
Abstract
The nuclear pore complex (NPC) is a large multimeric structure that is interspersed throughout the membrane of the nucleus and consists of at least 33 protein components. Individual components cooperate within the nuclear pore to facilitate selective passage of materials between the nucleus and cytoplasm while simultaneously performing pore-independent roles throughout the cell. NPC dysfunction is a hallmark of neurodegenerative disorders including Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (ALS). NPC components can become mislocalized or altered in expression in neurodegeneration. These alterations in NPC structure are often detrimental to the neuronal function and ultimately lead to neuronal loss. This review highlights the importance of nucleocytoplasmic transport and NPC integrity and how dysfunction of such may contribute to neurodegeneration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
One of the main distinctions between eukaryotic and prokaryotic cells is that the former can compartmentalize materials and biological processes within membrane-bound regions known as organelles. Perhaps the most well studied of these compartments is the nucleus, a double-membrane-bound organelle that contains the genetic material of the cell. One of the main functions of the nuclear envelope, the boundary between the nucleus and the cytoplasm, is to maintain separation between DNA and cytoplasmic processes. This separation allows for proper maturation of newly transcribed RNA prior to its translation [1], as well as protection of DNA from cytoplasmic damaging agents [2].
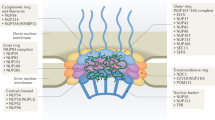
Though strict compartmentalization must be maintained, material transfer between the nucleus and cytoplasm is essential. Newly transcribed RNA (ribosomal, transfer, messenger, and non-coding) must be granted passage out of the nucleus, while nucleotides and newly translated nuclear proteins need to enter. Nuclear pores serve as numerous interspersed regulated tunnels between the nucleus and cytoplasm. Each nuclear pore is maintained by a nuclear pore complex (NPC), a massive structure composed of 33 known protein components, called nucleoporins (Fig. 1A), most of which are present in octagonal symmetry around the pore [3]. In aggregate, the total number of nucleoporins that compose the NPC is over 400, making each pore one of the largest protein complexes in the cell [3]. Notably, the half-lives of some nucleoporins are measured in years, an intriguing characteristic when one examines the biological basis of late onset human diseases [4]. This structure, as a whole or on a subunit basis, is responsible for the selective passage of materials in and out of the nucleus [5, 6].

The nuclear pore complex (NPC) is a massive structure comprised of 33 known protein components organized into five main structural domains–the cytoplasmic filaments (yellow), the inner and outer nuclear rings (green), the transmembrane components (purple), the nuclear core components (blue), and the nuclear basket (red) (A). Several nuclear pore components have been found to be disrupted in cases of ALS (colored in black)–Gle1, GP210, NDC1, Nup107, Nup133, Nup153, Nup50, Nup62, Nup98, POM121, RanBP2, and Tpr (B). Nup98 and Nup62 are disrupted in Alzheimer’s disease (C). Cases of Huntington’s disease show disruption of Nup62, Nup88, and Tpr (D). Miscellaneous juvenile neurodegenerative disorders also display disruption in nuclear pore components including ALADIN, Gle1, Nup214, Nup62, Nup88, and RanBP2, many involving a direct mutation in the gene encoding that NPC component (E)
Individual nucleoporins also play roles in a variety of nuclear and cytoplasmic processes, independent of their function in maintaining proper transport across the nuclear pore [7,8,9]. Nup98, for example, interacts with DHX9 in the nucleoplasm to regulate the transcription and splicing of certain mRNA transcripts [10]. SEH1, another nucleoporin, performs a cytoplasmic role in the regulation of MTORC1 as part of the GATOR2 complex [11, 12].
Here, we discuss the effect of disease-related nucleoporin mutations on NPC selectivity. Additionally, we examine numerous neurological diseases that present significant alterations in nuclear pore structure and function.
Disease-Related Mutations in Nuclear Pore Complex Components
Many studies over the past few decades have focused on disruptions to the NPC in neurodegeneration. Oftentimes, components of the NPC are mislocalized or dysregulated as a result of an unrelated or even an unknown genetic mutation. However, mutations in genes that directly encode for NPC components can also lead to dysregulation of the nuclear pore and sequential neurodegeneration in both human patients and cell and animal models of disease (Fig. 1E). Below, we review several nucleoporin (Nup) mutations and the associated cell-specific disorders.
RANBP2 (Nup358)
Genetic acute necrotizing encephalopathy (ANE1) is a rare disease resulting from viral infections in patients carrying mutations (most commonly a missense mutation, c.1880CT, p.Thr585Met [13]) in RanBP2 [14], though the precise mechanism of neuronal toxicity is unknown. ANE1 presents as an acute encephalopathy followed by an onset of neurological symptoms including deteriorating consciousness, progression to coma, and possible seizures. RanBP2 (also known as Nup358) is a major component of the cytoplasmic filaments of the NPC. RanBP2 regulates the Ran GTPase cycle which is necessary for nuclear import and export via the active transport receptors, importin β and exportin-1. Ran GTPase activity is required for the release of these receptors from Ran-GTP to initiate and terminate nuclear import and export, respectively [15,16,17,18,19,20]. Knockout of RanBP2 in mouse spinal cord motor neurons leads to the subcellular disorganization of the known RanBP2 substrates importin β, exportin 1, Ran GTPase, and Ran-GTP [21].
In control mice, importin β and exportin 1 are found predominantly at the nuclear rim, while Ran GTPase and Ran-GTP are found largely in the cytoplasm. However, in the RanBP2 knockout mice, importin β becomes sequestered to the nuclear compartment and is lost at the nuclear rim; exportin 1 is completely lost from the nuclear rim; and expressions of Ran GTPase and Ran-GTP are broadly distributed between the nucleus and cytoplasm. RanBP2 ablation in Thy+ neurons in the central nervous system and spinal cord is sufficient to mimic the disease progression of ALS with the mice displaying hypoactivity, hind limb paralysis, respiratory distress, and premature death. Interestingly RanBP2 is also necessary for cell viability in other cell types. For example, loss of the Ran GTPase binding domains 2 and 3 (RBD2/3) of RanBP2 leads to cone photoreceptor degeneration [22]. Whether these findings relate to the pathogenesis of ANE1 is unclear, though they provide interesting insight into the effect of neuronal RanBP2 loss or dysfunction.
Gle1
Gle1 mutations have been discovered in patients with sporadic and familial ALS [23]. The first mutation identified was a nonsense mutation that introduced a premature stop early on in Exon 2 of Gle1, while the other mutation was a splice site mutation that caused a shift in the reading frame and the replacement of the last 44 amino acids of the protein with 88 different amino acids. The NPC-associated factor Gle1 is a critical component of the mRNA export machinery [24, 25]. Humans have two isoforms of Gle1–Gle1A and Gle1B–that differ only in their 3’ untranslated regions, with Gle1B being more highly expressed and localized to the nuclear rim through an interaction with CG1 and Nup155 [26, 27]. In vitro analysis of these mutations revealed two distinct mechanisms of protein disruption depending on the mutation [23]. The nonsense mutation leads to a loss of the Gle1 mRNA via nonsense mediated decay and an overall loss of Gle1 protein. Alternatively, the splice site mutation, with the addition of the amino acids in its C-terminus, is unable to localize properly to the NPC and instead becomes biased towards the cytoplasm.
Interestingly, knockdown of Gle1 in zebrafish embryos using antisense morpholinos causes morphological anomalies in the jaw as well as cell death in the spinal cord, both phenotypes which are reminiscent of symptoms in patients with lethal congenital contracture syndrome-1 (LCCS1) [23, 28]. LCCS1 is an embryonic degenerative disease marked by the loss of the ventral spinal cord and anterior horn motor neurons [29] and is caused by numerous identified mutations in Gle1, including a splice site mutation and several missense mutations [30]. Distinct from the Gle1 mutations that cause ALS, Gle1 mutations in LCCS1 do not alter the localization of the protein from the NPC [31]. Instead, the disease-causing mutation inserts a proline-phenylalanine-glutamine group into the coil-coil domain of Gle1 which prevents its ability to oligomerize and therefore inhibits its role in mRNA export. A closely related disease, lethal arthrogryposis with anterior horn cell disease (LAAHD), has similar motor neuron loss in the anterior horn of the spinal cord and is also caused by a mutation in Gle1 [32].
ALADIN
The nuclear pore component, ALADIN, is anchored to the cytoplasmic side of the NPC through its interaction with NDC1, a nucleoporin required for NPC assembly [33, 34]. A myriad of mutations, including a number of point mutations, frameshift mutations, and nonsense mutations, in the AAAS gene that encodes ALADIN cause the disease triple A syndrome. Triple A syndrome is a multisystemic disorder that includes progressive neurodegeneration and neural impairments [35]. In cells harboring a triple A syndrome-causing AAAS mutation, ALADIN itself predominantly mislocalizes to the cytoplasm; however, there are no other overt morphological abnormalities to the nuclei, nuclear envelope, or nuclear pore complex [36,37,38]. How then does the NPC contribute to disease pathology in triple A syndrome? Fibroblasts from triple A syndrome patients have increased sensitivity to oxidative stress [39]. To further elucidate how mislocalized ALADIN could cause this susceptibility to oxidative stress, another group discovered that there is a decrease in the nuclear accumulation of import cargoes reliant on the karyopherin-α/β import pathway in triple A syndrome fibroblasts [40]. Additionally, these cells have increased sensitivity to oxidative stress and a decreased ability to repair damaged DNA, likely due to the negatively affected nuclear import. Indeed, ALADIN has been shown to interact with and regulate the translocation of ferratin heavy chain protein (FTH1) [41], a nuclear protein that has a protective role in oxidative damage of DNA [42,43,44]. Triple A syndrome patient fibroblasts display a complete absence of nuclear FTH [41]. It is therefore likely that AAAS mutations cause mislocalization of ALADIN, inhibit its ability to import FTH1 to the nucleus, and lead to increased stress from oxidative damage and eventual cell death.
Nup214, Nup88, and Nup62
While our understanding of how the NPC contributes directly to cell death in neurodegeneration is expanding, there are still many cases in which the mechanism underlying disease pathology is incompletely characterized, despite our knowledge of the genetic cause. As mentioned above, mutations to RanBP2 are causative of ANE1 [13, 14, 45]. How the viral infection exacerbates the potential nucleocytoplasmic transport defects due to a lack of RanBP2 function is poorly understood. Similarly, missense and frameshift mutations in Nup214, another component of the NPC cytoplasmic filaments, are causative of acute febrile encephalopathy [46, 47], a disease that is similar to ANE1 and is also very poorly understood mechanistically. A familial missense mutation causing infantile bilateral striatal necrosis (IBSN) was mapped to Nup62 [48]. How the mutation in Nup62 contributes to the disease is unknown; however, it could be through its role in maintaining chromosomal integrity [49]. Mutations in yet another member of the NPC cytoplasmic filaments, Nup88, cause fetal akinesia deformation sequence (FADS), a disease characterized by impaired fetal movement [50]. In a nup88-null zebrafish line, there is reduced association of Nup62 with the NPC as well as a loss of interaction between Nup88 and Nup214. While these impaired interactions may not be the sole cause of FADS, it is possible that it contributes to disease.
Huntington’s Disease
Huntington’s disease (HD) is a disorder with three distinct sets of symptoms including motor deficits, cognitive decline, and psychiatric abnormalities due to progressive degeneration mainly in the striatum and basal ganglia [51], although other distant brain regions also undergo alterations. HD is caused by a CAG trinucleotide repeat expansion in the huntingtin gene (HTT) which leads to a polyglutamine (polyQ) expansion in the N-terminal of the Huntingtin protein [52]. An interesting proteomics study in 2001 discovered that many NPC proteins, particularly Nup62, co-aggregate with polyQ aggregates [53]. These results initiated a hypothesis in which HD mutations may confer toxicity through altered NPC function (Fig. 1D).
Indeed, a subset of Nups (Nup62, Nup88, and RanGAP1) colocalize with mutant huntingtin (mHTT) intracellular aggregates in the striatum and cortex of an HD mouse model which is consistent with observations in postmortem tissue from HD patients [54, 55]. In the mouse cortex and striatum, mHTT exacerbated age-related nuclear dysfunction, including disruption of the nuclear envelope and accumulation of intranuclear mRNA [55]. As the mice age, accumulation of RanGAP1 and mHTT shifts to the perinuclear and cytoplasmic regions of the cell which is also observed in HD patient cortices. This cytoplasmic accumulation of mHTT could cause additional mislocalization of proteins containing disordered and low complexity sequences in neurons, including multiple factors of the nuclear transport machinery, similar to what has been demonstrated in immortalized cell line studies [56]. Whether such mislocalization of these NPC components leads to functional alterations in the NPC is yet uninvestigated.
Additionally, induced pluripotent stem cell (iPSC)-derived neurons from HD patients also display mislocalized RanGAP1 and Nup62 as well as a significant reduction in the nucleocytoplasmic ratio of endogenous Ran, suggesting deficient active transport between the nucleus and cytoplasm in HD. This observation was replicated in primary cortical neurons transduced with mHTT as well as in a Drosophila HD model–several Nups, including RanGAP1, Nup62, and Nup88, are mislocalized and these cells display deficits in nucleocytoplasmic transport. Overexpressing Ran and RanGAP1 confer neuroprotection in both of these systems [54] suggesting that restoring proper transport could be an effective way to mitigate mHTT-induced degeneration.
It is clear that mHTT causes disruptions in NPC components, but evidence connecting these observations to cellular toxicity is lacking. Several studies suggest that mHTT accumulation in the nucleus impairs nuclear export of mRNA and protein. mHTT was found to have preferential binding to RanGAP1 and the mRNA export factor RAE1 [57] which could account for the mRNA accumulation observed in the nucleus in previously described HD models [54, 55]. Interestingly, the N-terminal of wild-type HTT interacts with Tpr, another member of the NPC that facilitates mRNA and protein export [58]. The polyQ expansion in mHTT was shown to cause reduced nuclear export of the protein, accumulation within the nucleus, and subsequent nuclear membrane distortions. mHTT was also found to have increased phosphorylation at Serine-16 in its N-terminus in striatal neurons of an HD mouse model [59]. The increased phosphorylation promotes nuclear accumulation and decreases the interaction between HTT protein and Tpr, possibly accounting for the nuclear accumulations of both mRNA and mHTT protein. The N-terminal of HTT also includes a nuclear export signal which allows the protein to be transported out of the nucleus via exportin 1 [60]. It is therefore unclear whether the nuclear export of cleaved HTT is entirely functional, as the N-terminal fragment of HTT is well exported when overexpressed in HEK293T cells [60] but is also observed within intranuclear accumulations in mouse AD models [61].
Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive degenerative disease that contributes to 60–80% of the dementia cases [62]. Infamous hallmarks of AD include aggregation of amyloid-beta (Aβ) protein and/or phosphorylated Tau in neurons [63]. Early electron microscopy studies from AD biopsies revealed neurons containing neurofibrillary tangles (NFTs)–insoluble protein tangles consisting mostly of misfolded Tau–have irregular nuclear structure with the NFTs often associated with the nuclear lamina and nuclear pore complexes [64]. Further investigation of AD biopsies and postmortem tissues revealed that irregular nuclear pores often associate with NFTs [65]. While there are no apparent differences in Nup expression or distribution in these samples, hippocampal neurons with and without NFTs have increased accumulation of nuclear transport factor 2 (NTF2) which transports cargoes from the cytoplasm to the nucleus. Additionally, importin α1 mislocalizes to cytoplasmic inclusions in the hippocampus of AD patients [66]. Previous studies revealed inhibition of classical nuclear localization sequence (NLS)-mediated cargo transport via importin α1 as a result of cell stress [67, 68] and it is therefore likely that the importin α1 accumulation is a result of other unknown factors causing cell stress.
Drosophila models of Tau pathology highlighted the importance of nuclear integrity in the context of neurodegeneration. Transgenic expression of human Tau in Drosophila causes nuclear envelope invaginations and a significant decrease in the Drosophila B-type Lamin protein [69], and interestingly similar pathology is seen in AD brain samples [70]. Most disrupted nuclei are associated with extra-nuclear pathogenic Tau aggregates, suggesting a possible correlation between Tau accumulations and disruption of nuclear structure. This Tau-induced Lamin dysfunction impairs mRNA trafficking [71] and is sufficient to drive cell-cycle activation and subsequent apoptotic death [69]. While these studies provide us with a platform to further our understanding of nuclear dysfunction in tauopathies, the mechanisms connecting mRNA and protein trafficking directly to neurodegeneration are still poorly understood.
Using a combination of human, mouse, and cell models of AD, it was recently shown that phosphor-Tau can interfere with NPC integrity and nucleocytoplasmic transport through a variety of mechanisms [72]. As such, a few NPC components have been shown to specifically be altered in AD (Fig. 1C). Hippocampal tissue of AD patients displays a shallower nuclear-to-cytoplasmic Ran gradient than tissue from unaffected individuals. In primary neuron cultures, pathogenic Tau is sufficient to disrupt the Ran gradient. Using nuclear import and export reporters in a live cell assay, Eftekharzadeh and colleagues also demonstrated that neurons containing Tau protein show disruption in both nuclear import and export. In addition to an altered nucleocytoplasmic Ran gradient, Nup98 is mislocalized to the cytoplasm of neurons that contain misfolded Tau and phosphor-Tau in AD postmortem hippocampal tissue as well as in brain tissue from Tau-overexpressing transgenic mice. Nup98 appears to colocalize with the NFTs in the cytoplasm through a direct interaction with Tau and this phenomenon is specific to Nup98, and to a lesser extent Nup62, while other Nups remain unaffected. The mouse lines used in this study express the human Tau transgene that can be suppressed by doxycycline (DOX). Treating these mice with DOX leads to a reduction in soluble Tau, relatively few NFTs, and interestingly a rescue of Nup98 expression at the nuclear membrane, indicating a role for Tau in facilitating Nup98 localization. In primary mouse neuronal cultures, Nup98 facilitates Tau aggregation and also co-aggregates with Tau via a direct interaction [72]. Additionally, Nup98 and Tau colocalize to cytoplasmic accumulations in postmortem AD brain samples. Another group discovered that the repeat binding domain of Tau mediates its interaction with Nup98 [73]. Interestingly, the phosphorylation of Tau, but not its oligomerization, facilitates the association with Nup98, and once bound, the interaction is quite strong. Although unconfirmed, it is likely that the accumulation of phosphor-Tau and Nup98 at the outer surface of the nuclear membrane is a significant contributor to decreased nucleocytoplasmic transport.
ALS/FTD
Amyotrophic lateral sclerosis (ALS) is a progressive and invariably fatal neurodegenerative disease that affects upper and lower motor neurons [74]. Around 10% of cases are familial (fALS) via inheritance of a mutation in one of many known genes, while the remainder are sporadic (sALS), meaning the genetic cause is unknown [74, 75]. The first reported defect in NPC function arose from three independent investigations into the pathogenesis of the C9ORF72 mutation, a repeat expansion that accounts for a significant portion of fALS and sALS [74, 76,77,78]. These groups observed disruption of the Ran GTPase gradient in fixed cells [76] and reporter-based dysfunction of nuclear import [76], and identified numerous components of the nuclear transport machinery that modified toxicity resulting from the mutation [76,77,78]. Dipeptide repeat proteins (DPRs), one type of toxic product resulting from the C9ORF72 mutation, were subsequently linked to alterations in nucleocytoplasmic transport [79, 80]. Two groups demonstrated that arginine-rich DPRs bind to and inhibit the function of the nuclear import receptor, importin β [79, 80]. These experiments were performed using a variety of in vitro permeabilized cell and biochemical assays with synthetic DPRs. Interestingly, an independent investigation that utilized transiently overexpressed DPRs concluded that arginine-rich DPRs had no effect on nuclear transport function in numerous cell types, including Hela Kyoto, SH-SY5Y, and human-induced pluripotent stem cell-derived motor neurons (iPSNs) [81]. This discrepancy may be attributed to the identity of the nuclear localization signals present within the tested nuclear transport cargoes and reporters, and highlights the need to test nuclear transport of multiple classes of cargoes rather than relying on a single reporter. While all three groups assessed various aspects of nuclear transport, none of these investigations were focused on the effect of DPRs on nuclear pore complex structure itself.
While the C9ORF72 mutation is the most common known cause of fALS, mutations in FUS, TARDBP, and SOD1 are also causative of this disease. Pathology arising from these mutations has also been linked to deficits in the function of the nuclear pore complex. For example, overexpression of disease-linked TDP-43 (encoded by TARDBP) variants in Drosophila melanogaster photoreceptors or N2a cells leads to abnormal nuclear envelope morphology, while iPSNs harboring endogenous TDP-43 mutations demonstrate reduced nuclear import of an NLS-tdTomato-NES reporter [82]. Overexpression of various nucleoporins (Nup50, Nup93, Nup98-96, Nup107, and Nup214) rescue photoreceptor and motor defects in their fruit fly model; however, their specific neuroprotective function in this setting remains unclear [82]. Additionally, in female mice expressing an ALS-linked variant of SOD1, various nucleoporins and RanGAP1 exhibit altered spatiotemporal distributions compared to wild-type littermates [83]. While this may indicate at a potential role for altered NPC function in SOD1-ALS, no functional assays were performed to demonstrate whether these alterations led to any functional consequence. While minimal defects in the Ran GTPase gradient were observed in iPSNs harboring FUS mutations, there were evident abnormalities in nuclear lamina partially as a result of Nup62 association with FUS [84]. This interaction between FUS and Nup62 has the potential to form insoluble aggregates in the cytoplasm in vitro, so it is therefore possible that these aggregates sequester and trap Nup62 and other Nups in the cytoplasm, further disrupting the nuclear pore structure. Although numerous groups have reported association of various nucleoporins within aggregates, stress granules, or other cytoplasmic accumulations [80, 82, 85], it was not immediately clear whether this association was subsequent to loss of these nucleoporins from the NPC itself. A recent study demonstrated that in iPSNs harboring the C9ORF72 mutation, eight specific nucleoporins have decreased expression within the NPC (TPR, Nup98, NDC1, POM121, Nup107, Nup133, Nup50, GP210) [86]. This seminal work utilized structured illumination microscopy to comprehensively assess the presence of the 23 nucleoporins within NPCs [86]. Impressively, the same eight nucleoporins are depleted from NPCs in postmortem motor cortex [86], demonstrating the power of endogenous model systems. This loss of nucleoporins is linked to pathogenic hexanucleotide repeat RNA that results from the C9ORF72 mutation and aberrant degradation of nuclear pore components via an ESCRT-III pathway [87, 88], rather than sequestration in stress granules or other cytoplasmic accumulations [86].
Interestingly, loss of certain nucleoporins has also been observed in up to 90% of sporadic ALS patient-derived iPSNs. In iPSNs derived from sporadic ALS patients, five nucleoporins are consistently depleted from the NPC (Nup50, Nup153, TPR, POM121, Nup133) [87]. The loss of these nucleoporins has similarly been linked to degradation via an ESCRT-III pathway. While the primary injury resulting in this aberrant degradation is theoretically different in C9ORF72 ALS and sALS (only ~10% of sALS cases harbor the C9ORF72 mutation) [74], it is possible that the initiation of ESCRT-III overactivity and subsequent loss of nucleoporins occurs via a common event. Future investigations into the similarities between NPC dysfunction in fALS and sALS may yield interesting and highly impactful results.
Importantly, this NPC injury in sALS iPSNs appears to result in the loss of nuclear TDP-43 and its RNA metabolism function, as substantiated by an inverse correlation between aberrant ESCRT-II function and TDP-43 nuclear depletion [87]. Nuclear clearance and cytoplasmic accumulation of TDP-43 is a common pathological hallmark of ALS [89, 90]. Approximately ninety-seven percent of postmortem cases exhibit motor neurons with reduced nuclear and increased cytoplasmic presence of TDP-43, though only a small percentage of neurons within each case demonstrate this pathology [74].
Due to the prominence of this protein pathology, recent investigations have focused on understanding the downstream effects of TDP-43 nuclear clearance. Indeed, recent RNA sequencing studies have uncovered hundreds of transcripts that are altered in abundance upon artificial global TDP-43 depletion, the most presently notable of which is Stathmin-2 [91, 92]. Although Stathmin-2 plays a significant role in the regeneration of axons [91, 92], it is yet unclear whether therapeutic targeting of a single dysregulated TDP-43 target will prove effective in patients.
On the other hand, reversal of NPC injury itself may prove to prevent or diminish TDP-43 nuclear clearance in affected motor neurons, effectively restoring all TDP-43 function. Antisense oligonucleotides (ASO) designed to deplete CHMP7, the identified pathogenic component of the ESCRT-III pathway in ALS, completely repaired the NPC injury and normalized aberrant TDP-43 function [87]. For these reasons, CHMP7 ASO may be an attractive candidate therapy in sporadic ALS.
Discussion
Nuclear structure and function are vital for the proper segregation of nuclear and cytoplasmic processes and materials. The NPC maintains precise passage of materials between these two compartments. When the NPC is disrupted functionally or structurally, a common consequence, especially in the context of neurons, is cellular degeneration.
NPC function can be disrupted in a variety of ways, the most obvious of which is via mutations in NPC components themselves. We discussed that mutations in different nucleoporins lead to distinct cellular consequences. Mutations in RANBP2 and GLE1, for example, have a significant impact on nuclear transport function, while those in ALADIN appear to induce toxicity via increased sensitivity to oxidative stress and DNA damage. This emphasizes that despite its main role as a nucleocytoplasmic gate, the NPC consists of numerous individual proteins, each of which have functions independent of their roles within the NPC itself. Such diversity in function of the numerous NPC components makes it understandably difficult to determine if (and how) mislocalization of these proteins leads to cellular toxicity.
Yet, it remains tempting to presume that altered cellular distribution of one or even many nucleoporins necessarily leads to altered NPC function as a whole. For example, one group overstates their observations as “nuclear pore destruction” in a single postmortem case of juvenile-onset ALS, entirely substantiated by altered nuclear rim staining of Nup62 [93]. In the burgeoning field of NPC-centric investigations of neurodegeneration, high-quality imaging and reproducibility of such phenotypes will be required to substantiate anecdotal observations, e.g., functional impact of mislocalized or absent NPC components on their distal functions, as well as their contribution to NPC function as a whole.
Numerous groups have utilized fluorescent reporter-based assays to quantitate nuclear import and export function [76, 84, 86]. These “shuttle reporters,” typically a GFP- or RFP-based fluorophore appended to an NLS and NES, serve as exogenous indicators of transport into and out of the nucleus [94]. Introduction of such reporters into a perturbed system (e.g., in iPSN harboring a disease mutation) can allow investigators to compare import and export rates across various conditions. However, not all shuttle reporters interrogate both nuclear import and export function. A shuttle reporter, such as the S-GFP reporter [85], with a strong NES and weak NLS, for example, will be mostly cytoplasmic under baseline conditions. This provides significant dynamic range to assess alterations in nuclear export, but alterations to nuclear import function may not be assessable. Meanwhile the S-tdTomato reporter, used in the same study [85], can effectively report on the opposite (it is useful for monitoring changes in nuclear import, but not export). Therefore, it is apparent that shuttle reporters should be carefully chosen to answer a specific question about NPC function, rather than as a catch-all attempt to interrogate multiple pore functions.
As reliable methods to investigate NPC structure and function have become more available and widely used by the neurodegeneration community, we have learned that loss of at least one nucleoporin is differentially associated with alterations to others. Reduction of neuronal POM121, for example, directly leads to a reduction in the NPC presence of 6 or more other nucleoporins [86]. Meanwhile, mislocalization of ALADIN in the context of triple A syndrome leaves the rest of the NPC structurally unaffected [36,37,38]. Future investigations into mislocalized NPC components would do well to test for inter-nucleoporin effects rather than assuming that observed alterations are restricted to a single component of this massive protein complex.
Conclusion
Extensive literature supports a role for NPC dysfunction in the pathogenesis of neurodegenerative diseases ranging from Huntington’s disease to Alzheimer’s disease to amyotrophic lateral sclerosis. However, with the exception of ALS, it is still unclear whether NPC dysfunction is a toxicity-initiating event, or a consequence of some other primary injury. Regardless of whether the event is disease initiation or downstream response to injury–the loss of the NPC and nucleocytoplasmic transport itself is an injurious event and may also be a future target for repair. Additional studies that comprehensively and mechanistically interrogate the structure and function of the NPC in relevant disease models will be crucial to further our attempts to identify useful therapeutic targets in these diseases.
References
Singer RH, Green MR. Compartmentalization of eukaryotic gene expression: causes and effects. Cell. 1997;91:291–4.
Fabrini R, Bocedi A, Pallottini V, Canuti L, De Canio M, Urbani A, Marzano V, Cornetta T, Stano P, Giovanetti A, Stella L, Canini A, Federici G, Ricci G. Nuclear shield: a multi-enzyme task-force for nucleus protection. PLoS ONE. 2010;5:e14125–e14125.
Lin DH, Hoelz A. The structure of the nuclear pore complex (an update). Annu Rev Biochem. 2019;88:725–83.
Savas JN, Toyama BH, Xu T, Yates JR 3rd, Hetzer MW. Extremely long-lived nuclear pore proteins in the rat brain. Science. 2012;335:942.
Nigg EA. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature. 1997;386:779–87.
Görlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–8.
Chatel G, Fahrenkrog B. Dynamics and diverse functions of nuclear pore complex proteins. Nucleus. 2012;3:162–71.
Khan AU, Qu R, Ouyang J, Dai J. Role of nucleoporins and transport receptors in cell differentiation. Front Physiol. 2020;11:239.
Guglielmi V, Sakuma S, D’Angelo MA. Nuclear pore complexes in development and tissue homeostasis. Development. 2020;147:dev183442.
Capitanio JS, Montpetit B, Wozniak RW. Human Nup98 regulates the localization and activity of DExH/D-box helicase DHX9. eLife. 2017;6:e18825.
Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–6.
Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, Abu-Remaileh M, Frankel WN, Sabatini DM. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543:438–42.
Neilson DE, Adams MD, Orr CM, Schelling DK, Eiben RM, Kerr DS, Anderson J, Bassuk AG, Bye AM, Childs AM, Clarke A, Crow YJ, Di Rocco M, Dohna-Schwake C, Dueckers G, Fasano AE, Gika AD, Gionnis D, Gorman MP, Grattan-Smith PJ, Hackenberg A, Kuster A, Lentschig MG, Lopez-Laso E, Marco EJ, Mastroyianni S, Perrier J, Schmitt-Mechelke T, Servidei S, Skardoutsou A, Uldall P, van der Knaap MS, Goglin KC, Tefft DL, Aubin C, de Jager P, Hafler D, Warman ML. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet. 2009;84:44–51.
Levine JM, Ahsan N, Ho E, Santoro JD. Genetic acute necrotizing encephalopathy associated with RANBP2: clinical and therapeutic implications in pediatrics. Mult Scler Relat Disord. 2020;43: 102194.
Hamada M, Haeger A, Jeganathan KB, van Ree JH, Malureanu L, Wälde S, Joseph J, Kehlenbach RH, van Deursen JM. Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J Cell Biol. 2011;194:597–612.
Vetter IR, Nowak C, Nishimoto T, Kuhlmann J, Wittinghofer A. Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: implications for nuclear transport. Nature. 1999;398:39–46.
Cautain B, Hill R, de Pedro N, Link W. Components and regulation of nuclear transport processes. Febs j. 2015;282:445–62.
Yokoyama N, Hayashi N, Seki T, Panté N, Ohba T, Nishii K, Kuma K, Hayashida T, Miyata T, Aebi U, et al. A giant nucleopore protein that binds Ran/TC4. Nature. 1995;376:184–8.
Singh BB, Patel HH, Roepman R, Schick D, Ferreira PA. The zinc finger cluster domain of RanBP2 is a specific docking site for the nuclear export factor, exportin-1. J Biol Chem. 1999;274:37370–8.
Villa Braslavsky CI, Nowak C, Görlich D, Wittinghofer A, Kuhlmann J. Different structural and kinetic requirements for the interaction of Ran with the Ran-binding domains from RanBP2 and importin-beta. Biochemistry. 2000;39:11629–39.
Cho KI, Yoon D, Qiu S, Danziger Z, Grill WM, Wetsel WC, Ferreira PA. Loss of Ranbp2 in motoneurons causes disruption of nucleocytoplasmic and chemokine signaling, proteostasis of hnRNPH3 and Mmp28, and development of amyotrophic lateral sclerosis-like syndromes. Dis Model Mech. 2017;10:559–79.
Patil H, Saha A, Senda E, Cho KI, Haque M, Yu M, Qiu S, Yoon D, Hao Y, Peachey NS, Ferreira PA. Selective impairment of a subset of Ran-GTP-binding domains of ran-binding protein 2 (Ranbp2) suffices to recapitulate the degeneration of the retinal pigment epithelium (RPE) triggered by Ranbp2 ablation. J Biol Chem. 2014;289:29767–89.
Kaneb HM, Folkmann AW, Belzil VV, Jao LE, Leblond CS, Girard SL, Daoud H, Noreau A, Rochefort D, Hince P, Szuto A, Levert A, Vidal S, André-Guimont C, Camu W, Bouchard JP, Dupré N, Rouleau GA, Wente SR, Dion PA. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:1363–73.
Murphy R, Wente SR. An RNA-export mediator with an essential nuclear export signal. Nature. 1996;383:357–60.
Kendirgi F, Barry DM, Griffis ER, Powers MA, Wente SR. An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J Cell Biol. 2003;160:1029–40.
Kendirgi F, Rexer DJ, Alcázar-Román AR, Onishko HM, Wente SR. Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: a conserved mechanism in the export of Hsp70 mRNA. Mol Biol Cell. 2005;16:4304–15.
Rayala HJ, Kendirgi F, Barry DM, Majerus PW, Wente SR. The mRNA export factor human Gle1 interacts with the nuclear pore complex protein Nup155. Mol Cell Proteomics. 2004;3:145–55.
Jao LE, Appel B, Wente SR. A zebrafish model of lethal congenital contracture syndrome 1 reveals Gle1 function in spinal neural precursor survival and motor axon arborization. Development. 2012;139:1316–26.
Herva R, Conradi NG, Kalimo H, Leisti J, Sourander P. A syndrome of multiple congenital contractures: neuropathological analysis on five fetal cases. Am J Med Genet. 1988;29:67–76.
Nousiainen HO, Kestilä M, Pakkasjärvi N, Honkala H, Kuure S, Tallila J, Vuopala K, Ignatius J, Herva R, Peltonen L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40:155–7.
Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD, Wente SR. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell. 2013;155:582–93.
Vuopala K, Ignatius J, Herva R. Lethal arthrogryposis with anterior horn cell disease. Hum Pathol. 1995;26:12–9.
Kind B, Koehler K, Lorenz M, Huebner A. The nuclear pore complex protein ALADIN is anchored via NDC1 but not via POM121 and GP210 in the nuclear envelope. Biochem Biophys Res Commun. 2009;390:205–10.
Yamazumi Y, Kamiya A, Nishida A, Nishihara A, Iemura S, Natsume T, Akiyama T. The transmembrane nucleoporin NDC1 is required for targeting of ALADIN to nuclear pore complexes. Biochem Biophys Res Commun. 2009;389:100–4.
Pogliaghi G, Cangiano B, Duminuco P, Vezzoli V, Bonomi M. Triple-A syndrome (TAS): an in-depth overview on genetic and phenotype heterogeneity. Protein Pept Lett. 2020;27:1192–203.
Cronshaw JM, Matunis MJ. The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc Natl Acad Sci USA. 2003;100:5823–7.
Huebner A, Kaindl AM, Knobeloch KP, Petzold H, Mann P, Koehler K. The triple A syndrome is due to mutations in ALADIN, a novel member of the nuclear pore complex. Endocr Res. 2004;30:891–9.
Krumbholz M, Koehler K, Huebner A. Cellular localization of 17 natural mutant variants of ALADIN protein in triple A syndrome - shedding light on an unexpected splice mutation. Biochem Cell Biol. 2006;84:243–9.
Kind B, Koehler K, Krumbholz M, Landgraf D, Huebner A. Intracellular ROS level is increased in fibroblasts of triple A syndrome patients. J Mol Med (Berl). 2010;88:1233–42.
Hirano M, Furiya Y, Asai H, Yasui A, Ueno S. ALADINI482S causes selective failure of nuclear protein import and hypersensitivity to oxidative stress in triple A syndrome. Proc Natl Acad Sci USA. 2006;103:2298–303.
Storr HL, Kind B, Parfitt DA, Chapple JP, Lorenz M, Koehler K, Huebner A, Clark AJ. Deficiency of ferritin heavy-chain nuclear import in triple a syndrome implies nuclear oxidative damage as the primary disease mechanism. Mol Endocrinol. 2009;23:2086–94.
Cai CX, Birk DE, Linsenmayer TF. Nuclear ferritin protects DNA from UV damage in corneal epithelial cells. Mol Biol Cell. 1998;9:1037–51.
Thompson KJ, Fried MG, Ye Z, Boyer P, Connor JR. Regulation, mechanisms and proposed function of ferritin translocation to cell nuclei. J Cell Sci. 2002;115:2165–77.
Surguladze N, Patton S, Cozzi A, Fried MG, Connor JR. Characterization of nuclear ferritin and mechanism of translocation. Biochem J. 2005;388:731–40.
Sell K, Storch K, Hahn G, Lee-Kirsch MA, Ramantani G, Jackson S, Neilson D, von der Hagen M, Hehr U, Smitka M. Variable clinical course in acute necrotizing encephalopathy and identification of a novel RANBP2 mutation. Brain Dev. 2016;38:777–80.
Fichtman B, Harel T, Biran N, Zagairy F, Applegate CD, Salzberg Y, Gilboa T, Salah S, Shaag A, Simanovsky N, Ayoubieh H, Sobreira N, Punzi G, Pierri CL, Hamosh A, Elpeleg O, Harel A, Edvardson S. Pathogenic variants in NUP214 cause “plugged” nuclear pore channels and acute febrile encephalopathy. Am J Hum Genet. 2019;105:48–64.
Shamseldin HE, Makhseed N, Ibrahim N, Al-Sheddi T, Alobeid E, Abdulwahab F, Alkuraya FS. NUP214 deficiency causes severe encephalopathy and microcephaly in humans. Hum Genet. 2019;138:221–9.
Basel-Vanagaite L, Muncher L, Straussberg R, Pasmanik-Chor M, Yahav M, Rainshtein L, Walsh CA, Magal N, Taub E, Drasinover V, Shalev H, Attia R, Rechavi G, Simon AJ, Shohat M. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol. 2006;60:214–22.
Chien ML, Lai JH, Lin TF, Yang WS, Juang YL. NUP62 is required for the maintenance of the spindle assembly checkpoint and chromosomal stability. Int J Biochem Cell Biol. 2020;128: 105843.
Bonnin E, Cabochette P, Filosa A, Jühlen R, Komatsuzaki S, Hezwani M, Dickmanns A, Martinelli V, Vermeersch M, Supply L, Martins N, Pirenne L, Ravenscroft G, Lombard M, Port S, Spillner C, Janssens S, Roets E, Van Dorpe J, Lammens M, Kehlenbach RH, Ficner R, Laing NG, Hoffmann K, Vanhollebeke B, Fahrenkrog B. Biallelic mutations in nucleoporin NUP88 cause lethal fetal akinesia deformation sequence. PLoS Genet. 2018;14: e1007845.
Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98.
Pradhan S, Gao R, Bush K, Zhang N, Wairkar YP, Sarkar PS. Polyglutamine expansion in huntingtin and mechanism of DNA damage repair defects in Huntington’s Disease. Front Cell Neurosci. 2022;16: 837576.
Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–94.
Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer JC, Pham JT, Ahmed I, Peng Q, Wadhwa H, Pletnikova O, Troncoso JC, Duan W, Snyder SH, Ranum LPW, Thompson LM, Lloyd TE, Ross CA, Rothstein JD. Mutant huntingtin disrupts the nuclear pore complex. Neuron. 2017;94:93–107.
Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, Wheeler VC, Bang AG, Cleveland DW, Lagier-Tourenne C. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron. 2017;94:48–57.
Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351:173–6.
Hosp F, Vossfeldt H, Heinig M, Vasiljevic D, Arumughan A, Wyler E, Landthaler M, Hubner N, Wanker EE, Lannfelt L, Ingelsson M, Lalowski M, Voigt A, Selbach M. Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 2015;11:1134–46.
Cornett J, Cao F, Wang CE, Ross CA, Bates GP, Li SH, Li XJ. Polyglutamine expansion of huntingtin impairs its nuclear export. Nat Genet. 2005;37:198–204.
Havel LS, Wang CE, Wade B, Huang B, Li S, Li XJ. Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum Mol Genet. 2011;20:1424–37.
Zheng Z, Li A, Holmes BB, Marasa JC, Diamond MI. An N-terminal nuclear export signal regulates trafficking and aggregation of Huntingtin (Htt) protein exon 1. J Biol Chem. 2013;288:6063–71.
Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–9.
Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, Nixon RA, Jones DT. Alzheimer disease. Nat Rev Dis Primers. 2021;7:33.
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12.
Metuzals J, Robitaille Y, Houghton S, Gauthier S, Leblanc R. Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer’s disease. J Neurocytol. 1988;17:827–33.
Sheffield LG, Miskiewicz HB, Tannenbaum LB, Mirra SS. Nuclear pore complex proteins in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:45–54.
Lee HG, Ueda M, Miyamoto Y, Yoneda Y, Perry G, Smith MA, Zhu X. Aberrant localization of importin alpha1 in hippocampal neurons in Alzheimer disease. Brain Res. 2006;1124:1–4.
Miyamoto Y, Saiwaki T, Yamashita J, Yasuda Y, Kotera I, Shibata S, Shigeta M, Hiraoka Y, Haraguchi T, Yoneda Y. Cellular stresses induce the nuclear accumulation of importin alpha and cause a conventional nuclear import block. J Cell Biol. 2004;165:617–23.
Kodiha M, Chu A, Matusiewicz N, Stochaj U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 2004;11:862–74.
Frost B, Bardai FH, Feany MB. Lamin dysfunction mediates neurodegeneration in tauopathies. Curr Biol. 2016;26:129–36.
Frost B, Bardai FH, Feany MB. Lamin dysfunction mediates neurodegeneration in tauopathies. Curr Biol. 2016;26:129–36.
Cornelison GL, Levy SA, Jenson T, Frost B. Tau-induced nuclear envelope invagination causes a toxic accumulation of mRNA in Drosophila. Aging Cell. 2019;18: e12847.
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, Grima JC, Bennett RE, Tepper K, DeTure M, Vanderburg CR, Corjuc BT, DeVos SL, Gonzalez JA, Chew J, Vidensky S, Gage FH, Mertens J, Troncoso J, Mandelkow E, Salvatella X, Lim RYH, Petrucelli L, Wegmann S, Rothstein JD, Hyman BT. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron. 2018;99:925–40.
Diez L, Kapinos LE, Hochmair J, Huebschmann S, Dominguez-Baquero A, Vogt A, Rankovic M, Zweckstetter M, Lim RYH, Wegmann S. Phosphorylation but not oligomerization drives the accumulation of tau with nucleoporin Nup98. Int J Mol Sci. 2022;23:3495.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–38.
Nguyen HP, Van Broeckhoven C, van der Zee J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018;34:404–23.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61.
Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–9.
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–33.
Hutten S, Usluer S, Bourgeois B, Simonetti F, Odeh HM, Fare CM, Czuppa M, Hruska-Plochan M, Hofweber M, Polymenidou M, Shorter J, Edbauer D, Madl T, Dormann D. Nuclear import receptors directly bind to arginine-rich dipeptide repeat proteins and suppress their pathological interactions. Cell Rep. 2020;33: 108538.
Hayes LR, Duan L, Bowen K, Kalab P, Rothstein JD. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. Elife. 2020;9:e51685.
Vanneste J, Vercruysse T, Boeynaems S, Sicart A, Van Damme P, Daelemans D, Van Den Bosch L. C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci Rep. 2019;9:15728–15728.
Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, Seyfried NT, Powers MA, Kukar T, Hales CM, Gearing M, Cairns NJ, Boylan KB, Dickson DW, Rademakers R, Zhang YJ, Petrucelli L, Sattler R, Zarnescu DC, Glass JD, Rossoll W. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21:228–39.
Shang J, Yamashita T, Nakano Y, Morihara R, Li X, Feng T, Liu X, Huang Y, Fukui Y, Hishikawa N, Ohta Y, Abe K. Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience. 2017;350:158–68.
Lin YC, Kumar MS, Ramesh N, Anderson EN, Nguyen AT, Kim B, Cheung S, McDonough JA, Skarnes WC, Lopez-Gonzalez R, Landers JE, Fawzi NL, Mackenzie IRA, Lee EB, Nickerson JA, Grunwald D, Pandey UB, Bosco DA. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat Neurosci. 2021;24:1077–88.
Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, Bowen KE, Wadhwa H, Yang P, Rigo F, Taylor JP, Gitler AD, Rothstein JD, Lloyd TE. Stress granule assembly disrupts nucleocytoplasmic transport. Cell. 2018;173:958–71.
Coyne AN, Zaepfel BL, Hayes L, Fitchman B, Salzberg Y, Luo EC, Bowen K, Trost H, Aigner S, Rigo F, Yeo GW, Harel A, Svendsen CN, Sareen D, Rothstein JD. G4C2 repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron. 2020;107:1124–40.
Coyne AN, Baskerville V, Zaepfel BL, Dickson DW, Rigo F, Bennett F, Lusk CP, Rothstein JD. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci Transl Med. 2021;13:eabe1923.
Coyne AN, Rothstein JD. The ESCRT-III protein VPS4, but not CHMP4B or CHMP2B, is pathologically increased in familial and sporadic ALS neuronal nuclei. Acta Neuropathol Commun. 2021;9:127.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22:167–79.
Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22:180–90.
Aizawa H, Teramoto S, Hideyama T, Kato H, Terashi H, Suzuki Y, Kimura T, Kwak S. Nuclear pore destruction and loss of nuclear TDP-43 in FUS mutation-related amyotrophic lateral sclerosis motor neurons. J Neurol Sci. 2022;436:120187.
Semmelink MFW, Steen A, Veenhoff LM. Measuring and interpreting nuclear transport in neurodegenerative disease-the example of C9orf72 ALS. Int J Mol Sci. 2021;22:9217.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Olivia Spead and Benjamin L Zaepfel contributed equally to this work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Spead, O., Zaepfel, B.L. & Rothstein, J.D. Nuclear Pore Dysfunction in Neurodegeneration. Neurotherapeutics 19, 1050–1060 (2022). https://doi.org/10.1007/s13311-022-01293-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01293-w