:500 in PBS; Santa Cruz Biotechnology, Inc., Dallas, TX), or Smi32 primaries (1:1000; BioLegend; San Diego, CA) at 4 °C. On day 2, secondary incubation for Iba1 or Smi32 lasted 1 h at room temperature using goat antirabbit AlexaFluor 488 secondary. Sections were rinsed, incubated with Hoescht dye (1:3000) for 3 min, rinsed, and coverslipped.
For each mouse, complete spinal cord mosaics were imaged at 20 × using Leica DMi8 Thunder Imager. The total area of Iba1 and the total number of Smi32+ neurons were manually counted by two researchers blinded to groups using FIJI (Image J 1.53c). Additionally, we assessed the ER chaperone Grp78/BiP as an indicator of ER stress [17].
Analysis of VLCFA
For all mice (Studies 1 and 2, dose response) fresh frozen lumbar spinal cord segments were homogenized in 10% w/v PBS and the total lipids were extracted by the method of Folch et al. [18]. Aliquots of the total lipid extract were taken for the measurement of C26:0 LPC by liquid-chromatography tandem mass spectrometry (LC–MS/MS by AB Sciex QTRAP 4500) and the total lipid fatty acids were measured by gas-chromatography mass spectrometry (GC–MS) [19, 20]. Final C26:0-LPC and total lipid C26:0 concentrations were normalized to total protein.
RNAScope
To assess efficacy of D-4PBA to induce changes in Abcd2 expression, RNAscope® in situ hybridization was used to quantify and localize Abcd2 mRNA within mouse spinal cord tissues. RNA transcripts were visualized by RNAscope per manufacturer’s instructions, and samples were costained with Smi32 as described above, but were incubated 72 h in primary antibody. Sections were imaged using a Leica SP8 confocal microscope. Puncta were counted using FIJI ImageJ by analyzed particle, and were subtracted from the Smi32 channel to quantify only transcripts present within Smi32-expressing neurons.
Data Analysis
Mouse behavioral endpoints were averaged by group and analyzed by mixed model repeated measures Analysis of Variance (ANOVA) using group and time as factors. Histology data, LPC, and RNAscope data were similarly averaged by group and assessed by one-way ANOVA. Quantification was done by FIJI ImageJ 1.53c and data were analyzed by GraphPad Prism 8. Data are represented as boxplots showing the mean and 5–95 percentile; significance is reached when p ≤ 0.05.
Results
Dendrimer-4PBA (D-4PBA) Synthesis, Characterization, and Drug Release
The synthesis of D-4PBA began with the modification of 4PBA to attach a linker with a carboxylic acid end group. The purpose of linker attachment was to extend the distance between the conjugated drug and the dendrimer surface, enabling drug release. We have previously observed that the drug release from the dendrimer depends upon the linker length and if the drug is too close to the dendrimer surface, the drug release is hindered [21]. To facilitate optimal drug release, a 3-carbon long linker was used. The synthesis of 4PBA linker was achieved by reacting 4PBA (1) with tert-butyl 3-hydroxypropanoate (2) in the presence of coupling agents to afford a tert-butyl protected 4PBA linker (3) that was deprotected under mild acidic conditions to afford 4PBA linker (4; Fig. 1A). The 4PBA linker was further reacted with generation 5 hydroxyl PAMAM dendrimer (5) using esterification conditions to produce D-4PBA conjugate (6). The structure of conjugate was evaluated using 1H NMR (Fig. 1B), which showed the peaks corresponding to both 4PBA and dendrimer protons. The loading of 4PBA was calculated using a proton integration method comparing the internal amide protons of dendrimer to the 4PBA protons in the aromatic region of the NMR spectrum. Each conjugate has ~14 molecules of 4PBA attached with a loading of ~14 wt%. We have previously published that drug loading of less than 20wt% does not affect dendrimer’s brain penetration or microglia/macrophage targeting ability [21,22,23]. The HPLC purity of this dendrimer conjugate was >98% (Fig. 1C). We also evaluated the drug release and conjugate stability under conditions which mimic the extracellular environment of pH 7.4 and 37 °C. The D-4PBA conjugate is very stable and showed minimum release (<1%) in 48 h at pH 7.4. We further evaluated the in vitro drug release of 4PBA from the dendrimer conjugate at intracellular conditions, mimicking the endosomal environment (pH 5.5 in the presence of esterase at 37 °C) [24], which revealed a fast release of 4PBA from the dendrimer with ~50% drug release in first 5 h and more than 80% release over a period of 1 week (Fig. 1D). Even though part of the drug is released relatively fast under intracellular conditions, we hypothesize that some of the hydrophobic PBA drug molecules may fold interior to the dendrimer which is hydrophobic, enabling a sustained release of the remaining drug. HPLC data of D-4PBA and its starting materials are shown in Supplemental Fig. 1.

Synthesis and characterization of D-4PBA. A Schematic representation of step-wise synthesis of D-4PBA conjugate. B 1H NMR characterization of D-4PBA representing characteristic protons from dendrimer and 4-PBA. C HPLC profile of D-4PBA conjugate demonstrating a purity > 98%. D In vitro drug release profile of conjugate to mimic intracellular and extracellular pH
Biodistribution
Qualitative analysis of Cy5 signal within the ventral horn of Abcd1 KO and WT mouse spinal cord indicates uptake of dendrimer within target cells (Fig. 2). Uptake within KO Smi32+ neurons and Iba1+ microglia were visibly greater, compared to uptake observed within WT tissues.

Biodistribution of Cy5-labeled dendrimer (D-Cy5; red) delivered to 8-month-old WT and KO male mice. Ventral horn uptake is shown in both neurons (Smi32, green; arrow heads) and microglial cells (Iba1, white; arrows). D-Cy5 (50 mg/kg; i.p.) uptake is shown after 24 h. Scale bar represents 20 μm
D-4PBA Dose Response
To pinpoint the optimal dose for long-term therapy, Abcd1 KO mice were administered 0, 50, 100, or 200 mg/kg D-4PBA weekly for 8 weeks with 5 mice per group. After the short-term treatment, VLCFA analysis showed increasing doses of D-4PBA resulted in a concomitant decrease in C26:0 LPC (Fig. 3A). Both 100 and 200 mg/kg showed decreased C26:0 LPC compared to untreated mice and showed similarities in maintaining body mass; therefore, we chose 100 mg/kg, the lowest dose showing early efficacy (one-way ANOVA, p = 0.39).

D-4PBA dose response. D-4PBA (0 n = 3, 50 n = 5, 100 n = 5, or 200 mg/kg n = 3) was delivered weekly to Abcd1 KO mice for 8 weeks. A Analysis of spinal cord VLCFA LPC reveals a dose-dependent decrease in C26:0 after 8 weeks and B no significant change to body weight during the treatment period. For all, error bars indicate standard error of the mean
Study 1
D-4PBA treatment was started at 7 months of age, and Abcd1 KO mice were monitored for overall health and weight at each drug administration and during monthly behavior. After 9 months of administration with 50 mg/kg every other week and a failure to observe a behavioral change, the dose was increased to 100 mg/kg per week. Behavior improved thereafter until 1 year of drug dosing when the study was ended (mixed model ANOVA, main effect of time, p = 0.055; main effect of group, p = 0.057; post hoc Abcd1 KO Veh vs Abcd1 KO D4PBA at time 12, p = 0.028; Fig. 4A). Proportional target area (PTA) of Iba1-expressing microglia in the gray matter of spinal cord was increased in Abcd1 KO mice, though not significantly different from WT mice, and unchanged compared to D-4PBA treated mice (data not shown).

Study 1 behavior and histology. In parallel to WT and Abcd1− vehicle-treated mice, Abcd1 KO mice were treated with 50 mg/kg D-4PBA biweekly starting at 7 months of age. A Latency (s) to fall from the Rotor Rod was measured monthly. After nearly 9 months of dosing, D-4PBA was increased to 100 mg/kg per month. Mice were dosed for 12 full months. B Representative images of Smi32-stained neurons within the ventral horn of spinal cord sections (scale bar represents 50 μm). C The number of Smi32.+ neurons within the spinal cord gray matter were used as an index of overall neuronal and axonal survival. Although not significant, D-4PBA-treated mice showed an increase in the number of neurons detected. D Quantification of ER stress marker BiP within spinal cord gray matter. E C26:0 LPC was elevated as expected in Abcd1 KO vehicle-treated mice (p = 0.0030) and treatment with D-4PBA significantly reduced VLCFA accumulation within the spinal cord (p = 0.0245). *p ≤ 0.05. At study’s end, WT n = 7, Abcd1 KO n = 6, and Abcd1 D-4PBA n = 6
Smi32, which labels the non-phosphorylated epitope of neurofilament proteins, identified neurons in the spinal cord. Smi32-positive cells within Abcd1 KO vehicle-treated mice were numerically decreased in the gray matter compared to WT, while treatment with D-4PBA increased counts (Fig. 4B, C). BiP, an ER chaperone and regulator of ER stress, which is induced in response to ER stress, showed a trend towards increased levels in Abcd1 KO vehicle-treated mice, and a slight reduction in KO mice treated with D-4PBA with early treatment (Fig. 4D). However, both Smi32 expression and BiP induction did not reach significance with these numbers.
C26:0 LPC analysis by LC–MS/MS showed a significant increase of VLCFA in Abcd1 KO vehicle-treated mice compared to WT (F(2,13) = 9.461, p = 0.0029; WT vs Abcd1 KO Veh, p = 0.0030), and a significant reduction of D-4PBA-treated mice compared to Abcd1 KO vehicle-treated (KO Veh vs KO + D-4PBA, p = 0.0245; Fig. 4E). Total lipid C26:0 findings share a similar pattern of change and are shown in Table 1.
Study 2
In order to determine whether a beneficial effect is still present in older animals, and building from the previous study where we observed improvements at 100 mg/kg weekly, we tried a slightly delayed treatment paradigm starting therapy at 1 year of age. Abcd1 KO mice began weekly administration of 100 mg/kg D-4PBA, until 19 months of age. Treatment began immediately following baseline behavioral testing and behavior showed rapid improvement compared to untreated Abcd1 KO mice (mixed model ANOVA main effect of group, p = 0.048; Fig. 5A).

Study 2 behavior and histology. In parallel to WT and Abcd1− vehicle-treated mice, Abcd1 KO mice were treated with 100 mg/kg D-4PBA weekly starting at 1 year of age. A Latency (s) to fall from the Rotor Rod was measured monthly and mice showed a marked improvement in latency shortly after treatment initiation. B Representative images of Smi32-stained neurons within the ventral horn of the spinal cord (scale bar represents 50 μm). C Smi32.+ neurons were decreased within Abcd1 KO mice with no significant effect of D-4PBA treatment. D Quantification of ER stress marker BiP within spinal cord gray matter. E C26:0 LPC was again elevated as expected in Abcd1 KO vehicle-treated mice (p = 0.0002); however, in this study, treatment with D-4PBA had no effect. At study’s end, WT n = 9, Abcd1 KO n = 7, and Abcd1 D-4PBA n = 7
Similar to Study 1, PTA of Iba1-expressing cells within the gray matter of spinal cord sections was unchanged (data not shown). Although Abcd1 KO mice showed decreased numbers of Smi32+ neurons which was slightly elevated by treatment with D-4PBA, these findings failed to reach significance (Fig. 5B, C). Unlike in Study 1, levels of the ER stress marker, BiP, were unchanged in Abcd1 KO mice, compared to WT or D-4PBA-treated mice (Fig. 5D). Again, C26:0 LPC was elevated in Abcd1 KO mice compared to WT (F(2,17) = 20.56, p < 0.0001; WT vs Abcd1 KO Veh, p = 0.0002); however, in contrast with Study 1, spinal cord C26:0 from D-4PBA-treated mice was unchanged compared to Abcd1 vehicle-treated mice, perhaps owing to the advanced age at the start of treatment, the shortened duration of therapy, or that VLCFA alone is not predictive of behavioral outcome (Fig. 5D). These findings were again reflected in total lipid C26:0 and are shown in Table 1.
RNAScope
An in situ hybridization approach was used to quantify changes in Abcd2 gene expression as a result of D-4PBA treatment. Tissue sections from mouse spinal cord were hybridized with probes targeting 1 kb of Abcd2 transcripts and quantified. Although counts across both studies show decreased Abcd2 expression in Abcd1 KO Veh compared to WT mice and a correction by D-4PBA, findings only reached significance in Study 2 (main effect: F(2,13) = 3.846, p = 0.0487 and WT vs Abcd1 KO Veh, p = 0.0394; Fig. 6A, B), indicating that a higher dose may be required for upregulating Abcd2 expression.

RNAScope was used to quantify and localize gene expression of Abcd2 within mouse spinal cord. Abcd2 transcripts as quantified by the number of red-labeled puncta in Study 1 (A) and Study 2 (B) were decreased in Abcd1 KO mice compared to WT, and treatment with D-4PBA increased Abcd2 across both studies reaching significance in Study 2 (main effect, p = 0.0487). C Representative images of Smi32 staining and Abcd2 transcripts are shown; scale bar represents 50 μm
Discussion
AMN remains a progressively debilitating disorder with no treatment options. While newborn screening for ALD has recently been implemented across several states within the USA resulting in the identification of many new families with multiple affected members across multiple generations, several challenges remain in predicting phenotypic progression in these patients. Loss of ABCD1 transporter function in ALD can be partly rescued through activation of ABCD2; however, the translational potential of the ABCD2-activator 4PBA has previously been limited by high doses necessary to counteract the rapid clearance of the drug [8, 9]. Nanoparticle systems, such as PAMAM dendrimer, not only allow for trafficking across the disease-affected blood–brain barrier and intracellular delivery, but also extend bioavailability of drugs otherwise rapidly cleared from the system [25,26,27]. Here, dendrimer conjugated to 4PBA showed dose-dependent effects on VLCFA within the spinal cord of treated Abcd1 KO mice, slightly improved histopathological findings within spinal cord neurons, and improved neurobehavior following long-term treatment. Compared to previous studies testing 4PBA as a therapy in the ALD mouse, dendrimer-enabled delivery significantly enhances the efficiency of drug delivery, and data presented here reflect the use of a 140-fold reduced dose compared to previous mouse studies [8]. Importantly, this efficacy at a lower dose due to improved intracellular delivery as demonstrated in other dendrimer-drug conjugates, and the relatively rapid elimination of dendrimer-drug conjugates from off-target organs through the kidney (mostly intact) in 24–48 h, significantly reduce the side effects of free 4-PBA [13]. While a weakness of our study is that a lack of direct comparison of effectiveness of D-4PBA to “free,” or unconjugated, 4PBA, we believe that such direct comparison would be impossible since free 4PBA therapy would require high doses administered orally through chow while D-4PBA is injected weekly. In addition, the objective of this study was not to reproduce old results of free 4PBA since a pilot human trial of 4PBA failed to decrease plasma VLCFA, but rather to determine if this novel formulation can in fact be effective in ameliorating the neurobehavioral and pathological phenotypes in the disease model.
The Abcd1 KO mouse harbors biochemical abnormalities in line with ALD patients, including early elevations of VLCFA, and reduced β-oxidation. In patients, increased exposure to VLCFA in cerebral ALD patient-derived macrophages increases the release of pro-inflammatory cytokines while severely depleting the potent antioxidant, glutathione [28], and these markers of oxidative damage have been detected within demyelinating lesions of ALD patient brain tissue [29]. While the major inflammatory components of the disease, and the role of infiltrative macrophages in the progression of myelin pathology in late disease, contribute greatly to the pathogenesis of AMN and certainly cerebral ALD (cALD), the Abcd1 KO mouse fails to develop this severe inflammatory cerebral disease. Still, ALD mice share many other hallmarks of the disease, including neuromotor impairment in late adulthood representing the dying back axonopathy of the long spinal cord tracts and thus serve mainly as an appropriate model of AMN [30]. The dying back axonopathy characteristic to ALD results from a multitude of metabolic changes including changes to complex lipid networks observed across all ALD phenotypes. These complex lipid networks, namely, VLCFA-induced destabilization of membranes, disrupt homeostasis and activate the unfolded protein response (UPR) as well as endoplasmic reticulum (ER) stress. Multiple groups have identified ER stress as directly correlating to VLCFA, and even fatty acid chain length, within ALD patient fibroblasts [31, 32] and mitigating this stress using tauroursodeoxycholic acid was shown to halt axonal degeneration and improve locomotion in the Abcd1−/Abcd2−/− knockout mouse [33]. Further studies identify VLCFA as playing a role in protein quality and membrane homeostasis, with dysregulated membrane composition being a trigger of protein misfolding [34]. Interestingly, ER stress and inflammation are intricately linked, as prolonged ER stress activates inflammatory cell death pathways, predominately mediated through nuclear factor kappa-light chain enhancer of activated B cells (NFκB), mitogen-activated protein kinase (MAPK), and toll-like receptor signaling [35, 36]. In our study, long-term D-4PBA therapy, when started early, showed increased BiP in Abcd1 KO mice compared to WT, a nominal reversal of BiP levels following D-4PBA treatment, and significant reduction in VLCFA. While small, control of these VLCFA mediated processes may bring some relief to ALD patients and potentially stave off accumulation of inflammatory factors that exacerbate and potentiate severity of disease later in its course.
In addition to being a known activator of ABCD2, 4PBA promotes protein folding thereby repressing the UPR mediated through the ER. Studies in a mouse model of cerebral ischemia also show that pretreatment with 4PBA reduces ER stress and linked pro-inflammatory cytokines such as tumor necrosis factor (Tnf), interleukin (Il)-6, and Il-1β [36]. We observed increases in Abcd2 when treatment was started at the higher dose and delayed; however, there were no changes to VLCFA and minimal detection of ER stress, suggesting a complex underlying interaction of these systems that is not dependent on VLCFA, and requires a careful treatment strategy. Furthermore, long-term treatment beginning at 7, but not 12, months of age offered partial protection against behavioral deficits and neuronal loss. In contrast, López-Eruaskin et al. report that high-dose and prolonged antioxidant treatment in the Abcd1− mouse beginning at 12 or 16 months of age was sufficient to reduce both oxidative damage and axonal degeneration [37]. These findings of early efficacy targeting oxidative damage and inflammation align with recent work by Bergner et al., who showed in autopsy samples from cALD patients that acute axonal injury early in lesion development occurs independently of inflammatory infiltration; however, later disease progression may be accompanied, and exacerbated, by activated macrophages/microglia [38]. These findings may suggest that long-term D-4PBA treatment at 7 months of age is an appropriate window for intervention of ER stress-related pathways in the ALD KO mouse, yet due to later activation of inflammatory pathways this same strategy via D-4PBA alone is insufficient to interrupt pathology by 12 months of age. While the timing of the late treatment may be the underlying reason for this lack of response, it is certainly possible that an even longer trial, higher dose trial, or a combination of 4PBA with antioxidants could still be beneficial in older mice. Future studies will be needed to address these questions.
Studies described here beg the comparison to trials testing 4PBA in adult men with AMN. In the small aforementioned trial of seven patients in which 20 g of 4PBA was administered daily for 6 weeks, followed by a 2-week break and additional 6 weeks of treatment (unpublished study discussed within Moser et al. [10]), treatment was ineffective in altering any measure of the VLCFA, C26:0. We highlight differences which would suggest significant improvements in the dendrimer-conjugated version compared to free drug alone: (1) prolonged bioavailability following conjugation allowing for one dose per week — a 20-fold reduction in dose for a 70-kg adult and (2) targeted delivery, uptake, and washout of excess compound. Importantly, while intraperitoneal administration is appropriate for the study of drug effects in rodent models, patients would receive such compounds via other routes [39]. The safety and tolerability of both intravenously and subcutaneously delivered dendrimer compounds have recently been demonstrated by a lead compound (dendrimer-NAC, OP-101) in clinical trials for severe COVID-19 (NCT04458298) in which survival benefits including reduced blood biomarkers of neurological injury were observed [40].
Together, these preliminary studies show promise for the use of the hydroxyl PAMAM dendrimer platform for targeted delivery of 4PBA where targeting and bioavailability of the compound can be specified and preserved, respectively. Certainly, a weakness to this study is the variability of several histopathological endpoints within the ALD mouse. Deficits in the Abcd1− mouse are subtle and indeed develop very late in life, making the detection of robust effects challenging. Other metrics, such as VLCFA quantification and functional behavior, showed more sensitivity to the effects of D-4PBA and lay the foundation for future work using D-4PBA. Our findings demonstrate that increasing Abcd2 expression offered only partial neuromotor recovery and reduction of ER stress concurrent with a significant reduction of VLCFA, in mice treated at a younger age, despite a failure to reduce this biochemical abnormality when older mice were treated. This caveat may be addressed through the use of a combinatorial antioxidant/4PBA therapy on the dendrimer platform, and we have previously shown that dendrimer conjugated to the potent antioxidant N-acetylcysteine reverses oxidative stress and the pro-inflammatory cytokine profile of AMN and cALD patient macrophages [28]. Using this approach, and considering findings reported here, such therapies may require enrollment of younger AMN patients who are in early disease stage.
References
Aubourg P, Blanche S, Jambaque I, Rocchiccioli F, Kalifa G, Naud-Saudreau C, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322:1860–6.
Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377:1630–8.
Mallack EJ, Turk B, Yan H, Eichler FS. The landscape of hematopoietic stem cell transplant and gene therapy for X-linked adrenoleukodystrophy. Curr Treat Options Neurol. 2019;21.
Tawbeh A, Gondcaille C, Trompier D, Savary S. Peroxisomal abc transporters: an update. Int J Mol Sci. 2021;22.
Fourcade S, Ruiz M, Camps C, Schlüter A, Houten SM, Mooyer PAW, et al. A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am J Physiol - Endocrinol Metab. 2009;296:211–21.
Kawaguchi K, Morita M. ABC Transporter Subfamily D: Distinct differences in behavior between ABCD1–3 and ABCD4 in subcellular localization, function, and human disease. Biomed Res Int. 2016;2016.
Hartley MD, Kirkemo LL, Banerji T, Scanlan TS. A thyroid hormone-based strategy for correcting the biochemical abnormality in X-linked adrenoleukodystrophy. Endocrinology. 2017;158:1328–38.
Kemp S, Wei H, Lu J, Braiterman L, McGuinness M, Moser A, et al. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med. 1998;4:1261–8.
Gondcaille C, Depreter M, Fourcade S, Lecca MR, Leclercq S, Martin PGP, et al. Phenylbutyrate up-regulates the adrenoleukodystrophy-related as a nonclassical peroxisome proliferator. J Cell Bio. 2005;169:93–104.
Moser HW, Smith KD, Watkins PA, Powers J, Moser AB. X-linked adrenoleukodystrophy. Metab Mol Bases Inherit Dis. 2002: 3257–301.
Kannan S, Dai H, Navath RS, Balakrishnan B, Jyoti A, Janisse J, et al. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci Transl Med. 2012;4:130ra46.
Mishra MK, Beaty CA, Lesniak WG, Kambhampati SP, Zhang F, Wilson MA, et al. Dendrimer brain uptake and targeted therapy for brain injury in a large animal model of hypothermic circulatory arrest. ACS Nano. 2014;8:2134–47.
Liaw K, Zhang F, Mangraviti A, Kannan S, Tyler B, Kannan RM. Dendrimer size effects on the selective brain tumor targeting in orthotopic tumor models upon systemic administration. Bioeng Transl Med. 2020;5:1–12.
Sharma R, Kambhampati SP, Zhang Z, Sharma A, Chen S, Duh EI, et al. Dendrimer mediated targeted delivery of sinomenine for the treatment of acute neuroinflammation in traumatic brain injury. J Control Release. 2020;323:361–75.
Galea E, Launay N, Portero-Otin M, Ruiz M, Pamplona R, Aubourg P, et al. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim Biophys Acta - Mol Basis Dis. 2012;1822:1475–88.
Fourcade S, Ferrer I, Pujol A. Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: a paradigm for axonal degeneration. Free Radic Biol Med. 2015;88:18–29.
Ibrahim IM, Abdelmalek DH, Elfiky AA. GRP78: A cell’s response to stress. Life Sci. 2019;226:156–63.
Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509.
Hubbard WC, Moser AB, Liu AC, Jones RO, Steinberg SJ, Lorey F, et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab. 2009;97:212–20.
Lagerstedt SA, Hinrichs DR, Batt SM, Magera MJ, Rinaldo P, McConnell JP. Quantitative determination of plasma C8–C26 total fatty acids for the biochemical diagnosis of nutritional and metabolic disorders. Mol Genet Metab. 2001;73:38–45.
Sharma R, Kim S-Y, Sharma A, Zhang Z, Kambhampati SP, Kannan S, et al. Activated microglia targeting dendrimer-minocycline conjugate as therapeutics for neuroinflammation. Bioconjug Chem. 2017;28:2874–86.
Sharma A, Liaw K, Sharma R, Spriggs T, Appiani La Rosa S, Kannan S, et al. Dendrimer-mediated targeted delivery of rapamycin to tumor-associated macrophages improves systemic treatment of glioblastoma. Biomacromol. 2020;21:5148–61.
Sharma A, Liaw K, Sharma R, Thomas AG, Slusher BS, Kannan S, et al. Targeting mitochondria in tumor-associated macrophages using a dendrimer-conjugated TSPO ligand that stimulates antitumor signaling in glioblastoma. Biomacromol. 2020;21:3909–22.
Perumal OP, Inapagolla R, Kannan S, Kannan RM. The effect of surface functionality on cellular trafficking of dendrimers. Biomaterials. 2008;29:3469–76.
Sharma R, Sharma A, Kambhampati S, Reddy R, Zhang Z, Cleland J, et al. Scalable synthesis and validation of PAMAM dendrimer-N-acetyl cysteine conjugate for potential translation. Bioeng Transl Med. 2018;3:87–101.
Sharma A, Liaw K, Sharma R, Zhang Z, Kannan S, Kannan RM. Targeting mitochondrial dysfunction and oxidative stress in activated microglia using dendrimer-based therapeutics. Theranostics. 2018;8:5529–47.
Zhang F, Lin YA, Kannan S, Kannan RM. Targeting specific cells in the brain with nanomedicines for CNS therapies. J Control Release. 2015;240:212–26.
Turk BR, Nemeth CL, Marx JS, Tiffany C, Jones R, Theisen B, et al. Dendrimer-N-acetyl-L-cysteine modulates monophagocytic response in adrenoleukodystrophy. Ann Neurol. 2018;84:452–62.
Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW, et al. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol. 2005;64:1067–79.
Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet. 2002;11:499–505.
Raas Q, van de Beek M-C, Forss-Petter S, Dijkstra IME, Deschiffart A, Freshner BC, et al. Metabolic rerouting via SCD1 induction impacts X-linked adrenoleukodystrophy. J Clin Invest. 2021;131.
van de Beek M-C, Ofman R, Dijkstra I, Wijburg F, Engelen M, Wanders R, et al. Lipid-induced endoplasmic reticulum stress in X-linked adrenoleukodystrophy. Mol Basis Dis. 2017;2255–65.
Launay N, Ruiz M, Grau L, Ortega FJ, Ilieva EV, Martínez JJ, et al. Tauroursodeoxycholic bile acid arrests axonal degeneration by inhibiting the unfolded protein response in X-linked adrenoleukodystrophy. Acta Neuropathol. 2017;133:283–301.
Micoogullari Y, Basu SS, Ang J, Weisshaar N, Schmitt ND, Abdelmoula WM, et al. Dysregulation of very-long-chain fatty acid metabolism causes membrane saturation and induction of the unfolded protein response. Mol Biol Cell. 2020;31:7–17.
Han Y, Yuan M, Guo Y-S, Shen X-Y, Gao Z-K, Bi X. Mechanism of endoplasmic reticulum stress in cerebral ischemia. Front Cell Neurosci. 2021;15.
Dong L, Tan CW, Feng PJ, Liu FB, Liu DX, Zhou JJ, et al. Activation of TREM-1 induces endoplasmic reticulum stress through IRE-1α/XBP-1s pathway in murine macrophages. Mol Immunol Elsevier Ltd. 2021;135:294–303.
López-Erauskin J, Fourcade S, Galino J, Ruiz M, Schlüter A, Naudi A, et al. Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann Neurol. 2011;70:84–92.
Bergner CG, Genc N, Hametner S, Franz J, Van Der Meer F, Mitkovski M, et al. Concurrent axon and myelin destruction differentiates X-linked adrenoleukodystrophy from multiple sclerosis. Glia. 2021;69:2362–77.
Al Shoyaib A, Archie SR, Karamyan VT. Intraperitoneal route of drug administration: should it be used in experimental animal studies? Pharm Res. 2020;37.
Gusdon A, Faraday N, Aita J, Kumar S, Mehta I, Choi H, et al. Phase2a trial of dendrimer nanotherapy attenuates inflammation, neurologic injury markers and improves outcomes in severe COVID-19. Sci Transl Med. 2022.
Acknowledgements
The authors would like to thank Ann Snowden and Richard Jones, PhD for their assistance with LPC analysis and interpretation. Furthermore, we thank George McNamara PhD and the Johns Hopkins University School of Medicine Department of Anesthesiology and Critical Care Medicine for the access to their Leica SP8 confocal microscope.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This research was funded by the Kennedy Krieger Institute Intellectual and Developmental Disabilities Research Center (NIH P50 HD103538), and through the thoughtful donations of patients and families through The Brian’s Hope Foundation.
Author information
Authors and Affiliations
Contributions
Study conceptualization, A. F., R. M. K., and S. K.; methodology, S. N. T., C. L. N., A. B. M., O. G., and A. S.; formal analysis, C. L. N., O. G., and A. S.; writing—original draft preparation, C. L. N. and A. S.; writing—review and editing, C. L. N., A. S., A. B. M., S. K., R. M. K., and A. F.; visualization, C. L. N., O. G., and A. S.; supervision, C. L. N., R. M. K., and A. F.; project administration, A. F.; funding acquisition, R. M. K., S. K., and A. F. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Under license agreements between Ashvattha Therapeutics Inc. and the Johns Hopkins University relating to the dendrimer platform, A. Fatemi, S. Kannan, and R. M. Kannan, the Kennedy Krieger Institute and the Johns Hopkins University are entitled to royalty distributions related to technology involved in the study discussed in this publication. S. Kannan (co-founder) and Rangaramanujam Kannan (co-founder) are co-inventors on patents relating to the platform, and hold equity in Ashvattha Therapeutics Inc., and Rangaramanujam Kannan serves on the Board of Directors. This arrangement has been reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nemeth, C.L., Gӧk, Ö., Tomlinson, S.N. et al. Targeted Brain Delivery of Dendrimer-4-Phenylbutyrate Ameliorates Neurological Deficits in a Long-Term ABCD1-Deficient Mouse Model of X-Linked Adrenoleukodystrophy. Neurotherapeutics 20, 272–283 (2023). https://doi.org/10.1007/s13311-022-01311-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01311-x