
Abstract
We assessed the frequency of pediatric monogenic epilepsies and precision therapies at a tertiary epilepsy center. We analyzed medical records of children, born in 2006–2011 and followed at the Danish Epilepsy Center from January to December 2015; 357 patients were identified, of whom 27 without epilepsy and 35 with acquired brain damage were excluded. Of the remaining 295 children, 188 were consented for study inclusion and genetic testing. At inclusion, 86/188 had a preexisting genetic diagnosis and did not undergo further genetic testing. The 102 genetically unsolved patients underwent WES, which identified a (likely) pathogenic variant in eight patients and a highly relevant variant of unknown significance (VUS) in seven additional patients. Single nucleotide polymorphism array was performed in the remaining 87 patients and revealed no (likely) pathogenic copy number variants (CNVs). Patients with a genetic diagnosis had a significantly lower median age at seizure onset and more often had febrile seizures, status epilepticus, or neurodevelopmental impairment compared to those who remained genetically unsolved. Most common epilepsies were focal or multifocal epilepsies and developmental and epileptic encephalopathies (DDEs). Fifty-three patients, with a putative genetic diagnosis, were potentially eligible for precision therapy approaches. Indeed, genetic diagnosis enabled treatment adjustment in 32/53 (60%); 30/32 (93%) patients experienced at least a 50% reduction in seizure burden while only 4/32 (12.5%) became seizure-free. In summary, a genetic diagnosis was achieved in approximately 50% of patients with non-acquired epilepsy enabling precision therapy approaches in half of the patients, a strategy that results in > 50% reduction in seizure burden, in the majority of the treated patients.
Similar content being viewed by others

Introduction
Epilepsy can derive from well-defined structural or metabolic disorders, some of which can have a monogenic cause. It is estimated that genetic factors can play a role approximately 70–80% of incidences of epilepsy [1]. These range from monogenic disorders caused by rare or ultra-rare variants with a high effect size to polygenic disorders with a complex genetic architecture [1]. During the past years, genetic testing for epilepsy has been increasingly incorporated into everyday clinical practice. This advance has primarily been driven by technological developments in next-generation sequencing, which has led to rapid discoveries in the etiology of rare monogenic epilepsies. Currently, at least 400 known genes have been associated with these disorders [2]. However, a recent modeling study of 31,058 parent–offspring trios estimated that more than 1000 genes associated with developmental disorders remain to be discovered, although their detection is increasingly difficult due to reduced penetrance and high pre- or perinatal mortality [3].
Obtaining a genetic diagnosis is of great importance for patients and their families. It provides an explanation and certainty and enables a more targeted genetic counseling, including knowledge about the prognosis and recurrence risk. Furthermore, it allows the patient and family to be connected to gene-specific networks of families with the same genetic condition. Last but definitely not least, in some cases the genetic diagnosis can help guide treatment. This type of personalized medication in epilepsy is constantly evolving, and knowledge about the pathomechanisms underlying difficult-to-treat epilepsies facilitates the development of new drugs and the repurposing of already available compounds [4]. Repurposing of already available drugs that are used for entirely different disorders means we can use compounds with known safety and tolerability profiles, thus shortening the time needed to set up clinical trials, as they require less time and resources compared to the development of new drugs [5]. One such example is the use of fenfluramine in Dravet syndrome [6]. Currently established personalized treatments based on knowledge about the pathophysiological effects of genetic variants include the use of sodium channel blockers in patients with disease-causing gain-of-function variants in SCN2A and SCN8A [7], ketogenic diet in patients with GLUT1 deficiency, and mammalian target of rapamycin (mTOR) inhibitors in mTORopathies [8].
A recently published Scottish population study tested children presenting with epilepsy before 36 months of age with a custom-designed 104-gene epilepsy panel [9]. The authors found that 80/333 (24%) children had a diagnostic genetic finding and the overall estimated annual incidence of monogenic epilepsies in their cohort was around 1 per 2120 live births [9]. The study also reported that a specific treatment approach was theoretically possible for 64/80 (80%) children with a monogenic diagnosis [9]. Peng et al. [10] used either exome or panel testing on 273 genetically unsolved children with drug-resistant epilepsy and achieved a genetic diagnosis in 86 patients (31%), of whom 34 (39%) benefited from an adjustment of their medication according to the genetic finding [10]. A recent multicenter systematic survey of 293 patients with a monogenic epilepsy found that a rational precision therapy approach was available for only 56 patients (19%) [11]. Such treatments were applied in 33/56 (59%) but were only successful (i.e., > 50% seizure reduction) in 10/33 (30%) patients [11]. The authors recommended “greater caution in raising expectation in people with epilepsy, clinicians and healthcare providers about the current impact of genetic findings in epilepsy” [11].
The present study explores the utility of stepwise genetic testing in children diagnosed with epilepsy and followed at the only tertiary epilepsy center in Denmark. The primary aim was to examine how often this would lead to a precision genetic diagnosis and thus facilitate precision therapy approaches.
Methods
Ethical Aspects
The study was approved by the local ethics committee in the Zealand region of Denmark (number SJ-91). Written informed consent was obtained from parents or legal guardians of each patient. Clinical information was collected from the patients’ hospital records and their family members.
The Clinical Setting
The Danish Epilepsy Center (DEC), Filadelfia, is the only tertiary hospital in Denmark that is specialized in the treatment of epilepsy. The hospital has approximately 2600 annual outpatient consultations with children aged 0–18 years. The vast majority of patients referred to the DEC have intractable epilepsy, and thus, the patient population is biased towards the most difficult-to-treat cases.
Patient Analysis
This cross-sectional study was conceptualized in 2016 but was first initiated on May 1, 2019. The study population was children followed at DEC in 2015. During 2015, patients were referred to the DEC from all 18 Danish regional pediatric departments. Inclusion criteria for the study were as follows: (i) patients born in 2006–2011 (both years included) and followed at the DEC from January 1 to December 31, 2015; (ii) patients with a diagnosis of epilepsy (recurrent unprovoked seizures); and (iii) study consent provided by caregivers. Exclusion criteria were as follows: (i) a non-genetic etiology that would fully explain the seizures (e.g., hypoxic ischemic encephalopathy (HIE), perinatal stroke, or meningitis) and (ii) lack of study consent. Patients reaching a satisfying genetic diagnosis prior to referral to DEC and who met the inclusion criteria were also included. Patients fulfilling the study criteria were approached electronically in June 2019 and offered to be included in the study; non-responders were electronically approached a second time in November 2020.
Exome sequencing was performed from August 2019 to June 2021 and single nucleotide polymorphism (SNP) array analysis was carried out from August 2021 to December 2021. When possible, treatments were adjusted based on the genetic diagnosis. This was carried out continuously until December 2021; the effect of treatment adjustment on seizure burden was evaluated in February 2022. At this point, the burden of seizures was compared to the burden at time of reaching a genetic diagnosis.
The records of included patients were systematically reviewed for data on seizure type(s), epilepsy type, EEG, epilepsy syndrome, cognitive skills, comorbidities, neuroimaging, surgery, preexisting genetic diagnosis, and medical treatment adjustments as a result of a genetic diagnosis.
Seizures and epilepsies were classified according to the International League Against Epilepsy (ILAE) 2017 position papers [12, 13]. Seizure types were classified as focal, generalized, or unknown while epilepsy types were classified as focal, generalized, combined focal and generalized, or unknown. Seizure types were determined by the treating epileptologist at the DEC, based on videos and parental descriptions in addition to ictal (video)EEG recordings. When possible, an electroclinical epilepsy syndrome was determined based on age at seizure onset, seizure and epilepsy types, (video)EEG recordings, and the neurodevelopmental trajectory. The diagnostic criteria used are available on the ILAE website http://www.epilepsydiagnosis.org and the ILAE 2022 position papers [14,15,16]. The term “developmental and epileptic encephalopathies” (DEE) embraced disorders where the developmental impairment is related to both the epileptiform activity and the underlying genetic etiology [12, 17, 18]. The term “early infantile developmental and epileptic encephalopathy” (EI-DEE) was used for patients categorized as either Ohtahara syndrome or early myoclonic encephalopathy [14]. “Genetic generalized epilepsy” was classified accordingly to the ILAE recommendations from 2022 [15, 19] and included “genetic epilepsy with febrile seizures plus” (GEFS +).
Global developmental delay (GDD) was based on both formal and functional assessments and classified according to the Diagnostic and Statistical Manual of Mental Disorders 5th edition [20]. GDD was classified as mild, moderate, severe, or profound by a pediatric neurologist (AB). The term N/A (not available) was used if the level of GDD was unknown or difficult to determine. Antiseizure medications (ASM) were considered to have efficacy if they resulted in > 50% reduction in seizures for a period longer than 6 months. Patients were classified as treatment-resistant if they failed to become and remain seizure-free for at least 6 months with adequate trials of two ASMs.
Variant Identification
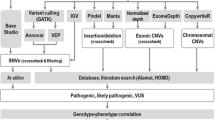
Some patients had already received a genetic diagnosis that could fully explain their symptoms. This diagnosis had been reached as part of a clinical workup undertaken prior to study inclusion (Figs. 1 and 2) using chromosomal karyotyping, comparative genomic hybridization (CGH) array, targeted panel sequencing with 45–600 genes, or whole exome sequencing (WES) (Fig. 2). We offered stepwise genetic testing to patients without a genetic diagnosis at study inclusion. This was done using DNA from peripheral blood lymphocytes, first with WES and then SNP microarray if WES did not provide a genetic diagnosis. Segregational analysis was performed if parental DNA was available. Figure 1 visualizes the different steps of the present study.

Overview of the different steps in the present study, starting with selection of the study cohort at the tertiary epilepsy center and followed by stepwise genetic testing and identification of disease-causing variants. The diagram illustrates how identification of genetic epilepsies can contribute to improved genetic counseling and precision therapy approaches

Detailed overview of the outcome of participants included in the present study. Full lines show the patients’ path through the study while dotted lines indicate procedures performed prior to study inclusion. Number of patients at each step is indicated by “n”. SNP, single nucleotide polymorphism; VUS, variant of unknown significance
WES
Singleton WES was initially performed, and segregational analysis was used for (likely) pathogenic and highly relevant variants of uncertain significance (VUSs). If a subject harbored three or more variants of interest, then parental WES was performed. DNA from peripheral blood lymphocytes was extracted using standard procedures. Library preparation and enrichment for WES was performed on PBL DNA following the standard TWIST procedure for the Twist Library Preparation EF Kit 1.0, Enzymatic Fragmentation, Twist Universal Adapter System, and TWIST Human Core Exome Kit (TWIST Bioscience, San Francisco, CA, USA). The TWIST libraries were loaded to a S1 flow cell and sequenced in a paired end 110 cycles on a NovaSeq 6000 system (Illumina, Inc., San Diego, CA, USA). For all included subjects, the genomic regions targeted by the respective enrichment design had an average coverage of > 100 reads, and > 97% were covered by at least 20 reads. The NGS method and the variant analysis pipeline was validated internally by analyzing five control samples per every 350 samples and externally by participating in the EMQN program.
Bioinformatics Following WES
Sequences were mapped to hg19, and variant calling was achieved with BWA-MEM and Freebayes. The variant filtering was performed using VarSeq software (Golden Helix, Bozeman, USA). Common SNPs with a variant allele frequency ≥ 25% and SNPs observed in more than one sample for each analyzed sample batch were filtered out. All possible modes of inheritance (sporadic de novo, dominant, recessive, X-linked) were analyzed using sensible minor population allele frequency cutoffs ≤ 0.01% in the Genome Aggregation Database (gnomAD) [21]. Genetic nonsynonymous missense/inframe insertions/inframe deletions/frameshift/stop_lost/stop_grain/5-prime UTR (< 10 base pair upstream)/splice site variants were evaluated through database searches such as dbSNP155, ClinVar, the Exome Aggregation Consortium database (ExAC), gnomAD, and Human Gene Mutation Database (HGMD) Professional [22]. Missense variants were also submitted to prediction softwares such as PolyPhen2 [23], SIFT [24], MutationTaster [25], MutationAssessor [26], FATHMM or FATHMM MKL Coding [27], CADD [28], and pathogenic variant enriched regions viewer while splice site variants were evaluated by the PWM, MaxEntScan, GeneSplicer, and NNSplice prediction tools. Variants analyzed under a dominant inheritance model that were observed more than three times in ExAC [29] and gnomAD [21] were considered too common and discarded. Potentially pathogenic variants were validated through conventional Sanger sequencing and, if possible, parents were included for segregation analysis.
Criteria for Pathogenicity of Rare Variants Detected by WES
Variants were classified for pathogenicity using the 2015 American College of Medical Genetics (ACMG) and Genomics Guidelines [30]. The web-based tool WInterVar (https://wintervar.wglab.org) was used to classify missense variations according to the ACMG criteria. A test was considered diagnostic when the subject was found to have one or two (likely) pathogenic variants in a single gene, depending on the mode of inheritance. Variants were classified as pathogenic if they (1) caused nonsynonymous, splice-site altering, or truncating changes; (2) were predicted as damaging by three or more prediction software programs; (3) were not present in controls in the ExAC or gnomAD control cohorts [21]; (4) had previously been classified as (L)PATH in either ClinVar [31] or HGMD; (5) were de novo changes or were inherited from an unaffected mosaic parent or an affected parent; or (6) showed a strong and specific correlation between the gene and the patient’s phenotype. We used Sanger sequencing to confirm variants and to perform segregation analysis.
In case of genes not yet linked to a human disorder, a VUS was considered highly relevant if (1) the variant was de novo and either extremely rare or never reported before in gnomAD [21] or the patient was compound heterozygous, homozygous, or hemizygous for a variant never reported in homo- or hemizygous form in gnomAD; (2) in silico predictions supported pathogenicity or the amino acid position in question was highly conserved in mammals and evolutionary more distant species, suggesting that the position does not tolerate variation; (3) animal models showed phenotype similar to the human subjects; and/or (4) in vitro studies revealed a pivotal role of the gene in the growth, differentiation, and/or function of neurons.
SNP Array
Patients genetically unsolved after WES were investigated using Illumina GSA V2 SNP-array of genomic DNA. CNVs were called by the PennCNV [32] version 1.0.5. PennCNV is a hidden Markov model (HMM)-based CNV calling algorithm that incorporates LogR ratio, B-allele frequency, population allele frequency, and the distance between adjacent SNPs into the HMM model. We used the joint CNV calling algorithm for trios and the standard CNV calling algorithm for single individuals, duos, and quartets. CNV calls with fewer than 30 consecutive SNPs (disregarding multi-allelic calls) were excluded. We visually inspected de novo CNVs as well as CNV where inheritance was unknown as both parents were not available for genetic testing. Visual inspection was done depicting LogR ratio and the B-allele frequency by genomic positional mapping on hg 38. We excluded CNVs in which general fluctuation in LogR or B-allele frequency was inconsistent with that of a true CNV.
CNVs were classified as either pathogenic, VUS, or benign according to the AMCG guidelines [33]. A CNV was considered pathogenic if (1) it occurred de novo, (2) it harbored one or more known epilepsy genes, (3) the proband’s phenotype was similar to published cases, and (4) it was absent or rarely reported in public CNV databases. If a known epilepsy gene was not present, additional factors were taken into consideration such as gene function and tissue expression, established haploinsufficiency or triplosensitivity, and the phenotype of knockout animal model.
Statistical Analyses
Quantitative statistics were analyzed using IBM SPSS version 24. Two-sided T-test was used to determine the association between age at seizure onset and the ability to achieve a genetic diagnosis. P value of 0.05 was considered significant. χ2 analyses were used to explore differences in degree of (i) cognitive impairment between genetically solved and unsolved patients and (ii) diagnostic yield of genetic testing based on age at seizure onset.
Results
We identified 357 patients who were born in 2006–2011 and followed at DEC in 2015. Figure 2 gives an overview of the flow of these patients through the present study. Twenty-seven patients (7.5%) turned out not to have epilepsy, and 35 (11%) had epilepsy due to acquired brain damage; both these groups were excluded from further analysis. Within the 35 patients with presumed acquired brain damage, the majority were believed to have perinatal HIE (n = 20) while less common etiologies included neonatal hypoglycemia (n = 4), prematurity (n = 4), postnatal hypoxia (n = 3), brain tumor (n = 2), congenital herpes infection (n = 2), and fetal alcohol syndrome (n = 1). We considered the remaining 295 children to have a potential monogenic epilepsy and offered them genetic testing as part of the present study. Parents of 188 patients from 186 unrelated families were willing to participate and gave consent for study inclusion and genetic testing as part of this study.
The clinical characteristics of these 188 patients are given in Table 1. The male to female ratio was 1.2:1. Of the 188 patients, 140 (74%) had some degree of developmental or cognitive impairment ranging from mild to profound (Table 1). The most common epilepsies were focal epilepsies (49/188; 26%), multifocal DEEs (36/188; 19%), and electroclinical syndromes such as epileptic spasms syndrome (41/188; 22%) and Dravet syndrome (20/188; 11%) (Table 2). Associated seizure types were either focal or combined focal and generalized seizures. Of the genetically tested patients, only 8.5% (16/188) were categorized as generalized epilepsies as they exclusively experienced generalized seizures with generalized ictal EEG discharges (Table 2). These included nine patients with GEFS + , four with childhood absence epilepsies (CAE), one patient with myoclonic absence epilepsy, and six patients with a generalized epilepsy that could not be further classified.
At study inclusion, 86/188 patients already had a genetic diagnosis that could fully explain their symptoms; 69 patients had a SNV, 16 had a CNV, and one patient had a chromosomal aberration (Supplementary Table 1). These diagnoses had been established during prior clinical workup (Figs. 1 and 2) using primarily gene panel sequencing (in 124/188 patients), followed by karyotyping, CGH-array, or WES in a minority of patients (Fig. 2). Although these 86 patients were included in the study, they did not undergo further genetic testing.
The 102/188 genetically unsolved patients underwent WES, comprising singleton WES in 89 patients and trio sequencing in 13 patients. This approach identified a (likely) pathogenic variant in eight patients (CWF19L1, IQSEC2, IRF2BPL, KCNMA1, POU3F3, and STAMBP) and a highly relevant VUS in seven patients (ADGRL1, CELSR1, CUL4B, KCNH5, NEXMIF, SLITRK2, and TRA2B) (Table 3). SNP array was performed in the remaining 87 patients and revealed neither (likely) pathogenic nor any highly relevant CNVs.
Next-generation sequencing approaches (pre-study panels and study-WES) thus identified a clinically relevant SNV or indel explaining the clinical picture in 77 patients, in addition to the highly relevant indel or SNV of unknown significance detected in seven patients. This encompassed variants across 41 genes (Table 3), and none of the patients harbored a dual genetic diagnosis (Table 3). The most common genetic causes were pathogenic variants in SCN1A causing Dravet syndrome or TSC2 causing tuberous sclerosis. Genes encoding ion channels were commonly affected, and variants in these genes explained the phenotype in 34 of the 101 patients with a putative genetic diagnosis. Only one of the diagnosed patients (ID-36) had an inborn error of metabolism related to pyridoxine 5'-phosphate oxidase deficiency.
The highest diagnostic yield based on electroclinical syndromes was in patients with EI-DEE (100%), Dravet syndrome (90%), multifocal DEE (66%), and epileptic spasms syndrome (61%) (Table 2). The numbers were lower in groups with developmental and/or epileptic encephalopathy with Spike-Wave activation in sleep (40%) and epilepsy with myoclonic-atonic seizures (EMA) (37%). There was a significant difference in diagnostic yield based on age at seizure onset (p < 0.001). The yield was highest if seizures started before 2 months of life (93%) and reached 59% in patients having first seizures between 2 months and 2 years of life. If onset of first seizure was between 2 and 9 years of life, the yield fell to 29% (Table 2). Of the 16 patients with a genetic generalized epilepsy, nine were classified as GEFS + while four had a childhood absence epilepsy (CAE). Of those with GEFS + , 55% (5/9) achieved a genetic diagnosis due to variants in SCN1A (n = 2), SCN1B (n = 1), GABRD (n = 1), and CUL4B (n = 1) while one patient with CAE was found to harbor a likely pathogenic variant in SLC6A1 (Supplementary Table 1).
Amongst the presumed genetically solved cases, causative variants were almost exclusively SNVs and CNVs as only a single patient had a complex chromosomal rearrangement. Seventy-eight percent of these patients had variants consistent with an autosomal dominant inheritance, 13% with an X-linked inheritance, and 6% with an autosomal recessive inheritance. Amongst the 84 patients with SNVs or indels, the most common variant types were missense variants (56%), followed by 22% frameshift variants, 18% splice-altering variants, and 4% inframe deletions. Larger CNVs were identified in 16 patients and included deletions/duplications that affected a number of genes. The complex chromosomal rearrangement was caused by a ring chromosome 20 detected in a girl (ID-101) with profound DD and cognitive impairment and treatment-resistant early-onset atypical absence seizures that often progressed into status epilepticus. Supplementary Table 1 shows the genetic variants and associated clinical phenotypes at individual level.
After dividing patients into those who were genetically solved and those who were not, we found that the proportion of patients with developmental delay or cognitive impairment was 89% in the genetically solved group and 56% in the unsolved group. Development/cognition was often more severely affected in the genetically solved group but was often within normal boundaries in the unsolved group (Table 1). In the solved group, 90% (91/102) had an impairment and this was classified as severe-profound in 47 cases; in the unsolved group, 44% (38/86) were not reported to have an impairment. These represented statistically significant differences between the two groups (p < 0.001, Table 1). We also found significant differences between the two groups regarding presence of febrile seizures or status epilepticus (p = 0.002 and p < 0.001, respectively, Table 1). There was no significant difference between the two groups in terms of treatment resistant seizures (p = 0.787, Table 1) and also the frequency of brain MRI abnormalities, and the overall frequency of such abnormalities was around 32% (61/188) (p = 0.298, Table 1). The distribution of epilepsy syndromes differed between the genetically solved group and the unsolved group (Table 2). Although the distribution of GGEs was fairly similar in the two groups, the genetically solved group had a larger proportion of DEEs (63%, 78/123) and fewer focal (non-DEE) epilepsies (32%, 16/49).
For the full cohort of 188 patients, median age at seizure onset was 12 months (ranged from 1 day to 9 years of life). Median age at seizure onset was significantly lower in the group reaching a genetic diagnosis than in those who remained genetically unsolved (6 months vs 24 months, t(186) = 4.587, p = < 0.001) (Table 1). We also found a significantly higher proportion of patients with neurodevelopmental impairment of any kind in the solved group (χ2 (2188) = 58.18, p < 0.001).
Ultimately, (likely) pathogenic variant(s) were present in 94/188 patients and a highly relevant VUS was found in 7/188 patients. Of these 101/188 patients, 53 were potentially eligible for precision therapy approaches (Supplementary Table 1). Treatment was adjusted according to the genetic findings in 32/53 (60%). More than 50% seizure reduction was reported in 30/32 (93%) but only 4 of these 30 patients became seizure-free. Everolimus in the treatment of mTORopathies and fenfluramine in the treatment of Dravet Syndrome were the most commonly used precision therapies (Supplementary Table 1). In 12/53 (40%) patients, satisfactory seizure control was obtained prior to genetic diagnosis, thus preventing further treatment adjustment; however, the ASMs used are not considered precision therapy for the underlying genetic condition. An overview of these ASMs is available in Supplementary Table 1; they include levetiracetam, valproate, stiropentol, ethosuzimide, vigabatrin, and four different sodium channel blockers (oxcarbamazapine, lamotrigine, zonisamide, and rufinamide). In this context, sodium channel blockers were effective in five patients with a disease-causing variant in DEPDC5, NPRL3, PIGN, SLC6A1, and TSC2, respectively. Levetirazetam and valproate were most commonly used either alone or in combination with the other ASMs. Table 4 shows number of patients with specific genetic disorders, how often therapy was adjusted based on the genetic diagnosis and the efficacy of the therapeutic adjustment. Further clinical details on a patient specific level are available in Supplementary Table 1.
Discussion
Of the 357 patients, 101 (28%) reached a genetic result that prevented further genetic testing, corresponding to 28% of the entire cohort. We classified the variant(s) found in 94 patients as (likely) pathogenic according to the ACMG guidelines [30], while seven had a highly relevant SNV/indel. SNP-array analysis did not identify a genetic diagnosis for any of the participants. Comparing the group of genetically solved patients (101/188, 54%) with those who remained unsolved (87/188, 46%), we found significant differences in median age at seizure onset (p < 0.001), presence of febrile seizure (p = 0.002), status epilepticus (p < 0.001), and degree of developmental and/or cognitive impairment (Table 1). This difference is likely caused by the higher proportion of DEEs in the solved group (63%, 78/123) compared to the unsolved group (37%, 45/123). In contrast, the unsolved group had more patients with focal (non-DEE) epilepsies (68%, 33/49) compared to the solved group (32%, 16/49), and such epilepsies are more likely to have a more complex genetic background [34]. The diagnostic yield was particularly high amongst patients with seizure onset before the second year of life. Causative variants were almost exclusively SNVs/indels (82%) and CNVs (17%). Based on the 94 patients with a (likely) pathogenic variant(s) and the seven patients with a highly relevant VUS, precision therapy was available in 53 (52%) patients but was implemented in only 32/53 (60%). Satisfactory seizure control using alternative treatments was obtained prior to genetic diagnosis in 12/53 (40%) patients, thus preventing further treatment adjustments. Precision therapy was efficient in 30/32 (93%) patients, but only 4/30 (12.5%) became seizure-free.
We found that SCN1A or TSC2 were the most commonly affected genes, causing a clinical spectrum ranging from GEFS + to Dravet syndrome in 18 patients (SCN1A) and tuberous sclerosis in 10 patients (TSC2). Although less frequent, pathogenic variants in ten genes, including PCDH19, SCN2A, SCN8A, CACNA1A, DEPDC5, GABRB3, and SLC6A1, were identified in 2–4 patients each, while disease-causing variants in 29 genes were only detected in single patients. In comparison, the Scottish population study [9] reported PRRT2, SCN1A, and KCNQ2 to be the most prevalent genes followed by SLC2A1, CDKL5, PCDH19, SLC6A1, DEPDC5, CACNA1A, KCNA2, and KCNQ3. These findings are more representative of the epilepsies presenting at a general pediatric department and not at a tertiary epilepsy center. PRRT2 causes self-limiting and treatable infantile-onset epilepsies [18] while KCNQ2 is associated with self-limiting/benign neonatal-onset seizures as well as neonatal onset DEEs [18]. In the Scottish study [9], only 2/10 patients with a pathogenic variant in KCNQ2 were diagnosed with a DEE; this is compatible with our findings as we only found a single patient with KCNQ2-related DEE. Self-limiting neonatal or infantile seizures are not expected to be followed at a tertiary epilepsy center, and we found no patients with PPRT2- and only one patient with KCNQ2-DEE. Precision therapy approaches used in the present study included mTOR-inhibitors in epilepsies caused by pathogenic variants in the mTOR pathway such as TSC1, TSC2, DEPDC5, NPRL2, and NPRL3 [8]; repurposing of fenfluramine (a serotonin re-uptake inhibitor) in the treatment of Dravet syndrome [6]; pyridoxine in GPI anchoring disorders [35]; avoiding ASMs that may worsen seizure activity (i.e., sodium channel blockers in epilepsies due to loss-of-function (LoF) variants in SCN1A); or deploying sodium channel blockers in epilepsies caused by gain-of-function variants in SCN2A and SCN8A [7].
Our data support a role for early genetic testing to provide a diagnosis that can enable personalized therapies. However, there is still a gap between reaching a genetic diagnosis and getting a disease-specific treatment. Often, a precision therapy does not exist or is limited to the option of avoiding certain drugs (e.g., sodium channel blockers in LoF SCN2A-related disorders or gaba-potentiating drugs in GoF GABA-related disorders) [18, 36]. Sometimes promising drugs, such as fenfluramine or ganaxolone, are in the horizon but remain difficult or impossible to prescribe. Ganaxolone reduces seizure frequency in CDKL5-related epilepsies [37] but is currently only approved by the Food and Drug Administration and not the European Medicines Agency. Fenfluramine is authorized in all EU countries but still needs to be approved in Denmark (unless a “compassionate use” permit is obtained from the Danish Medicines Agency). Finally, it must be pointed out that when available, the treatment often only targets seizures while leaving the developmental problems and comorbidities unchanged.
The obvious questions are as follows: How to bridge the gap and how do we develop therapies that not only target seizures but also tackle neurodevelopmental issues? A possible solution may come from promising therapies targeting DNA, RNA translation, and protein modulation. Such treatments are more likely to treat seizures as well as comorbidities since they are acting directly on the underlying pathomechanism causing the different phenotypic features [18, 38]. Gene therapies include DNA targeting treatments but also single-stranded antisense oligonucleotides (ASOs) that bind and alter RNA translation and ultimately protein expression [39, 40]. Several ASOs are in preclinical development in translational mice models including SCN1A- and SCN8A-encephalopathy and DS [41, 42] and MECP2 duplication syndrome [43]. Some have reached clinical trials, including an ASO designed to treat patients with DS by preventing inclusion of a poison exon thereby upregulating the will-type allele (https://clinicaltrials.gov/ct2/show/NCT04442295).
A recent meta-analysis showed that for diagnostic purposes the highest yield was in whole genome sequencing (WGS) (48%, 95% CI = 28–70%) followed by WES (24%, 95% CI = 18–30%), panel sequencing (19%, 95% CI = 16–24%), and array CGH (9%, 95% CI = 7–11%) [44]. Although any patient with treatment-refractory epilepsy could potentially benefit from genetic testing, it would probably be of most importance in those with seizures starting before 3 years of age, and in our cohort, a genetic diagnosis was reached in 86/137 (67%) of patients whose seizures started within this age. It would also likely benefit those with a family history of seizures or those with associated neurological deficit such as developmental and/or cognitive impairment. Several studies have shown that the overall diagnostic yield of targeted panels and WES is dependent on the epilepsy phenotype and age at seizure onset [44,45,46]. The highest yield is in DEEs (45, 46), and the lowest is in cohorts with focal epilepsy [44]. In 2016, Møller et al. [47] reported on 216 patients consecutively referred for genetic testing for epilepsies ranging from benign neonatal seizures to DEEs. Patients underwent genetic testing using a panel with 46 genes, and a presumed disease-causing variant was detected in 23% of all cases. Neonatal-onset epilepsies were associated with the highest rate of positive findings (57%), followed by 26% amongst those who started seizures between 2 months and 2 years of life, and only 14% in those with onset between 2 and 9 years of life [47]. Although the two cohorts from the current study are not directly comparable with this previous study, and our data may be biased as they are obtained from a tertiary epilepsy center, we found that an overall diagnostic yield of about 60% if seizures started within the first 2 years of life, and 29% when seizure onset was between the 2nd and 9th years of life. In the Møller et al. study [47], the yield for patients with genetic generalized epilepsies was 17% compared to 43% (7/16, Table 1) found in the present study; the genetically solved generalized epilepsy syndromes in our study included GEFS + (5 patients), myoclonic absences (1 patient), and childhood absence epilepsy (1 patient). This was mostly attributed to the high numbers of children with GEFS + in the present study as this electroclinical syndrome is presumed to have an underlying monogenic architecture [18]. The number of CNVs in our study was approximately 9% while the diagnostic yield of panel combined with WES reached 44% (84/188); the high number of mendelian inherited disorders was likely attributed by the high number of DEEs, as these are more likely to have a monogenic etiology [48]. Although our study design was not able to compare the yield of multigene panels to that of WES, we found that WES detected (likely) pathogenic variant(s) in nine patients (9/188; 4.8%) who were genetically unsolved at study inclusion despite having been tested with a multigene panel.
WGS and WES are the most comprehensive tests in genetic diagnostics of monogenic epilepsies, and WGS is currently considered the ultimate diagnostic tool. Although increasingly considered as first-line tests, some clinicians still consider exome and genome sequencing strategies as a last resort [49]. Early use of these sequencing approaches enable a precise genetic diagnosis and can potentially end the diagnostic odyssey that many patients face. On average, a molecular cause of complex neurological disorders of suspected genetic origin in the field of pediatric neurology is determined after three misdiagnoses and 16 physician visits over several years [50, 51]. A clear benefit of WES/WGS is the possibility of reanalyzing the data annually or every second year, especially in an era when new epilepsy genes are constantly being identified [45]. WGS has a higher diagnostic yield compared to WES [2, 44], firstly, due to a more evenly distributed coverage depth [52], and secondly, because WGS captures SNVs and indels in coding and noncoding regions, CNVs, and chromosomal alterations including inversions and transpositions [45]. A clear disadvantage is the large volume of data provided by these methods, most of which can be misleading or useless.
One of the strengths of the current study was the availability of parental samples and the access to clinical data on probands and their families. This meant we could ensure that variants of interest occurred de novo and could also compare the phenotypes with those described for the implicated genes. Furthermore, all genetically unsolved patients underwent a stepwise genetic investigation; the rationale behind this approach is that although WES is a powerful diagnostic tool, it might miss CNVs that are otherwise easily detectable by microarray. Chromosomal karyotyping and FMR1 testing were not included in the genetic workup of the present study as their diagnostic yield in a tertiary epilepsy center is limited to 2% and 0.6%, respectively [53]. A limitation of our study was that a trio WES was not offered to all patients. We suspect that this could have increased the likelihood of detecting further potential de novo disease-causing variants.
Of the 35 patients with a presumed acquired brain damage, 20 where believed to have a perinatal HIE and were subsequently not offered genetic testing. A subset of patients with perinatal HIE may actually have an underlying genetic diagnosis. A recent study exome sequenced 113 encephalopathic neonates with an acute peripartum/intrapartum event or Apgar score ≤ 7 and found that 19 patients carried disease causing genetic variations [54]. If we extrapolate these results to our study, less than five patients with HIE may have an underlying monogenic disorder; although, this is important for individual patients, it is unlikely to affect our overall results.
We considered that 295 children could potentially have a monogenic epilepsy and offered them genetic testing by electronically approaching their parents; despite two attempts to achieve consent parents of 107 patients never responded. The first study invitation was sent in 2019 and at that time patients were 10–13 years old; it is possible that many parents, after enduring a long diagnostic and therapeutic journey, were less anxious for the future and had settled with not knowing the underlying cause of the illness. Some parents might previously have received negative or uncertain results, leaving them unwilling to pursue updated genetic testing. Some patients may have had a milder and treatable epilepsy and parents might have reached a point where they felt, they no longer needed genetic testing in order to provide the best care for their child, to get information for their family, and to gather information for childbearing decisions.
Although the yield of genetic testing in difficult-to-treat childhood-onset epilepsies has been extensively investigated [2, 44], the impact of genetic testing on therapeutic decision-making has been less studied [9, 11, 53, 55]. Study results are not directly comparable due to varying study designs and study cohorts, but all papers offer an estimate of how often genetic testing enables a precision therapy approach, i.e., 44% [11], 47% [53], 63% [55], and 80% [9] of genetically solved patients. The high availability of precision therapy in the Symonds et al. study [9] was due to the high number of patients with self-limiting familial seizures caused by variants in PRRT2 and KCNQ2. The impact of genetic diagnosis on precision therapy implementation and efficacy has only been studied once; although a targeted therapy was available in approximately half of the patients, a treatment change was prompted in 36% (106/293) and was effective in only 32% (34/106) [11]. A younger age at genetic testing was also associated with a more favorable seizure outcome, suggesting that the chance of improving outcome could diminish with increasing age [11]. An age effect was also reported in precision medicine studies where a drug-repurposing approach showed that better results and even seizure freedom could be achieved when the targeted treatment was started in early childhood rather than adolescence or adulthood [56]. Before we can fully understand the failure of some precision therapies, we first need to explore their efficacy as first-line treatments in prospective and preferably nationwide studies once a genetic diagnosis has been established.
Data Availability
Anonymized data not published in this article will be made available by request from any qualified investigator.
References
Thomas RH, Berkovic SF. The hidden genetics of epilepsy-a clinically important new paradigm. Nat Rev Neurol. 2014;10(5):283–92.
Stefanski A, Calle-Lopez Y, Leu C, Perez-Palma E, Pestana-Knight E, Lal D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: a systematic review and meta-analysis. Epilepsia. 2021;62(1):143–51.
Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586(7831):757–62.
Johannessen Landmark C, Potschka H, Auvin S, Wilmshurst JM, Johannessen SI, Kasteleijn-Nolst Trenite D, et al. The role of new medical treatments for the management of developmental and epileptic encephalopathies: Novel concepts and results. Epilepsia. 2021;62(4):857–73.
Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18(1):41–58.
Sullivan J, Scheffer IE, Lagae L, Nabbout R, Pringsheim M, Talwar D, et al. Fenfluramine HCl (Fintepla((R)) ) provides long-term clinically meaningful reduction in seizure frequency: analysis of an ongoing open-label extension study. Epilepsia. 2020;61(11):2396–404.
Musto E, Gardella E, Moller RS. Recent advances in treatment of epilepsy-related sodium channelopathies. Eur J Paediatr Neurol. 2020;24:123–8.
Schubert-Bast S, Strzelczyk A. Review of the treatment options for epilepsy in tuberous sclerosis complex: towards precision medicine. Ther Adv Neurol Disord. 2021;14:17562864211031100.
Symonds JD, Zuberi SM, Stewart K, McLellan A, O’Regan M, MacLeod S, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019;142(8):2303–18.
Peng J, Pang N, Wang Y, Wang XL, Chen J, Xiong J, et al. Next-generation sequencing improves treatment efficacy and reduces hospitalization in children with drug-resistant epilepsy. CNS Neurosci Ther. 2019;25(1):14–20.
Balestrini S, Chiarello D, Gogou M, Silvennoinen K, Puvirajasinghe C, Jones WD, et al. Real-life survey of pitfalls and successes of precision medicine in genetic epilepsies. J Neurol Neurosurg Psychiatry. 2021.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21.
Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475–82.
Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022.
Wirrell EC, Nabbout R, Scheffer IE, Alsaadi T, Bogacz A, French JA, et al. Methodology for classification and definition of epilepsy syndromes with list of syndromes: Report of the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022.
Specchio N, Wirrell EC, Scheffer IE, Nabbout R, Riney K, Samia P, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022.
Scheffer IE, Liao J. Deciphering the concepts behind “Epileptic encephalopathy” and “Developmental and epileptic encephalopathy.” Eur J Paediatr Neurol. 2020;24:11–4.
Bayat A, Bayat M, Rubboli G, Moller RS. Epilepsy syndromes in the first year of life and usefulness of genetic testing for precision therapy. Genes (Basel). 2021;12(7).
Hirsch E, French J, Scheffer IE, Bogacz A, Alsaadi T, Sperling MR, et al. ILAE definition of the idiopathic generalized epilepsy syndromes: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022.
Diagnostic and statistical manual of mental disorders American Psychiatric Association. 2013.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD((R))): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139(10):1197–207.
Yu H, He X, Liu X, Zhang H, Shen Z, Shi Y, et al. A novel missense variant in cathepsin C gene leads to PLS in a Chinese patient: a case report and literature review. Mol Genet Genomic Med. 2021:e1686.
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6.
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17): e118.
Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31(10):1536–43.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Landrum MJ, Kattman BL. ClinVar at five years: Delivering on the promise. Hum Mutat. 2018;39(11):1623–30.
Wang K, Chen Z, Tadesse MG, Glessner J, Grant SF, Hakonarson H, et al. Modeling genetic inheritance of copy number variations. Nucleic Acids Res. 2008;36(21): e138.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22(2):245–57.
Leu C, Stevelink R, Smith AW, Goleva SB, Kanai M, Ferguson L, et al. Polygenic burden in focal and generalized epilepsies. Brain. 2019;142(11):3473–81.
Tanigawa J, Nabatame S, Tominaga K, Nishimura Y, Maegaki Y, Kinosita T, et al. High-dose pyridoxine treatment for inherited glycosylphosphatidylinositol deficiency. Brain Dev. 2021.
Absalom NL, Liao VWY, Kothur K, Indurthi DC, Bennetts B, Troedson C, et al. Gain-of-function GABRB3 variants identified in vigabatrin-hypersensitive epileptic encephalopathies. Brain Commun. 2020;2(2):fcaa162.
Knight EMP, Amin S, Bahi-Buisson N, Benke TA, Cross JH, Demarest ST, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2022;21(5):417–27.
McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15(3):304–16.
Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19(10):673–94.
Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14(1):9–21.
Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12(558).
Lenk GM, Jafar-Nejad P, Hill SF, Huffman LD, Smolen CE, Wagnon JL, et al. Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann Neurol. 2020;87(3):339–46.
Shao Y, Sztainberg Y, Wang Q, Bajikar SS, Trostle AJ, Wan YW, et al. Antisense oligonucleotide therapy in a humanized mouse model of MECP2 duplication syndrome. Sci Transl Med. 2021;13(583).
Sheidley BR, Malinowski J, Bergner AL, Bier L, Gloss DS, Mu W, et al. Genetic testing for the epilepsies: A systematic review. Epilepsia. 2021.
Moller RS, Hammer TB, Rubboli G, Lemke JR, Johannesen KM. From next-generation sequencing to targeted treatment of non-acquired epilepsies. Expert Rev Mol Diagn. 2019;19(3):217–28.
Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18(9):898–905.
Moller RS, Larsen LH, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol. 2016;7(4):210–9.
Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. 2017;101(5):664–85.
Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Correction: Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2020;22(10):1731–2.
Turro E, Astle WJ, Megy K, Graf S, Greene D, Shamardina O, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583(7814):96–102.
Vissers L, van Nimwegen KJM, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017;19(9):1055–63.
Burdick KJ, Cogan JD, Rives LC, Robertson AK, Koziura ME, Brokamp E, et al. Limitations of exome sequencing in detecting rare and undiagnosed diseases. Am J Med Genet A. 2020;182(6):1400–6.
Zacher P, Mayer T, Brandhoff F, Bartolomaeus T, Le Duc D, Finzel M, et al. The genetic landscape of intellectual disability and epilepsy in adults and the elderly: a systematic genetic work-up of 150 individuals. Genet Med. 2021.
Xiao TT, Yang L, Wu BB, Peng XM, Wang HJ, Cheng GQ, et al. Genotype and phenotype analysis of neonates with neonatal encephalopathy complicated with perinatal hypoxic event. Zhonghua Er Ke Za Zhi. 2021;59(4):280–5.
Hoelz H, Herdl C, Gerstl L, Tacke M, Vill K, von Stuelpnagel C, et al. Impact on clinical decision making of next-generation sequencing in pediatric epilepsy in a tertiary epilepsy referral center. Clin EEG Neurosci. 2020;51(1):61–9.
Hedrich UBS, Lauxmann S, Wolff M, Synofzik M, Bast T, Binelli A, et al. 4-Aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci Transl Med. 2021;13(609):eaaz4957.
Acknowledgements
The authors would like to thank the participants and their families for their participation in our research. Illustrations were created with BioRender.com. AB received financial support through the Disposition foundation (D51), the A.P. Møller foundation (20-L-0241), the Beckett foundation (21-2-7182), and the Grosserer L.F. Foghts foundation (22.030). The authors thank Dr. Claire Gudex for her contribution to editing the final draft of the article. The Danish Cytogenetic Central Registry study group consists of the following members: Mathilde Faurholdt Lauridsen, Christina Fagerberg, Tina Duelund Hjortshøj, Iben Bache, and Ida Vogel.
Author information
Authors and Affiliations
Contributions
Conceptualization, AB and RSM; methodology, AB, RSM, and TFH; software, CDF, IN, and TRT; formal analysis, AB and CDF; data curation, AB, CDF, and TRT; writing—original draft preparation, AB; writing—review and editing, AB, CDF, TRT, IN, TFH, GR, and RSM; supervision, GR, RSM, and TFH; project administration, AB; funding acquisition, AB and RSM. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosure
CDF and IN were employed by the company Amplexa. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bayat, A., Fenger, C.D., Techlo, T.R. et al. Impact of Genetic Testing on Therapeutic Decision-Making in Childhood-Onset Epilepsies—a Study in a Tertiary Epilepsy Center. Neurotherapeutics 19, 1353–1367 (2022). https://doi.org/10.1007/s13311-022-01264-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01264-1