
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that primarily affects motor neurons and causes muscle atrophy, paralysis, and death. While a great deal of progress has been made in deciphering the underlying pathogenic mechanisms, no effective treatments for the disease are currently available. This is mainly due to the high degree of complexity and heterogeneity that characterizes the disease. Over the last few decades of research, alterations to bioenergetic and metabolic homeostasis have emerged as a common denominator across many different forms of ALS. These alterations are found at the cellular level (e.g., mitochondrial dysfunction and impaired expression of monocarboxylate transporters) and at the systemic level (e.g., low BMI and hypermetabolism) and tend to be associated with survival or disease outcomes in patients. Furthermore, an increasing amount of preclinical evidence and some promising clinical evidence suggests that targeting energy metabolism could be an effective therapeutic strategy. This review examines the evidence both for and against these ALS-associated metabolic alterations and highlights potential avenues for therapeutic intervention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by selective degeneration of upper and lower motor neurons, leading to muscle atrophy, paralysis, and death [1]. ALS is a remarkably progressive disease, with death occurring typically 3–5 years from symptom onset, usually due to respiratory failure [1]. Furthermore, no therapeutics capable of extending survival more than a few months are currently available. This is mainly due to the high level of complexity and heterogeneity that characterizes ALS. Most cases (90–95%) are sporadic, meaning they present with no family history of the disease and with little to no indication of the underlying cause [1]. The remaining 5–10% present with family history and are termed familial [1]. These are often caused by genetic mutations, and indeed, mutations in dozens of different genes have been found to independently cause ALS, the most common of which (accounting for over 50% of familial cases) is a G4C2 hexanucleotide repeat expansion mutation in the C9orf72 gene [1]. Others include mutations in superoxide dismutase 1 (SOD1), TAR DNA binding protein (TAR-DBP), and fused in sarcoma (FUS) [1]. Interestingly, the proteins these genes encode often have wildly different cellular roles or functions. Furthermore, a subset of patients — especially those with the C9orf72 repeat expansion — also present with cognitive impairment and are co-diagnosed with frontotemporal dementia (FTD) [2]. Despite these added layers of complexity, the presence of a common disease phenotype among all disease subtypes — the selective degeneration of upper and lower motor neurons — suggests that distinct pathogenic mechanisms of these different ALS subtypes may eventually converge, or at least share certain features. Identifying these mechanistic commonalities or points of convergence will enhance the field’s understanding of this disease. It will also be critical in determining therapeutic agents that will be efficacious across all forms of ALS.
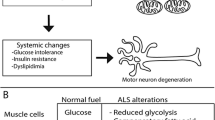
One such common disease feature appears to be energy imbalance. Neurons (and motor neurons in particular) are highly energy-dependent, mainly due to the high ATP cost associated with neurotransmission and axonal transport (motor neurons) [3]. Therefore, motor neurons are highly vulnerable to any disruptions to the energy balance. Indeed, ALS is broadly characterized by several forms of energy imbalance, including (but not limited to) hypermetabolism, mitochondrial dysfunction, abnormal carbohydrate metabolism, insulin resistance, and dyslipidemia. Furthermore, these metabolic abnormalities tend to be associated with a worse disease outcome, suggesting that they are involved in the pathogenic mechanism and may be a primary driver of disease. In many cases, these metabolic abnormalities precede symptom onset [4,5,6], indicating that a critical window for therapeutic intervention exists. This review examines the evidence both for and against each of these energy imbalances and highlights potential avenues for therapeutic intervention.
General Energy Imbalances: Low Body Mass Index (BMI) and Systemic Hypermetabolism
One of the most apparent energy imbalances in ALS patients is low body mass index (BMI) and weight loss [7,8,9,10]. While this could be explained by the loss of lean body mass due to disease-related muscle atrophy, other factors are likely at play (Fig. 1). For one, degeneration of bulbar muscles often leads to dysphagia, i.e., difficulty swallowing, which can make it difficult for patients to obtain sufficient nutrition and lead to weight loss [11]. However, remarkably, patients without dysphagia still exhibit severe weight loss [9, 12], which suggests the involvement of other factors. Patients’ difficulty consuming enough food is often exacerbated by a lack of appetite, a common ALS occurrence that worsens as the disease progresses [13, 14]. In support of this, a clear association is seen between lack of appetite, decreased caloric intake, and weight loss in patients [15]. Another potential factor is atrophy of the hypothalamus, which was recently identified in the brains of ALS patients (as measured by histological staining of post-mortem tissue and MRI of live subjects) [16,17,18]. The hypothalamus is the primary regulator of whole-body energy homeostasis, including feeding behavior and energy expenditure (among many other things) [19]. Thus, its atrophy could also decrease caloric intake and ultimately lead to weight loss. Indeed, a correlation has been identified between hypothalamic volume and BMI in ALS patients [16]. And finally, some ALS patients could be genetically predisposed towards weight loss. A recent meta-analysis of ALS GWAS datasets identified an association between several SNPs in the ACSL5 gene and ALS [20]. ACSL5 encodes a long-chain fatty-acid-coenzyme A ligase that plays a crucial role in lipid biosynthesis and fatty acid degradation [21]. Furthermore, mutations in ACSL5 have been associated with rapid weight loss in humans [21] and thus could contribute to weight loss in ALS.

Potential mechanisms in ALS patients leading to systemic hypermetabolism, low body mass index, and weight loss
Another form of energy imbalance consistently observed in ALS is systemic hypermetabolism [11, 22,23,24,25,26,27,28], simply defined as an excessive increase in resting energy expenditure (REE). For example, a recent study found that ALS patients had a daily metabolic rate of 1500 kilocalories (kcal) per day, compared to 1230 kcal per day in age-matched controls [28]. This, combined with the multitude of other energy imbalances outlined in this review, makes obtaining sufficient energy a truly daunting task for ALS patients. Furthermore, this phenomenon is also seen in the human mutant SOD1G93A mouse model of ALS, suggesting that it is intrinsic to the ALS disease mechanism [29]. It is worth noting that, with muscle being the highest energy-consuming tissue in the body, the loss of muscle mass that occurs in ALS would be expected to cause a decrease in REE, not an increase. Thus, the hypermetabolic phenotype is quite paradoxical — so what could explain it? There are several potential factors (Fig. 1). Firstly, it has been postulated that increased respiratory activity (deriving from the deterioration of diaphragmatic function) could contribute. However, in a study of 62 ALS patients, REE was not found to be associated with forced vital capacity [23], so this is likely not the case. It has also been suggested that muscle fasciculations, which require energy, could play a role. However, no correlation between fasciculations and REE has been identified in patients, either [23]. A third explanation is that mitochondrial dysfunction — which, as discussed at length in the next section, is widely seen in ALS patients and ALS disease models [30] — could decrease the efficiency of energy production and lead to hypermetabolism. Indeed, certain mitochondrial disturbances, including uncoupling of oxidative phosphorylation, have been shown to contribute to hypermetabolism [31]. Hypothalamic atrophy (discussed earlier in this section) could also be involved. And finally, cortical hyperexcitability — a well-established feature of ALS [32] — could increase energy requirements within the nervous system and lead to increased energy expenditure.
Regardless of how or why they occur, these energy imbalances do not bode well for patients. Many studies have linked low BMI to lower ALS-FRS score, faster disease progression, and worse survivability [10, 33,34,35,36,37,38], as well as an increased risk of developing ALS in the first place [39]. Furthermore, while not all patients are hypermetabolic, those that tend to have a worse disease outcome [27]. Conversely, overweight individuals have a lower likelihood of developing ALS, and when they do, they tend to have a better disease outcome [37, 40, 41]. Therefore, preserving patients’ energy balance appears essential for maximizing patient survival and may present an opportunity for therapeutic intervention. Indeed, implementing a high-calorie/high-fat diet is beneficial in both patients and animal models and will be discussed at greater length in the final section of this review.
Mitochondrial Dysfunction
Mammalian cells generate the bulk of their ATP via oxidative phosphorylation, which occurs exclusively in the mitochondrion [42]. During this process, a series of enzymatic reactions known collectively as the Kreb’s cycle facilitates the oxidation of the high-energy molecule acetyl-CoA (among other potential substrates) into carbon dioxide [42]. The energy released during this process converts NAD+ molecules into the higher-energy NADH molecules, which serve as the electron donor during the electron transport chain to fuel ATP synthesis [42]. The brain is one of the most highly ATP-dependent organs in the entire body, which renders the nervous system particularly vulnerable to mitochondrial perturbations or dysfunctions [3].
A growing body of literature suggests that various aspects of mitochondrial physiology are perturbed in ALS. When examined at high resolution using cryo-EM, postmortem samples from sporadic ALS patients tend to exhibit swollen and aggregated mitochondria in both the spinal cord and skeletal muscle [43,44,45,46,47,48]. Likewise, abnormal mitochondrial morphology has been consistently detected in several mouse models of familial ALS, including the SOD1G93A model [49,50,51,52], the TDP-43A315T model [53, 54], and the FUSP525L model [55]. Similar results have also been obtained with in vitro studies, with over-expression of human mutant SOD1 [56, 57], TDP-43 [58, 59], or FUS [60, 61] leading to mitochondrial damage; furthermore, both fibroblasts and iPSC-derived neurons from patients with the C9orf72 mutation show evidence of abnormal mitochondrial morphology [62, 63]. Interestingly, over-expression of wild-type TDP-43, or even just the C-terminal fragment of wild-type TDP-43, also leads to mitochondrial damage [58, 64, 65]. It should be noted that TDP-43 contains internal mitochondrial targeting sequences and has been found to localize to the mitochondria in patient neurons [48, 66] and overexpression systems [48, 58, 59, 64]. Mis-localization of TDP-43 to the cytoplasm — a hallmark of almost all forms of ALS [67] — could give TDP-43 greater opportunity to localize to and damage mitochondria, which could contribute to the mitochondrial dysfunction seen in sporadic cases.
Other ALS proteins have also been found to localize to and play critical roles within the mitochondria. In SOD1G93A transgenic animals, the ALS-causative SOD1G93A mutant protein accumulates in various compartments of the mitochondria [68,69,70,71]. Likewise, wild-type and ALS-linked mutant FUS proteins interact with mitochondria when overexpressed in a neuron-like cell line [60]. A recent study found that, under normal conditions, the C9orf72 protein localizes to the inner mitochondrial membrane (IMM), where it stabilizes assembly of respiratory chain complex I through its specific interaction with translocase of internal mitochondrial membrane domain containing 1 (TIMMDC1) [72]. Another study found that C9orf72 interacts with many other mitochondrial proteins, especially those localized to the IMM, highlighting its role as an IMM-localized mitochondrial protein [73]. And finally, poly-glycine-arginine (poly-GR) — one of the dipeptide repeats made from RAN translation of C9orf72-ALS linked G4C2 repeat expansion — was found to interact with mitochondrial ribosomal proteins [74]. Poly-GR also interacted with the mitochondrial contact site and cristae organization system (MICOS), impairing mitochondrial ion homeostasis and IMM structure [75].
In addition to mitochondrial morphological defects, alterations to mitochondrial function have also been reported in ALS and ALS model systems. Indeed, spectrophotometric enzymatic assays have demonstrated decreases in the specific activities of respiratory chain components in patient spinal cord samples [76, 77] and skeletal muscle cells [78,79,80]. Likewise, both SOD1G93A mice and cell culture models of mutant SOD1 overexpression consistently exhibit decreased mitochondrial respiration and oxygen consumption [51, 52, 69, 81, 82]. Similarly, the expression of TDP-43 in various cell types causes a reduction in respiratory chain complex activity [48, 58]. Interestingly, TDP-43 binds to and impairs the expression of mitochondrial-transcribed mRNAs encoding complex I subunits, which causes complex I disassembly and could potentially underlie the respiratory chain defects seen in TDP-43 models [66]. Coincidentally, haploinsufficiency in C9orf72 was also found to cause complex I disassembly, in this case via disruption of the interaction between C9orf72 and TIMMDC1, ultimately leading to impaired respiratory chain function and ATP production [72]. Therefore, the loss of complex I integrity is a common disease feature across several forms of ALS, although the specific mechanism by which this could lead to neurodegeneration remains unclear. Still, the potential importance of complex I integrity was highlighted by a 2012 study, which identified a correlation between lymphocyte complex I activity and patient disease progression [83], which suggests that mitochondrial function (and complex I function in particular) may be a key determinant of disease outcome. It is worth noting that additional studies have obtained conflicting results, identifying either no difference or an increase in respiratory chain activity [84,85,86]. The reasons for the discrepancy between these studies remain unknown.
Impairments to respiratory chain complex activity would be expected to reduce the driving force for ATP production — i.e., the transmembrane proton gradient — which can be reflected as a decrease in mitochondrial membrane potential (MMP) and a decrease in ATP production [42]. Indeed, there is evidence that both MMP and ATP production are compromised in ALS. Overexpression of various ALS-associated proteins, including TDP-43, SOD1, and FUS, as well as loss of C9orf72, has all been found to decrease MMP [57, 58, 60, 64, 72] and ultimately reduce ATP levels [48, 57, 87]. Furthermore, C9orf72 patient iPSC-derived neurons cultured in galactose (to force reliance on oxidative metabolism) or treated with glutamate (to induce hyperexcitability, which is highly energy-dependent) were found to have reduced ATP levels compared to similarly treated iPSC-derived neurons from healthy control subjects [72]. Lymphocytes from ALS patients were also found to have a reduced ATP:ADP ratio [83].
In addition to causing an energy imbalance, defective mitochondrial respiration can lead to several other downstream consequences, one of which is oxidative stress [88]. Under normal conditions, oxidative phosphorylation is the most prominent cellular source of reactive oxygen species (ROS), including superoxide (O−2) and hydrogen peroxide (H2O2) [88]. When produced at “normal” levels, these molecules serve critical signaling roles [88]. However, when made excessively, which can be caused by inhibition of the respiratory chain (as is seen in ALS), they can cause oxidative damage to various cellular components, including the respiratory chain enzymes and mitochondrial DNA (mtDNA) [89]. Indeed, evidence of oxidative damage has been identified in ALS patient postmortem tissue and biofluids, as well as in various animal models, and has been extensively studied (reviewed in [90]). Accordingly, alterations to mtDNA, including diminished overall levels and an increased frequency of mutation to mtDNA, have also been found in patients [77, 91]. However, this is just one of many potential downstream consequences of ROS overproduction. Furthermore, Edaravone, one of just two FDA-approved drugs for ALS treatment, functions as a free radical scavenger and thus works to mitigate oxidative damage [92].
Alterations to Glucose Metabolism
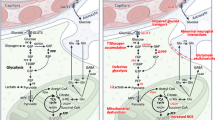
The brain is highly reliant on glucose as a source of energy. Indeed, although the brain constitutes less than ~2% of total body weight, it consumes ~20% of all glucose that enters the bloodstream [93]. Furthermore, there is strong evidence to suggest that cerebral glucose uptake is impaired in ALS patients. Glucose is imported into the nervous system at the blood–brain barrier — specifically, glucose in the bloodstream passes through GLUT1 transporters localized on the membrane of neurovascular endothelial cells [93]. In patients, the rate of cerebral glucose import can be approximated by infusing them with the radiolabeled glucose analog [18F]-2-deoxy-D-glucose (FDG) and then measuring its uptake into various regions of the brain via positron emission tomography (PET). Like glucose, FDG is taken up into the brain via GLUT1 and imported into brain cells by either GLUT1 or GLUT3, where it is then phosphorylated by hexokinase [94]. But then, unlike glucose, it cannot be further metabolized and subsequently accumulates in the cell [94]. Therefore, the accumulation of FDG in various cells or tissues, which can be measured by PET imaging, serves as a proxy for glucose utilization in that region. This technique was first applied to ALS patients in two studies performed in 1987 and 1992, both of which identified a decrease in FDG uptake throughout the cerebral cortex (as well as in some subcortical structures) of ALS patients compared to healthy controls [95, 96]. Two larger-scale studies confirmed these findings, one that included 195 patients and another 81 patients, both of which identified glucose hypometabolism throughout the cerebral cortex of ALS patients compared to healthy controls [97, 98]. Remarkably, discriminant analyses of these datasets could distinguish ALS patients from controls with up to 95% accuracy, suggesting that the pattern of cerebral glucose utilization could serve as a diagnostic or prognostic marker for ALS. Unsurprisingly, the SOD1G93A mouse model also exhibits a progressive decline of glucose metabolism during disease progression [99]. Furthermore, carriers of the ALS/FTD-linked C9orf72 repeat expansion mutation exhibit cortical glucose hypometabolism up to 10 years prior to symptom onset, suggesting that decreases in glucose uptake may be an early and critical disease modifier event, particularly in the case of the C9orf72 [5, 100].
While glucose uptake into cortical brain regions appears to be reduced in ALS, glucose uptake into peripheral regions may be increased. Indeed, mid-symptomatic SOD1G93A mice were recently found to have increased glucose uptake in several peripheral regions, including liver, skeletal muscle, adipose tissue, and others, which interestingly was found to occur in an insulin-independent manner [101]. In line with this, enhanced glucose metabolism in peripheral regions has also been identified in ALS patients, specifically in denervated forearm muscle [102]. The mechanisms underlying these tissue-specific alterations in glucose metabolism, whether increased or decreased, are unclear and require further investigation.
Evidence also suggests that glycolysis may be downregulated in ALS. Once within the nervous system, glucose is taken up by glia via GLUT1 or by neurons via GLUT3 and then either metabolized via glycolysis, which is the other main pathway (in addition to oxidative metabolism) through which mammalian cells can generate ATP [93], or converted to glycogen for storage. Downregulation of essential glycolytic genes, including PFKB3 (which encodes the rate-limiting glycolytic enzyme), has been identified in ALS patients’ motor cortex [103, 104] and fibroblasts from sporadic ALS patients [105]. Accordingly, the spinal cords from ALS patients (and SOD1G93A mice) were found to have elevated glycogen concentrations [106], presumably caused by decreased glycolytic flux. Alternatively, elevated glycogen concentrations could be caused by reduced expression of the enzymes responsible for glycogen breakdown (namely glycogen phosphorylase and glycogen phosphoglucomutase), as observed in induced astrocytes derived from C9orf72 patients [107]. Somewhat surprisingly, several in vitro models of SOD1-ALS exhibit increased glycolytic activity [108, 109]; similarly, overexpression of TDP-43 in Drosophila led to an upregulation of PFK [110]. These observations could reflect a compensatory mechanism that has adapted to “correct” the energy imbalance caused by overexpression of these mutant proteins. Indeed, enhancing glucose metabolism in the same flies overexpressing TDP-43 — either by feeding them a high-sugar diet or further upregulating PFK — led to the significant rescue of locomotor function [110]. It should also be noted that riluzole, the other FDA-approved drug for the treatment of ALS, has been found to increase glucose metabolism both in the rat brain and in motor neuron-like cells, likely by increasing translocation of glucose transporters to the plasma membrane [111, 112]. Collectively, these studies point towards significant aberrations in brain glucose metabolism in ALS and suggest that enhancing brain glucose metabolism could be a viable therapeutic approach.
Impaired Metabolic Support from Glia
Brain metabolism is not restricted only to neuronal cells. In addition to neurons, the brain is also made up of glial cells, including astrocytes, oligodendroglia, and microglia, all of which are known to play supportive roles (or, in some cases, pathological roles) both in health and in disease [113]. Notably, glia are known to provide metabolic support to neurons [113]. One of the primary ways is through lactate shuttling, which can be defined as the transfer of glycolysis-derived lactate from one cell type to another as a supplemental energy source. Cells generate lactate by converting it from pyruvate (the end product of glycolysis) via lactate dehydrogenase [113]. This lactate can then be released via monocarboxylate transporters (MCTs) and subsequently taken up by recipient cells, also by MCTs [113]. The lactate is then re-converted by recipient cells into pyruvate, which can be utilized as an energy substrate for oxidative metabolism [113]. There is evidence that, in the brain, glia adopt a more glycolytic profile compared to neurons and that glia (including both astrocytes and oligodendroglia) provide lactate to neurons as a supplemental form of energy [114,115,116].
Furthermore, a loss of glial MCT expression, which could impair brain lactate shuttling and contribute to disease, is seen in ALS. First and foremost, genetic excision of mutant SOD1 specifically in oligodendroglia extends survival in the SOD1G37R mouse model [117], suggesting a non-cell-autonomous role for oligodendroglia in the pathogenesis of ALS. Additional work found that oligodendroglia are the primary cell type that expresses MCT1 in the brain and that oligodendroglial precursor cells (OPCs) specifically exhibit reduced expression of MCT1 in SOD1G93A mice compared to wild type animals [116]. A loss of MCT1 was also detected in the motor cortex of patients [116] and in both mice and HEK cells overexpressing mutant SOD1 [118]. These findings suggest that, in the context of SOD1-ALS, oligodendroglia have a reduced capacity to extrude lactate. Accordingly, spinal cord lactate levels were significantly reduced in the SOD1G93A mouse [119]. But what might be the downstream consequences of this? In organotypic slice cultures, the loss of oligodendroglial MCT1 expression was found to cause axonal damage and neuron loss [116]; furthermore, genetic ablation of oligodendroglial lineage-specific MCT1 caused axonal degeneration, including swollen mitochondria and concomitant hypomyelination [120]. Thus, a loss of MCT1 expression appears to have profound and deleterious consequences on neighboring neurons. However, the proposed mechanism by which this occurs — i.e., a loss of metabolic support towards neurons — requires further evidence. It should also be noted that a loss of MCT4, which is primarily expressed by astrocytes, has also been observed in ALS [116], and that toxicity of SOD1G93A-derived astrocytes towards motor neurons can be rescued by supplementing the media with lactate [119], suggesting that astrocytic lactate shuttling may also be perturbed in ALS. Therefore, the contribution of astrocytes to this lactate shuttling paradigm should not be overlooked. Collectively, these data suggest that compromised glial MCT expression — whether it be MCT1, MCT4, or both — may contribute to energy imbalances in ALS and that MCTs may be a viable therapeutic target for ALS.
Insulin Resistance
Insulin resistance is also a consistently observed phenomenon in ALS [121,122,123,124,125]. Insulin is a hormone released by beta cells of the pancreas following the consumption of carbohydrates [126]. Under normal conditions, insulin release stimulates glucose uptake from the blood by tissues including the brain, muscle, and fat [126]. Insulin resistance — a defining characteristic of diabetes mellitus (DM) type II — is a state in which abnormally high insulin levels are required to elicit a proper glucose uptake response [126]. Remarkably, the incidence of DM type II is associated with a 4-year delayed onset of ALS, thus appearing to be protective [127]. Conversely, the incidence of DM type I is associated with an increased risk of ALS [128]. The reasons underlying these conflicting correlations are unknown.
The high prevalence of insulin resistance in ALS is generally assumed to be a consequence of muscle atrophy since muscle is a primary site of glucose uptake in the body. However, this may not be a safe assumption. In one study identifying insulin resistance in ALS, the ALS patients weighed more than the healthy controls [123]. In another, no correlation was found between insulin resistance and the degree of muscle atrophy nor locomotor activity [124], together suggesting that insulin resistance is intrinsic to ALS itself rather than simply a byproduct of muscle wasting.
Interestingly, overexpression of TDP-43 was found to cause altered translocation of the insulin-sensitive glucose transporter GLUT4 in muscle fibers, as well as abnormal insulin-mediated glucose uptake [129]. Thus, the mis-localization of TDP-43, a feature common to almost all forms of ALS, could contribute to insulin resistance in patients. TDP-43 mis-localization was also identified in islet cells of postmortem ALS pancreatic tissue [130]. Furthermore, in vitro and in vivo, a loss of TDP-43 reduced beta cell exocytosis and insulin secretion, suggesting a novel mechanism by which TDP-43 pathology could cause aberrant insulin signaling [130]. In the context of C9orf72 ALS, overexpression of the G4C2 repeat expansion in Drosophila was found to cause a reduction in several insulin receptor ligands, suggesting that the mutation could perturb the insulin signaling cascade [131]. Moreover, insulin administration to these flies significantly extended survival, suggesting that modulation of insulin signaling could be an effective therapeutic intervention [131]. Additional work needs to be done to clarify these potential mechanisms and the role of insulin signaling in ALS.
Dyslipidemia
Mixed evidence for lipidic abnormalities has been identified in ALS patients. The types of lipids in question include (1) cholesterol, which is an essential constituent of cell membranes and serves as a precursor for the synthesis of hormones, and (2) triglycerides, which are the primary form of fat stored in adipocytes and collectively serve as an energy reservoir into which the body can tap during starvation conditions [132]. A French study of 369 ALS patients and 286 healthy controls found that the ALS patients were around twice as likely to have elevated cholesterol (specifically LDL) levels in the blood [133]. Interestingly, this effect seems to be protective — when those same 369 ALS patients were stratified into high- vs. low-cholesterol groups, the high-cholesterol group was found to have significantly better survival than the low-cholesterol group [133]. Similar results were obtained in a German study of 488 ALS patients, in which patients with elevated serum cholesterol or triglyceride levels survived significantly longer than those with normal levels [134]. However, these observations were non-significant on multivariate analysis. Furthermore, an Italian study of 658 ALS patients reported no difference in the prevalence of hyperlipidemia compared to an equal number of healthy controls, with no effect on survival [135]. And finally, a longitudinal UK study of 512 ALS patients found no significant link between serum lipid levels and survival in ALS [136]. The reasons underlying the discrepancies between these studies are unknown but could reflect regional genetic or dietary differences between each ALS patient cohort.
Lipid metabolism alterations have also been seen in ALS model systems. In particular, the SOD1G93A mouse exhibits a shift in mitochondrial fuel preference from glucose to fat (in muscle tissue) [137, 138] and an elevated rate of lipolysis throughout the disease course [106]. The causes of this are unknown but could potentially reflect a compensatory response to disease-related glucose intolerance and glucose hypometabolism. Glucose is the ideal fuel source for muscle metabolism, and so this switch towards lipid metabolism can be considered maladaptive. Also, in the SOD1G93A mouse, the activity of carnitine palmitoyltransferase 1 (CPT1), a central component of the carnitine shuttle, is associated with disease progression [139]. The carnitine shuttle facilitates the import of membrane-impermeable long-chain fatty acids into the mitochondria for metabolism in the Kreb’s cycle [140]. Thus, upregulation of this pathway may be a compensatory shift towards lipid metabolism. Interestingly, pharmacological and genetic downregulation of CPT1 activity ameliorated mitochondrial dysfunction and oxidative stress, improved motor and cognitive function, and delayed disease progression in the SOD1G93A mouse [139]. Similarly, RNAi-mediated knockdown of CPT1 mitigated locomotor dysfunction in TDP-43G298S-expressing flies [141]. These effects could potentially reflect a metabolic shift towards preferential use of glucose — indeed, knockdown of CPT1 led to over a 50% reduction in serum glucose levels in the SOD1G93A mouse [139], indicating an increase in glucose metabolism — however, additional evidence is required to establish this as the protective mechanism.
AMP Kinase, PGC-1α, and Aberrant Energy Signaling in ALS
The AMP-activated protein kinase (AMPK) is a key energy sensor in mammalian cells and is activated in ALS [142,143,144]. When a cell enters a state of sustained energy deficit, it will eventually begin to deplete its local ATP stores, leading to an increase in the AMP:ATP ratio, which in turn serves as a molecular trigger for the phosphorylation and activation of AMPK [145]. Generally speaking, activation of AMPK promotes catabolic pathways (which lead to ATP production) and inhibits anabolic pathways (which lead to ATP consumption), with the net effect being a restoration of the cellular energy balance back to baseline levels [145]. Therefore, AMPK activation serves as a cellular safeguard that helps to correct local energy imbalances as they occur. Given the plethora of energy imbalances in ALS that have already been outlined in this review, it would be expected for AMPK to be activated in patient cells. Indeed, evidence of AMPK activation has been identified in both motor neurons of ALS patients [146] and the spinal cord of SOD1G93A mice [143, 144]. Interestingly, both in vitro and in vivo evidence suggest that AMPK activation in the context of ALS is deleterious rather than protective — pharmacological inhibition of AMPK was found to rescue toxicity in NSC-34 motor neuron-like cells expressing mutant SOD1; furthermore, in C. elegans overexpressing either mutant SOD1 or mutant TDP-43, genetic ablation of the AMPK orthologue aak-2 rescued locomotor deficits [143]. Similarly, treating the SOD1G93A mouse model with metformin, which increases AMPK activation, worsened the survival in female animals [147]. Given that AMPK activation is generally corrective, these findings are somewhat counterintuitive. However, a potential explanation is that the subcellular localization of TDP-43 appears to be linked to AMPK activation status. Specifically, activation of AMPK activity has been found to cause mislocalization of TDP-43 to the cytoplasm, while inhibition of AMPK activity has been found to reduce it [142]. Assuming that mislocalization of TDP-43 is indeed a central pathogenic event, it could conceivably be the downstream mechanism through which AMPK activation exerts toxic effects. However, additional evidence of this is lacking.
Interestingly, several other lines of evidence suggest that AMPK activation — when timed correctly — could be beneficial in ALS. Pre-conditioning of SOD1G93A mice with latrepiridine (a potent activator of AMPK) from postnatal day 70 to 120 delayed symptom onset and improved survival [148]. Similarly, the treatment of SOD1G93A mice with resveratrol (a polyphenol known to activate AMPK) from 8 or 12 weeks of age onwards delayed the symptom onset, extended the lifespan, and ameliorated the locomotor deficits [149]. However, while both of these compounds activate AMPK, neither are entirely selective for AMPK — for example, resveratrol is also known to activate Sirt1 [150]. Therefore, effects on different pathways cannot be ruled out. Still, these studies interestingly point towards a “window of opportunity” during which activation of AMPK could benefit disease outcomes.
Downregulation of peroxisome proliferator-activated receptor (PPAR γ) coactivator PGC-1α has also been demonstrated in the motor cortex of ALS patients, as well as in transgenic SOD1 and FUS mice [151, 152]. PGC-1α is a transcriptional coactivator that, similarly to AMPK, is activated by a cellular energy deficit [153]. Once activated, PGC-1α interacts with a wide variety of transcription factors which primarily elicit metabolic responses, such as mitochondrial biogenesis and glucose/fatty acid metabolism, and help to reestablish cellular energy balance [153]. The loss of PGC-1α in ALS may, at least in part, underlie some of the metabolic imbalances already outlined in this review. In line with this, overexpression of PGC-1α in SOD1G93A animals improved the motor performance, mitigated the neuronal loss, and extended the survival [154,155,156], suggesting that PGC-1α expression is neuroprotective in the context of ALS. The precise mechanism by which this occurs is unknown, and indeed, a wide array of downstream targets could presumably be responsible for the neuroprotection mediated by PGC-1α.
Interestingly, PPARγ, one of the main targets of PGC-1α, was predicted to be one of the primary transcription coactivators differentially expressed between mutant SOD1 and wild type astrocytes [157]. Furthermore, several studies have found that selective positive regulation of PPARγ by treatment with pioglitazone, a classic anti-diabetic drug, could extend survival and improve disease symptoms in the SOD1-G93A mouse [158, 159], as well as in both TDP-43 and FUS Drosophila models [160], which suggests that the PPARγ axis could be a primary downstream pathway through which PGC-1α mediates neuroprotection. With these data in mind, pioglitazone would appear to be a promising candidate for clinical trials. But disappointingly, when tested in clinical trials, pioglitazone had no beneficial effect on ALS patient survival [161], nor did it affect patient BMI [162]. One potential explanation is that pioglitazone’s mechanism of action is via inhibition of the hypothalamic melanocortin system [162]. As mentioned earlier, hypothalamic atrophy has been identified in ALS patients [16,17,18]. Moreover, altered expression of two peptides that regulate the melanocortin system — specifically proopiomelanocortin (POMC) and agouti-related peptide (AGRP) — was observed in the hypothalamus of several ALS mouse models, which could impair the responsiveness of ALS patients to drugs like pioglitazone and could explain its lack of efficacy in clinical trials [162]. Still, the modulation of metabolic pathways that are subject to transcriptional control by PGC-1α, including the PPARγ pathway, remains a promising therapeutic approach for restoring the energy balance in ALS.
Additional Opportunities for Therapeutic Intervention
As we have seen in this review, numerous energy imbalances are observed in ALS patients and model systems. This leads to a situation in which ALS patients have insufficient energy, which likely contributes to disease pathogenesis and neurodegeneration. Therapeutic interventions that tip the scale back towards energy abundance would likely mitigate disease severity and prolong patient survival. Since brain glucose uptake is primarily driven by neuronal activity [3], simply increasing glucose concentration in the blood through diet or other means is likely not enough. In fact, high intake of carbohydrates has been associated with an increased risk of ALS [163]. Therefore, there is a clear need for other approaches to correct patients’ energy imbalances, which are outlined in Table 1 and explored in depth below.
High-Fat Diet
Dietary intervention is the simplest yet most powerful method to preserve patients’ energy balance. As mentioned in the first section, patients with a high BMI tend to have a better disease outcome than those with a lower BMI [7, 9, 10, 12, 33,34,35,36,37,38, 40, 41], and the primary way to promote a high BMI is through diet. Indeed, a 2016 study identified a correlation between increased eating behavior and increased survival, suggesting that simply modifying food intake can profoundly impact disease course [164]. Interestingly, the macronutrient content does seem to matter — in a cohort of 153 ALS patients, reduced intake of dietary fats (but not carbohydrates or protein) was correlated with an increased risk of ALS [163]. Furthermore, two other studies identified intake of polyunsaturated fats to be associated with a reduced risk of ALS [165, 166]. The reasons for these macronutrient-specific effects are unclear, although fats are more calorie-dense than other macronutrients and maybe more protective because they simply provide more energy [167]. With these findings in mind, a high-calorie diet, but more specifically a high-fat diet, should be protective in the context of ALS. Indeed, in the SOD1G93A mouse model, a high-fat diet corrected metabolic abnormalities, prevented motor neuron loss, and extended the survival [29, 168, 169], while calorie restriction worsened the survival [169, 170]. Similar results were obtained in the TDP-43A315T mouse model [171], suggesting that this intervention may be widely applicable to different forms of ALS. But what about in patients? In a recent study of 201 ALS patients, half were assigned to a high-calorie fatty diet (HCFD) and the other half were assigned to a placebo diet. Remarkably, fast-progressing patients who received the HCFD exhibited a decrease in neurofilament light chain serum levels (a general biomarker for neurodegeneration) and survived longer compared to placebo [172, 173]. For unclear reasons, these effects were not observed in slow-progressing patients — additional follow-up studies are warranted. Furthermore, since the study did not test different macronutrient compositions, it could not distinguish whether the HCFD diet was protective because it was high in fat or simply high in calories. Still, the results are promising and suggest that a high-calorie, and perhaps a high-fat diet specifically, is indeed therapeutically beneficial in ALS patients.
Re-energizing Mitochondria (Pyruvate, Dichloroacetate, Acetyl-l-carnitine, Triglycerides, Ketones, Olesoxime)
Mitochondria are the primary energy production site in cells [42], and therefore, maintaining their function is critical in maintaining proper energy homeostasis. However, to produce ATP, mitochondria need sufficient energy substrates as a starting point. In ALS, between the previously-discussed loss of body mass and decreases in glucose uptake, the availability of these energy substrates is not optimal. Providing mitochondria with additional or supplemental fuel sources may restore ATP production and improve energy balance. Perhaps, the most obvious of these is pyruvate, the end-product of glycolysis and one of the primary fuels for mitochondrial metabolism [42]. In the context of the SOD1G93A mouse model, pyruvate supplementation has produced mixed effects: in one study, it delayed disease progression and improved motor performance [174], while in another, it had no effect [175]. The reasons underlying this discrepancy are unclear. However, simply supplementing with pyruvate may not be sufficient. Before mitochondria can utilize pyruvate, it must first be converted into acetyl-CoA [42]. This conversion is catalyzed by pyruvate dehydrogenase (PDH), which is in turn subject to negative regulation by pyruvate dehydrogenase kinase (PDK). Phosphorylation of PDH by PDK decreases PDH-mediated conversion of pyruvate into acetyl-CoA. Interestingly, overexpression of mutant SOD1 in NSC-34 motor neuron-like cells led to an increase in PDK expression, which may impair their ability to convert pyruvate into acetyl-CoA [108]. Inhibiting PDK activity to boost acetyl-CoA production may be a viable approach to enhance the functional status of mitochondria in ALS. Indeed, treatment of SOD1G93A mice with dichloroacetate, a PDK inhibitor, greatly improved the mitochondrial function in the spinal cord, mitigated the motor neuron loss, improved the motor function, and extended the survival [137, 176], suggesting that enhancing the conversion of pyruvate to acetyl-CoA is indeed neuroprotective, at least in the context of mutant SOD1 [137]. Similar approaches should be tested in other models of familial ALS. Furthermore, it would be interesting to test the effect of administering both pyruvate and dichloroacetate simultaneously — perhaps, it would have a synergistic impact on acetyl-CoA levels and lead to even better mitochondrial function and disease outcome.
Supplying mitochondria with alternative fuels, including ketone bodies and medium-chain triglycerides, may serve as another strategy to boost mitochondrial energy production. When glucose is in a short supply, ketone bodies are produced from body fat by the liver (i.e., ketogenesis) — this can be elicited by consumption of a ketogenic (i.e., a very low-carbohydrate, high-fat) diet [177]. The resulting ketone bodies are transported throughout the body, taken up by cells, and used directly in the Kreb's cycle [177]. Interestingly, administering SOD1G93A mice a ketogenic diet partially rescued motor neuron loss and delayed disease onset, suggesting it was protective to some degree — however, it did not affect survival [178]. Another potential mitochondrial fuel is bodies of medium-chain triglycerides, which are converted into acetyl-CoA by β-oxidation once taken up by cells, either directly or by first being converted into ketone bodies by the liver [178, 179]. Two different medium-chain triglycerides, namely caprylic triglyceride [180] and triheptanoin [179], have both shown efficacy in reducing motor neuron loss and delaying symptom onset in SOD1G93A mice. However, they are similar to the ketogenic diet in that they had no significant effect on survival. Thus, these alternative mitochondrial fuels appear to have a measurable yet limited benefit towards energy balance and disease outcome in the context of mutant SOD1. Similar approaches should be tested in other models of familial ALS.
Supplementation with acetyl-L-carnitine is another potential strategy for boosting mitochondrial energy production. Acetyl-L-carnitine is a naturally occurring molecule formed by enzymatic acetylation of carnitine and has two primary roles in metabolism [140]. Firstly, as a critical component of the carnitine shuttle, it facilitates the import of membrane-impermeable long-chain fatty acids into the mitochondria for β-oxidation [140]. And secondly, it serves as a source of acetyl groups for the regeneration of acetyl-CoA when needed [140]. Both roles promote mitochondrial energy production, especially in the metabolism of fatty acids, which should be protective in the context of ALS. Indeed, the treatment of SOD1G93A mice with L-carnitine delayed the disease onset, delayed the motor impairment, and extended the survival [181]. And remarkably, in phase II clinical study, ALS patients who took 3 g per day of acetyl-L-carnitine survived over twice as long compared to those who took a placebo [182]. A more extensive phase III trial should be carried out to confirm these exciting results.
And lastly, another compound that has demonstrated some efficacy in ALS preclinical models is olesoxime, an experimental mitochondria-targeting drug that is thought to stabilize the mitochondrial permeability transition pore (mPTP) [183]. Specifically, in the SOD1G93A mouse, olesoxime improved survival, prevented motor neuron loss, and ameliorated motor dysfunction [183, 184]. However, olesoxime showed mixed results in ALS patient–derived induced pluripotent stem cells [185]. Furthermore, when tested in a phase II–III clinical trial, olexosime did not benefit ALS patients [186].
Creatine Supplementation
Supplementation of ALS patients with creatine has also been explored as a potential therapeutic strategy. Creatine is an energy-buffering molecule produced by the liver and transported peripherally for use primarily within brain and muscle cells, where it gets phosphorylated into phosphocreatine [187]. During high energy consumption periods, an enzyme known as creatine kinase uses the phosphoryl group from phosphocreatine to regenerate ATP from ADP rapidly [187]. Thus, phosphocreatine serves as a cellular reserve of phosphoryl groups that, when needed, helps to buffer ATP levels. Furthermore, dietary supplementation with exogenous creatine has been shown to increase tissue phosphocreatine stores, thereby increasing the cells’ ability to regenerate ATP from ADP and satisfy energy demands when needed [187]. For this reason, creatine supplementation was thought to be a promising approach to improve energy balance in ALS. Indeed, depletion of ATP stores in the spinal cord and cerebral cortex of SOD1G93A mice was partially rescued by supplementation with creatine [188]. Furthermore, in SOD1G93A mice, creatine supplementation mitigated the neurodegeneration, improved the motor function, and extended the survival [189]. However, when tested in three ALS patient clinical trials, creatine supplementation had no beneficial effects [190,191,192]. The reasons for this are unknown, although the timing of the creatine supplementation is likely a key factor. In the animal experiments, creatine supplementation was initiated before symptom onset. In contrast, it was initiated well into disease progression in the patients. At that point, metabolic imbalances are pretty severe, and the body’s responsiveness to creatine may be limited. Therefore, administration sufficiently early in patients’ disease course (or, in an ideal scenario, before symptom onset) may significantly improve the clinical efficacy of creatine supplementation.
Conclusion
Although the underlying pathogenic mechanisms remain to be elucidated, numerous alterations to bioenergetic and metabolic homeostasis are observed in both ALS/FTD patients and model systems, including low BMI, hypermetabolism, hypothalamic abnormalities, mitochondrial dysfunction, glucose hypometabolism, impaired metabolic support from glia, dyslipidemia, insulin resistance, and aberrant energy signaling. These alterations culminate to produce a state of energy imbalance in patients, which likely contributes to motor neurons’ selective vulnerability and death. It is worth noting that the majority of the in vivo studies linking energy imbalances to ALS — especially the early studies — were done in the context of the SOD1G93A mouse model. This is primarily because this model accurately and reliably recapitulates ALS-like phenotypes, while many others do not. However, it only accounts for one subset of the disease. Furthermore, there are known discrepancies between the SOD1G93A mouse and human sporadic ALS — for example, the SOD1G93A mouse lacks robust TDP-43 pathology [193]. Therefore, it would be inappropriate to extend findings obtained exclusively with this model to other mutations, or to sporadic ALS, which accounts for ~90% of cases in humans. Thankfully, the advent of other model systems (including FUS, TDP-43, and C9orf72 transgenic mice) as well as patient-derived in vitro systems (including iPS technology) has allowed for findings obtained in the SOD1G93A mouse to be confirmed in the context of other mutations, or even in the context of sporadic disease. When looked at in totality and alongside patient data, these model systems confirm one another and paint a clear picture of altered metabolic homeostasis in ALS. Interestingly and even more broadly, several of the same energy imbalances observed in ALS are also seen in patients with other, distinct neurodegenerative disorders — for example, both weight loss and glucose hypometabolism are also associated with Alzheimer’s disease, Parksinson’s disease, and Huntington’s disease, which suggests that energy imbalances may also be involved in the degeneration of neuronal subtypes other than motor neurons (e.g., dopaminergic neurons in the case of Parkinson’s) [194].
The fact that energy imbalances are conserved across a variety of ALS subtypes suggests that targeting metabolic pathways as a therapeutic intervention would be broadly efficacious, despite the heterogeneity and complexity of ALS. Indeed, several therapeutics that aim to restore energy balance have shown promise in ALS clinical trials. In particular, both high-fat diet and supplementation with acetyl-L-carnitine have been shown to extend survival in ALS patients. Furthermore, several other compounds/strategies have demonstrated preclinical efficacy and deserve to be tested in clinical trials. With enough research, we believe that one (or some combination) of these strategies will emerge as an optimal approach to maintaining energy balance and will prove invaluable in slowing disease progression in ALS patients.
References
Hardiman O, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3.
Abramzon YA, Fratta P, Traynor BJ, Chia R. The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. 2020;14:42. https://doi.org/10.3389/fnins.2020.00042.
Watts ME, Pocock R, Claudianos C. Brain energy and oxygen metabolism: emerging role in normal function and disease. Front Mol Neurosci. 2018;11:216. https://doi.org/10.3389/fnmol.2018.00216.
Huisman MH, et al. Effect of Presymptomatic body mass index and consumption of fat and alcohol on amyotrophic lateral sclerosis. JAMA Neurol. 2015;72:1155–62. https://doi.org/10.1001/jamaneurol.2015.1584.
Popuri K, et al. FDG-PET in presymptomatic C9orf72 mutation carriers. Neuroimage Clin. 2021;31: 102687. https://doi.org/10.1016/j.nicl.2021.102687.
Pharaoh G, et al. Metabolic and stress response changes precede disease onset in the spinal cord of mutant SOD1 ALS mice. Front Neurosci. 2019;13:487. https://doi.org/10.3389/fnins.2019.00487.
Marin B, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2011;82:628–34. https://doi.org/10.1136/jnnp.2010.211474.
Nagel G, et al. Adipokines, C-reactive protein and amyotrophic lateral sclerosis - results from a population- based ALS registry in Germany. Sci Rep. 2017;7:4374. https://doi.org/10.1038/s41598-017-04706-5.
van Mantgem MRJ, et al. Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. 2020;91:867–75. https://doi.org/10.1136/jnnp-2020-322909.
Wei QQ, et al. Early weight instability is associated with cognitive decline and poor survival in amyotrophic lateral sclerosis. Brain Res Bull. 2021;171:10–5. https://doi.org/10.1016/j.brainresbull.2021.02.022.
Desport JC, et al. Nutritional assessment and survival in ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:91–6. https://doi.org/10.1080/14660820050515386.
Moglia C, et al. Early weight loss in amyotrophic lateral sclerosis: Outcome relevance and clinical correlates in a population-based cohort. J Neurol Neurosurg Psychiatry. 2019;90:666–73. https://doi.org/10.1136/jnnp-2018-319611.
Holm T, et al. Severe loss of appetite in amyotrophic lateral sclerosis patients: online self-assessment study. Interact J Med Res. 2013;2: e8. https://doi.org/10.2196/ijmr.2463.
Ngo ST, et al. Loss of appetite is associated with a loss of weight and fat mass in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:497–505. https://doi.org/10.1080/21678421.2019.1621346.
Mezoian T, et al. Loss of appetite in amyotrophic lateral sclerosis is associated with weight loss and decreased calorie consumption independent of dysphagia. Muscle Nerve. 2020;61:218–42.
Gorges M, et al. Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2017;88:1033–41. https://doi.org/10.1136/jnnp-2017-316382.
Bayer D, et al. Disruption of orbitofrontal-hypothalamic projections in a murine ALS model and in human patients. Transl Neurodegener. 2021;10:17. https://doi.org/10.1186/s40035-021-00241-6.
Gabery S, et al. Loss of the metabolism and sleep regulating neuronal populations expressing orexin and oxytocin in the hypothalamus in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2021;47:979–89. https://doi.org/10.1111/nan.12709.
Saper CB, Lowell BB. The hypothalamus. Curr Biol. 2014;24:R1111-1116. https://doi.org/10.1016/j.cub.2014.10.023.
Iacoangeli A, et al. Genome-wide meta-analysis finds the ACSL5-ZDHHC6 locus is associated with ALS and links weight loss to the disease genetics. Cell Rep. 2020;33: 108323. https://doi.org/10.1016/j.celrep.2020.108323.
Rajkumar A, et al. Acyl-CoA synthetase long-chain 5 genotype is associated with body composition changes in response to lifestyle interventions in postmenopausal women with overweight and obesity: a genetic association study on cohorts Montreal-Ottawa New Emerging Team, and Complications Associated with Obesity. BMC Med Genet. 2016;17:56. https://doi.org/10.1186/s12881-016-0320-4.
Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ. Nutritional status of patients with amyotrophic lateral sclerosis: Relation to the proximity of death. Am J Clin Nutr. 1996;63:130–7.
Desport JC, et al. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr. 2001;74:328–34.
Bouteloup C, et al. Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol. 2009;256:1236–42. https://doi.org/10.1007/s00415-009-5100-z.
Vaisman N, et al. Do patients with amyotrophic lateral sclerosis (ALS) have increased energy needs? J Neurol Sci. 2009;279:26–9. https://doi.org/10.1016/j.jns.2008.12.027.
Jesus P, et al. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur J Neurol. 2018;25:97–104. https://doi.org/10.1111/ene.13468.
Steyn FJ, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry. 2018;89:1016–23. https://doi.org/10.1136/jnnp-2018-318428.
Fayemendy P, et al. Hypermetabolism is a reality in amyotrophic lateral sclerosis compared to healthy subjects. J Neurol Sci. 2021;420: 117257. https://doi.org/10.1016/j.jns.2020.117257.
Dupuis L, Oudart H, Rene F, de Aguilar JLG, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004;101:11159–64. https://doi.org/10.1073/pnas.0402026101.
Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2019;710: 132933. https://doi.org/10.1016/j.neulet.2017.06.052.
Porter C, et al. Uncoupled skeletal muscle mitochondria contribute to hypermetabolism in severely burned adults. Am J Physiol Endocrinol Metab. 2014;307:E462-467. https://doi.org/10.1152/ajpendo.00206.2014.
Bae JS, Simon NG, Menon P, Vucic S, Kiernan MC. The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J Clin Neurol. 2013;9:65–74. https://doi.org/10.3988/jcn.2013.9.2.65.
Desport JC, et al. Nutritional status is a prognostic factor for survival in ALS patients. Neurology. 1999;53:1059–63.
Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011;44:20–4. https://doi.org/10.1002/mus.22114.
Marin B, et al. Population-based evidence that survival in amyotrophic lateral sclerosis is related to weight loss at diagnosis. Neurodegener Dis. 2016;16:225–35.
Peter RS, et al. Life course body mass index and risk and prognosis of amyotrophic lateral sclerosis: results from the ALS registry Swabia. Eur J Epidemiol. 2017;32:901–8. https://doi.org/10.1007/s10654-017-0318-z.
O’Reilly EJ, et al. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:205–11. https://doi.org/10.3109/21678421.2012.735240.
Shimizu T, et al. Prognostic significance of body weight variation after diagnosis in ALS: a single-centre prospective cohort study. J Neurol. 2019;266:1412–20. https://doi.org/10.1007/s00415-019-09276-2.
Mariosa D, et al. Body mass index and amyotrophic lateral sclerosis: a study of US military veterans. Am J Epidemiol. 2017;185:362–71. https://doi.org/10.1093/aje/kww140.
O’Reilly EJ, et al. Prediagnostic body size and risk of amyotrophic lateral sclerosis death in 10 studies. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:396–406. https://doi.org/10.1080/21678421.2018.1452944.
Dorst J, et al. Prognostic factors in ALS: a comparison between Germany and China. J Neurol. 2019;266:1516–25. https://doi.org/10.1007/s00415-019-09290-4.
Martinez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. https://doi.org/10.1038/s41467-019-13668-3.
Hart MN, Cancilla PA, Frommes S, Hirano A. Anterior horn cell degeneration and bunina-Type inclusions associated with dementia. Acta Neuropathol. 1977;38:225–8.
Okamoto K, Hirai S, Shoji M, Senoh T, Yamazaki T. Axonal swellings in the corticospinal tracts in amyotrophic lateral sclerosis. Acta Neuropathol. 1990;80:222–6.
Sasaki S, Iwata M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci Lett. 1996;204:53–6.
Chung MJ, Suh YL. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct Pathol. 2002;26:3–7. https://doi.org/10.1080/01913120252934260.
Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66:10–6.
Wang P, et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019;15: e1007947. https://doi.org/10.1371/journal.pgen.1007947.
Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;146:1271–9.
Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–50.
Szelechowski M, et al. Metabolic reprogramming in amyotrophic lateral sclerosis. Sci Rep. 2018;8:3953. https://doi.org/10.1038/s41598-018-22318-5.
Kirkinezos IG, et al. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25:164–72. https://doi.org/10.1523/JNEUROSCI.3829-04.2005.
Stribl C, et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J Biol Chem. 2014;289:10769–84. https://doi.org/10.1074/jbc.M113.515940.
Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413–24. https://doi.org/10.1093/hmg/ddt528.
Sharma A, et al. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7:10465. https://doi.org/10.1038/ncomms10465.
Menzies FM, et al. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–33.
Coussee E, et al. G37R SOD1 mutant alters mitochondrial complex I activity, Ca(2+) uptake and ATP production. Cell Calcium. 2011;49:217–25. https://doi.org/10.1016/j.ceca.2011.02.004.
Lu J, et al. Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res Bull. 2012;89:185–90. https://doi.org/10.1016/j.brainresbull.2012.09.005.
Wang W, et al. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. 2013;22:4706–19. https://doi.org/10.1093/hmg/ddt319.
Deng J, et al. FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 2015;11: e1005357. https://doi.org/10.1371/journal.pgen.1005357.
Tradewell ML, et al. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet. 2012;21:136–49. https://doi.org/10.1093/hmg/ddr448.
Onesto E, et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol Commun. 2016;4:47. https://doi.org/10.1186/s40478-016-0316-5.
Dafinca R, et al. C9orf72 Hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells. 2016;34:2063–78. https://doi.org/10.1002/stem.2388.
Hong K, et al. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci Lett. 2012;530:144–9. https://doi.org/10.1016/j.neulet.2012.10.003.
Khalila B, et al. Enhancing mitofusin/marf ameliorates neuromuscular dysfunction in drosophila models of TDP-43 proteinopathies. Neurobiol Aging. 2017;54:71–83.
Wang W, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22:869–78. https://doi.org/10.1038/nm.4130.
Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3.
Higgins CMJ, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22:1–6.
Mattiazzi M, et al. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–33. https://doi.org/10.1074/jbc.M203065200.
Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–70. https://doi.org/10.1523/JNEUROSCI.4385-04.2005.
Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci USA. 2008;105:4022–7. https://doi.org/10.1073/pnas.0712209105.
Wang T, et al. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021;33:531–46 e539. https://doi.org/10.1016/j.cmet.2021.01.005.
Blokhuis AM, et al. Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol. 2016;132:175–96. https://doi.org/10.1007/s00401-016-1575-8.
Lopez-Gonzalez R, et al. Poly(GR) in C9ORF72-Related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016;92:383–91. https://doi.org/10.1016/j.neuron.2016.09.015.
Li S, et al. Altered MICOS Morphology and mitochondrial ion homeostasis contribute to poly(GR) toxicity associated with C9-ALS/FTD. Cell Rep. 2020;32: 107989. https://doi.org/10.1016/j.celrep.2020.107989.
Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999;46:787–90.
Wiedemann FR, Manfredi G, Mawrin C, Beal F, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. 2002;80:616–25.
Echaniz-Laguna A, et al. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp Neurol. 2006;198:25–30. https://doi.org/10.1016/j.expneurol.2005.07.020.
Crugnola V, et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol. 2010;67:849–54.
Wiedemann F, et al. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci. 1998;156:65–72.
Jung C, Higgins CMJ, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem. 2002;83:535–45.
Ferri A, et al. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci USA. 2006;103:13860–5. https://doi.org/10.1073/pnas.0605814103.
Ghiasi P, Hosseinkhani S, Noori A, Nafissi S, Khajeh K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol Res. 2012;34:297–303. https://doi.org/10.1179/1743132812Y.0000000012.
Krasnianski A, et al. Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain. 2005;128:1870–6. https://doi.org/10.1093/brain/awh540.
Bowling AC, Schulz JB, Brown RH, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1993;61:2322–5.
Browne SE, et al. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J Neurochem. 1998;71:281–7.
Stoica R, et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016;17:1326–42. https://doi.org/10.15252/embr.201541726.
Pizzino G, et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763. https://doi.org/10.1155/2017/8416763.
Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003–14. https://doi.org/10.3969/j.issn.1673-5374.2013.21.009.
Cunha-Oliveira T, et al. Oxidative stress in amyotrophic lateral sclerosis: Pathophysiology and opportunities for pharmacological intervention. Oxid Med Cell Longev. 2020;2020:5021694. https://doi.org/10.1155/2020/5021694.
Vielhaber S, et al. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain. 2000;123:1339–48.
Yoshino H, Kimura A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph Lateral Scler. 2006;7:241–5. https://doi.org/10.1080/17482960600881870.
Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–97. https://doi.org/10.1016/j.tins.2013.07.001.
Kawada K, Iwamoto M, Sakai Y. Mechanisms underlying (18)F-fluorodeoxyglucose accumulation in colorectal cancer. World J Radiol. 2016;8:880–6. https://doi.org/10.4329/wjr.v8.i11.880.
Dalakas MC, Hatazawa J, Brooks RA, Di Chiro G. Lowered cerebral glucose utilization in amyotrophic lateral sclerosis. Ann Neurol. 1987;22:580–6.
Ludolph AC, et al. Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand. 1992;85:81–9.
Pagani M, et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurol. 2014;83:1067–74.
Van Laere K, et al. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. 2014;71:553–61. https://doi.org/10.1001/jamaneurol.2014.62.
Miyazaki K, et al. Early and progressive impairment of spinal blood flow-glucose metabolism coupling in motor neuron degeneration of ALS model mice. J Cereb Blood Flow Metab. 2012;32:456–67. https://doi.org/10.1038/jcbfm.2011.155.
De Vocht J, et al. Use of multimodal imaging and clinical biomarkers in presymptomatic carriers of C9orf72 repeat expansion. JAMA Neurol. 2020;77:1008–17. https://doi.org/10.1001/jamaneurol.2020.1087.
McDonald TS, Kumar V, Fung JN, Woodruff TM, Lee JD. Glucose clearance and uptake is increased in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis through an insulin-independent mechanism. FASEB J. 2021;35: e21707. https://doi.org/10.1096/fj.202002450R.
Poulton KR, Rossi ML. Peripheral nerve protein glycation and muscle fructolysis: Evidence of abnormal carbohydrate metabolism in ALS. Funct Neurol. 1993;8:33–42.
Wang XS, Simmons Z, Liu W, Boyer PJ, Connor JR. Differential expression of genes in amyotrophic lateral sclerosis revealed by profiling the post mortem cortex. Amyotroph Lateral Scler. 2006;7:201–10. https://doi.org/10.1080/17482960600947689.
Lederer CW, Torrisi A, Pantelidou M, Santama N, Cavallaro S. Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genom. 2007;8:26. https://doi.org/10.1186/1471-2164-8-26.
Raman R, et al. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol Appl Neurobiol. 2015;41:201–26. https://doi.org/10.1111/nan.12147.
Dodge JC, et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci USA. 2013;110:10812–7. https://doi.org/10.1073/pnas.1308421110.
Allen SP, et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain. 2019;142:3771–90. https://doi.org/10.1093/brain/awz302.
Valbuena GN, et al. Metabolomic analysis reveals increased aerobic glycolysis and amino acid deficit in a cellular model of amyotrophic lateral sclerosis. Mol Neurobiol. 2016;53:2222–40. https://doi.org/10.1007/s12035-015-9165-7.
Allen SP, et al. Superoxide dismutase 1 mutation in a cellular model of amyotrophic lateral sclerosis shifts energy generation from oxidative phosphorylation to glycolysis. Neurobiol Aging. 2014;35:1499–509. https://doi.org/10.1016/j.neurobiolaging.2013.11.025.
Manzo E, et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife. 2019;8. https://doi.org/10.7554/eLife.45114.
Chowdhury GM, et al. Chronic riluzole treatment increases glucose metabolism in rat prefrontal cortex and hippocampus. J Cereb Blood Flow Metab. 2008;28:1892–7. https://doi.org/10.1038/jcbfm.2008.78.
Daniel B, Green O, Viskind O, Gruzman A. Riluzole increases the rate of glucose transport in L6 myotubes and NSC-34 motor neuron-like cells via AMPK pathway activation. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:434–43. https://doi.org/10.3109/21678421.2013.808226.
Lago-Baldaia I, Fernandes VM, Ackerman SD. More than mortar: glia as architects of nervous system development and disease. Front Cell Dev Biol. 2020;8: 611269. https://doi.org/10.3389/fcell.2020.611269.
Machler P, et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 2016;23:94–102. https://doi.org/10.1016/j.cmet.2015.10.010.
Suzuki A, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–23. https://doi.org/10.1016/j.cell.2011.02.018.
Lee Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–8. https://doi.org/10.1038/nature11314.
Kang SH, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–9. https://doi.org/10.1038/nn.3357.
Philips T, et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain. 2013;136:471–82. https://doi.org/10.1093/brain/aws339.
Ferraiuolo L, et al. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134:2627–41. https://doi.org/10.1093/brain/awr193.
Philips T, et al. MCT1 deletion in oligodendrocyte lineage cells causes late-onset hypomyelination and axonal degeneration. Cell Rep. 2021;34: 108610. https://doi.org/10.1016/j.celrep.2020.108610.
Steinke J, Tyler HR. The association of amyotrophic lateral sclerosis (motor neuron disease) and carbohydrate intolerance, a clinical study. Metabolism. 1964;13:1376–81.
Ionasescu V, Luca N. Studies on carbohydrate metabolism in amyotrophic lateral sclerosis and hereditary proximal spinal musclar atrophy. Acta Neurol Scand. 1964;40:47–57.
Reyes ET, Perurena OH, Festoff BW, Jorgensen R, Moore WV. Insulin resistance in amyotrophic lateral sclerosis. J Neurol Sci. 1984;63:317–24.
Pradat PF, et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:166–71. https://doi.org/10.3109/17482960902822960.
Ahmed RM, et al. Eating peptides: Biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann Clin Transl Neurol. 2019;6:486–95. https://doi.org/10.1002/acn3.721.
Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: Review of the underlying molecular mechanisms. J Cell Physiol. 2019;234:8152–61. https://doi.org/10.1002/jcp.27603.
Jawaid A, et al. ALS disease onset may occur later in patients with pre-morbid diabetes mellitus. Eur J Neurol. 2010;17:733–9. https://doi.org/10.1111/j.1468-1331.2009.02923.x.
Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis - an epidemiologic study. Neurology. 2013;81.
Stallings NR, et al. TDP-43, an ALS Linked protein, regulates fat deposition and glucose homeostasis. PLoS One. 2013;8.
Araki K, et al. TDP-43 regulates early-phase insulin secretion via CaV1.2-mediated exocytosis in islets. J Clin Invest. 2019;129:3578–93. https://doi.org/10.1172/JCI124481.
Atilano ML, et al. Enhanced insulin signalling ameliorates C9orf72 hexanucleotide repeat expansion toxicity in Drosophila. Elife. 2021;10. https://doi.org/10.7554/eLife.58565.
Helkin A, et al. Dyslipidemia part 1–review of lipid metabolism and vascular cell physiology. Vasc Endovascular Surg. 2016;50:107–18. https://doi.org/10.1177/1538574416628654.
Dupuis L, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 2008;70:1004–9.
Dorst J, et al. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol. 2011;258:613–7. https://doi.org/10.1007/s00415-010-5805-z.
Chio A, et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology. 2009;73:1681–5.
Rafiq MK, Lee E, Bradburn M, McDermott CJ, Shaw PJ. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: Insights from the olesoxime clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:478–84. https://doi.org/10.3109/21678421.2015.1062517.
Palamiuc L, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7:526–46. https://doi.org/10.15252/emmm.201404433.
Steyn FJ, et al. Altered skeletal muscle glucose-fatty acid flux in amyotrophic lateral sclerosis. Brain Commun. 2020;2:fcaa154. https://doi.org/10.1093/braincomms/fcaa154.
Trabjerg MS, et al. Downregulating carnitine palmitoyl transferase 1 affects disease progression in the SOD1 G93A mouse model of ALS. Commun Biol. 2021;4:509. https://doi.org/10.1038/s42003-021-02034-z.
Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta. 1863;2422–2435:2016. https://doi.org/10.1016/j.bbamcr.2016.01.023.
Manzo E, et al. Medium-chain fatty acids, beta-hydroxybutyric acid and genetic modulation of the carnitine shuttle are protective in a drosophila model of ALS based on TDP-43. Front Mol Neurosci. 2018;11:182. https://doi.org/10.3389/fnmol.2018.00182.
Liu YJ, et al. Activation of AMP-activated protein kinase alpha1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:787–801. https://doi.org/10.1093/hmg/ddu497.
Lim MA, et al. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci. 2012;32:1123–41. https://doi.org/10.1523/JNEUROSCI.6554-10.2012.
Perera ND, et al. Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS One. 2014;9: e90449. https://doi.org/10.1371/journal.pone.0090449.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–23. https://doi.org/10.1038/ncb2329.
Liu YJ, Lee LM, Lai HL, Chern Y. Aberrant activation of AMP-activated protein kinase contributes to the abnormal distribution of HuR in amyotrophic lateral sclerosis. FEBS Lett. 2015;589:432–9. https://doi.org/10.1016/j.febslet.2014.12.029.
Kaneb HM, Sharp PS, Rahmani-Kondori N, Wells DJ. Metformin treatment has no beneficial effect in a dose-response survival study in the SOD1(G93A) mouse model of ALS and is harmful in female mice. PLoS One. 2011;6: e24189. https://doi.org/10.1371/journal.pone.0024189.
Coughlan KS, Mitchem MR, Hogg MC, Prehn JH. “Preconditioning” with latrepirdine, an adenosine 5’-monophosphate-activated protein kinase activator, delays amyotrophic lateral sclerosis progression in SOD1(G93A) mice. Neurobiol Aging. 2015;36:1140–50. https://doi.org/10.1016/j.neurobiolaging.2014.09.022.
Mancuso R, et al. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics. 2014;11:419–32. https://doi.org/10.1007/s13311-013-0253-y.
Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–95. https://doi.org/10.1074/jbc.M501250200.
Thau N, et al. Decreased mRNA expression of PGC-1a and PGC-1a-Yregulated factors in the SOD1-G93A ALS mouse model and in human sporadic ALS. J Neuropathol Exp Neurol. 2012;71:1064–74.
Bayer H, et al. ALS-causing mutations differentially affect PGC-1alpha expression and function in the brain vs. peripheral tissues. Neurobiol Dis. 2017;97:36–45. https://doi.org/10.1016/j.nbd.2016.11.001.
Miller KN, Clark JP, Anderson RM. Mitochondrial regulator PGC-1a-modulating the modulator. Curr Opin Endocr Metab Res. 2019;5:37–44. https://doi.org/10.1016/j.coemr.2019.02.002.
Liang H, et al. PGC-1alpha protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve. 2011;44:947–56. https://doi.org/10.1002/mus.22217.
Zhao W, et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1a) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener. 2011;6:1–8.
Da Cruz S, et al. Elevated PGC-1alpha activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 2012;15:778–86. https://doi.org/10.1016/j.cmet.2012.03.019.
Sun S, et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci USA. 2015;112:E6993-7002. https://doi.org/10.1073/pnas.1520639112.
Schutz B, et al. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–12. https://doi.org/10.1523/JNEUROSCI.2038-05.2005.
Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191:331–6. https://doi.org/10.1016/j.expneurol.2004.10.007.
Joardar A, et al. PPAR gamma activation is neuroprotective in a drosophila model of ALS based on TDP-43. Hum Mol Genet. 2015;24:1741–54. https://doi.org/10.1093/hmg/ddu587.
Dupuis L, et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS One. 2012;7.
Vercruysse P, et al. Alterations in the hypothalamic melanocortin pathway in amyotrophic lateral sclerosis. Brain. 2016;139:1106–22. https://doi.org/10.1093/brain/aww004.
Okamoto K, et al. Nutritional status and risk of amyotrophic lateral sclerosis in Japan. Amyotroph Lateral Scler. 2007;8:300–4. https://doi.org/10.1080/17482960701472249.
Ahmed RM, et al. Cognition and eating behavior in amyotrophic lateral sclerosis: effect on survival. J Neurol. 2016;263:1593–603. https://doi.org/10.1007/s00415-016-8168-2.
Veldink JH, et al. Intake of polyunsaturated fatty acids and vitamin E reduces the risk of developing amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007;78:367–71. https://doi.org/10.1136/jnnp.2005.083378.
Fitzgerald KC, et al. Dietary omega-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:1102–10. https://doi.org/10.1001/jamaneurol.2014.1214.
Stelmach-Mardas M, et al. Link between food energy density and body weight changes in obese adults. Nutrients. 2016;8:229. https://doi.org/10.3390/nu8040229.
Fergani A, et al. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J Lipid Res. 2007;48:1571–80. https://doi.org/10.1194/jlr.M700017-JLR200.
Zhao Z, Sui Y, Gao W, Cai B, Fan D. Effects of diet on adenosine monophosphate-activated protein kinase activity and disease progression in an amyotrophic lateral sclerosis model. J Int Med Res. 2015;43:67–79. https://doi.org/10.1177/0300060514554725.
Hamadeh MJ, Rodriguez MC, Kaczor JJ, Tarnopolsky MA. Caloric restriction transiently improves motor performance but hastens clinical onset of disease in the Cu/Zn-superoxide dismutase mutant G93A mouse. Muscle Nerve. 2005;31:214–20. https://doi.org/10.1002/mus.20255.
Coughlan KS, Halang L, Woods I, Prehn JH. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. Dis Model Mech. 2016;9:1029–37. https://doi.org/10.1242/dmm.024786.
Dorst J, et al. Effect of high-caloric nutrition on serum neurofilament light chain levels in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2020;91:1007–9.
Ludolph AC, et al. Effect of high-caloric nutrition on survival in amyotrophic lateral sclerosis. Ann Neurol. 2020;87:206–16. https://doi.org/10.1002/ana.25661.
Park JH, et al. Pyruvate slows disease progression in a G93A SOD1 mutant transgenic mouse model. Neurosci Lett. 2007;413:265–9. https://doi.org/10.1016/j.neulet.2006.11.058.
Esposito E, et al. Antioxidant strategies based on tomato-enriched food or pyruvate do not affect disease onset and survival in an animal model of amyotrophic lateral sclerosis. Brain Res. 2007;1168:90–6. https://doi.org/10.1016/j.brainres.2007.06.095.
Miquel E, et al. Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PLoS One. 2012;7: e34776. https://doi.org/10.1371/journal.pone.0034776.
Dowis K, Banga S. The potential health benefits of the ketogenic diet: a narrative review. Nutrients. 2021;13. https://doi.org/10.3390/nu1305165.
Zhao Z, et al. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006;7:29. https://doi.org/10.1186/1471-2202-7-29.
Tefera TW, et al. Triheptanoin protects motor neurons and delays the onset of motor symptoms in a mouse model of amyotrophic lateral sclerosis. PLoS One. 2016;11: e0161816. https://doi.org/10.1371/journal.pone.0161816.
Zhao W, et al. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS One. 2012;7: e49191. https://doi.org/10.1371/journal.pone.0049191.
Kira Y, Nishikawa M, Ochi A, Sato E, Inoue M. L-carnitine suppresses the onset of neuromuscular degeneration and increases the life span of mice with familial amyotrophic lateral sclerosis. Brain Res. 2006;1070:206–14. https://doi.org/10.1016/j.brainres.2005.11.052.
Beghi E, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:397–405. https://doi.org/10.3109/21678421.2013.764568.
Bordet T, et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322:709–20. https://doi.org/10.1124/jpet.107.123000.
Sunyach C, et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology. 2012;62:2346–52. https://doi.org/10.1016/j.neuropharm.2012.02.013.
Yang YM, et al. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 2013;12:713–26. https://doi.org/10.1016/j.stem.2013.04.003.
Lenglet T, et al. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur J Neurol. 2014;21:529–36. https://doi.org/10.1111/ene.12344.
Smith RN, Agharkar AS, Gonzales EB. A review of creatine supplementation in age-related diseases: more than a supplement for athletes. F1000Res. 2014;3:222. https://doi.org/10.12688/f1000research.5218.1.
Browne SE, et al. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol Dis. 2006;22:599–610. https://doi.org/10.1016/j.nbd.2006.01.001.
Klivenyi P, et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nature. 1999;5.
Groeneveld GJ, et al. A randomized sequential trial of creatinein amyotrophic lateral sclerosis. Ann Neurol. 2003;53.
Shefner JM, et al. A clinical trial of creatine in ALS. 2004;63(9):1656–61.
Rosenfeld J, et al. Creatine monohydrate in ALS: Effects on strength, fatigue, respiratory status and ALSFRS. Amyotroph Lateral Scler. 2008;9:266–72. https://doi.org/10.1080/17482960802028890.
Robertson J, et al. Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett. 2007;420:128–32. https://doi.org/10.1016/j.neulet.2007.03.066.
Cunnane SC, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609–33. https://doi.org/10.1038/s41573-020-0072-x.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This work was supported by the following sources: National Institutes of Health RF1-AG057882 (D.T.), Muscular Dystrophy Association grant 628389 (D.T.), and Family Strong for ALS & Farber Family Foundation (D.T.); F31-NS118838 (A.T.N.)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nelson, A., Trotti, D. Altered Bioenergetics and Metabolic Homeostasis in Amyotrophic Lateral Sclerosis. Neurotherapeutics 19, 1102–1118 (2022). https://doi.org/10.1007/s13311-022-01262-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01262-3