
Abstract
The emergence of ferroptosis as a cell death pathway associated with brain disorders including stroke and neurodegenerative diseases emphasizes the need to develop therapeutics able to target the brain and to protect neurons from ferroptotic death. Selenium plays an essential role in reducing lipid peroxidation generated during ferroptosis through its incorporation into the catalytic site of glutathione peroxidase 4. Here, we compared the anti-ferroptotic activity of several organic and inorganic selenium compounds: methylselenocysteine, selenocystine, selenomethionine, selenocystamine, ebselen, sodium selenite, and sodium selenate. All were effective against erastin- and RSL3-induced ferroptosis in vitro. We characterized the ability of the selenium compounds to release selenium and boost glutathione peroxidase expression and activity. Based on our results, we selected organic selenium compounds of similar characteristics and investigated their effectiveness in protecting against neuronal death in vivo using the cerebral ischemia–reperfusion injury mouse model. We found that pretreatment with methylselenocysteine or selenocystamine provided protection from ischemia–reperfusion neuronal damage in vivo. These data support the use of ferroptosis inhibitors for treatment and select selenium compounds for prevention of neuronal damage in ischemic stroke and other diseases of the brain where ferroptosis is implicated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Selenium is an essential element in humans and it is present in diet in multiple inorganic and organic forms. Selenomethionine is more bioavailable than inorganic forms of selenium such as selenite and selenate [1, 2]. Selenium is widely distributed in the body but animal studies suggested that selenium is preferentially retained in the brain under selenium-deficient conditions [3], and human studies showed that lower selenium levels are associated with cognitive decline and oxidative stress in the elderly [4,5,6]. Selenium supplementation has been investigated initially in multiple clinical trials mainly in the cancer prevention field with contradictory results generally attributed to the lack of information regarding selenium status of enrolled participants [7,8,9,10,11]. Selenium is transported in the body in the form of a selenocysteine-rich protein named selenoprotein P (SelP), which is taken up by cells through LRP2 and LRP8 receptors with LRP8 being the main selenium receptor expressed in neurons [12]. There are currently 25 selenoproteins in the human proteome classified into three subfamilies that include thioredoxin reductase, glutathione peroxidase, and iodothyronine deiodinases [13]. Since the discovery of ferroptosis in 2012 [14] and the emergence of the selenium-dependent glutathione peroxidase 4 (GPX4) as a master regulator of ferroptosis [15, 16], basic and clinical research on selenium-dependent glutathione peroxidase (GPX) has been linked to several brain diseases including neurodegenerative diseases and ischemic stroke [17,18,19,20,21].
Ferroptosis is a cell death pathway characterized by the build-up of lethal lipid hydroperoxides induced by iron-dependent catalytic mechanisms. Iron chelators such as deferiprone and lipophilic radical trapping agents such liproxstatin-1 protect against ferroptosis [14, 20]. The essential biological role of selenium as a major determinant of ferroptosis susceptibility is due to its functional incorporation in the active site of the GPX4 enzyme, which is the primary cellular defence against ferroptosis by detoxifying lipid hydroperoxides into lipid alcohols. Indeed, the specific incorporation of selenium as selenocysteine in GPX4 is critical for the survival of parvalbuminergic (PV+) interneurons, thereby preventing fatal epileptic seizures in mice [16]. Recently, the pharmacological supplementation of selenium (as a selenoprotein P fused to a transducing peptide) was demonstrated to drive a transcriptional response to augment GPX4 and other anti-ferroptotic genes to protect neurons from ferroptosis and prevent hemorrhagic and ischemic stroke in mice [22]. Here, we compare the anti-ferroptotic activities of several inorganic and organic forms of selenium using an in vitro neuronal cell culture model. We determine the protective effect of selected organic selenium compounds (selenocompounds) with similar anti-ferroptotic properties in transient middle cerebral artery occlusion (MCAO)–induced focal ischemia–reperfusion injury, an in vivo model of inducing ferroptosis in murine brains following ischemic stroke [19].
Results
Selenocompounds Confer a Potent Anti-ferroptotic Response in N27 Neuronal Cells
To evaluate the anti-ferroptotic activity of selenium, we selected a wide range of selenocompounds ranging from inorganic selenium (selenite, selenate), organic selenium (selenomethionine, selenocystine, methylselenocysteine, selenocystamine), and a synthetic selenocompound (ebselen). The effect of selenium on ferroptosis was evaluated in the N27 immortalized neuronal cell line model using liproxstatin-1 as the gold standard anti-ferroptotic drug for comparison and validation of ferroptosis (Fig. 1a). Ferroptosis was induced in two different modes using erastin or RSL3. Erastin, which is a potent inhibitor of system xCT, prevents cellular cystine import, leading to a concomitant blockade of glutathione synthesis, the cofactor of GPX4, ultimately inhibiting GPX4 activity. RSL3 is a direct inhibitor of GPX4 that induces ferroptosis by preventing GPX4-mediated clearance of lipid hydroperoxides.

Selenocompounds rescue ferroptosis in N27 cells. Ferroptosis was induced in N27 cells after incubation with either erastin (1 μM, blue line) or RSL3 (0.1 μM, red line) for 24 h. Toxicity of each compound was evaluated in the absence of ferroptosis inducers (control, black line). Dose–response curve of the standard anti-ferroptotic compound liproxstatin-1 a and the selenocompounds: sodium selenite b, sodium selenite c, selenomethionine d, selenocystine e, methylselenocysteine f, selenocystamine g, and ebselen h. Data are means ± SEM, n = 5 independent experiments with 4 replicates in each experiment. Data were fitted using a nonlinear regression curve to determine EC50 with 95% confidence interval as summarized in Table 1
All inorganic and organic selenocompounds were used at ≤ 1 μM, which was subtoxic but sufficient to provide full resistance to ferroptosis (11 < EC50 < 925 nM; Fig. 1b–g). Ebselen required higher (> 2 μM) concentrations to protect against ferroptosis (Fig. 1h). All selenocompounds exhibited dose-dependent rescue of cell death induced by erastin or RSL3; the EC50 of each compound against either toxin was similar. However, there was large variance in potencies between the different compounds (Table 1). The most potent selenocompounds were selenite (EC50erastin: 38 nM) and selenocystine (EC50erastin: 38 nM), which were slightly more potent than liproxstatin-1 (EC50erastin: 49 nM), followed by methylselenocysteine (EC50erastin: 146 nM) and selenocystamine (EC50erastin: 277 nM) with potencies 2- to fivefold lower than that of liproxstatin-1. Less potent compounds included selenomethionine (EC50erastin: 482 nM) and selenate (EC50erastin: 925 nM), while ebselen showed an atypical dose–response curve with a narrow micromolar concentration range (5–10 μM) exhibiting full anti-ferroptotic rescue concomitant with slight increase in toxicity in the absence of erastin/RSL3 (Fig. 1h).
Selenocompounds Increase Selenium Levels, GPX Expression, and Activity with Differing Responses
We surveyed the impact of the selenocompounds (at 1 μM for 24 h, except for ebselen which was also examined at 10 μM) on cellular selenium levels to test whether their impact on ferroptosis resistance was a simple product of selenium retention (Fig. 2a). Selenomethionine induced dramatically more selenium retention compared to the other organic selenocompounds and the degree of selenium retention did not readily account for ferroptosis resistance (Fig. 2a).

Selenocompounds increase intracellular selenium levels and GPX activities at different rates. a Total selenium levels measured by ICP-MS and b GPX activity were measured in N27 cells treated for 24 h with the different selenocompounds. All selenocompounds were used at the same concentration (1 μM), and ebselen was used at an additional higher dose (10 μM) as the determined EC50 was considerably higher than 1 μM. c Correlation between selenium level and GPX activity determined for the used selenocompounds indicate poor correlation for ebselen and selenomethionine (empty filled circles), Methyl-S-cys = methylselenocysteine. Data are means ± SEM, n = 3 independent experiments with 3 replicates in each experiment. P values were calculated using the Brown-Forsythe and Welch ANOVA test corrected for multiple comparison. *P < 0.05, ***P < 0.001
We next measured GPX activity in response to this supplementation with selenocompounds, since changes in selenium-containing GPX activity reflect the incorporation of selenium into the active sites of these enzymes. Inorganic and organic selenocompounds induced a comparable increase in GPX activity (Fig. 2b). Selenomethionine did not induce an increase in activity commensurate with the tenfold higher selenium levels that it induced (Fig. 2c). Therefore, although absorbed at a high rate, selenomethionine might not be contemporaneously converted into the selenocysteine that comprises the active site. In contrast, ebselen failed to increase GPX activity and only a slight increase in selenium was observed when a tenfold excess was used to treat the cells (Fig. 2b), indicating that ebselen does not rescue ferroptosis through a selenium-delivery mechanism (Fig. 2c).
We next analyzed the expression of three selenoproteins in response to selenocompound treatment (1 μM for 24 h): GPX1and GPX4 as the major selenium peroxidases and the selenium transport protein selenoprotein P (SelP) as a reference selenoprotein (Fig. 3). The selenocompounds induced an increase in GPX1 and GPX4 expression (Fig. 3a–c) commensurate with the increase in GPX activity (Fig. 2b). Expression of SelP was not altered by treatment (Fig. 3d). We repeated ebselen treatment using 1 and 10 μM and found that both concentrations were ineffective in increasing GPX1 levels while a slight increase in GPX4 expression was recorded when cells were treated with 10 μM (Fig. 3e, f).

Selenocompounds but not ebselen increase GPX protein levels. a Representative Western blot of selenoprotein expression in N27 cells after treatment with 1 μM of the selenocompounds for 24 h. Quantification of the expression of b GPX1, c GPX4, and d SelP was normalized to β-actin. e, f The effect of high doses of ebselen on the expression of selenoproteins in N27 cells. Data are means ± SEM, n = 3 independent experiments with 3 replicates in each experiment. P values were calculated using the Kruskal–Wallis test (b, c, d) and two-way ANOVA (f) corrected for multiple comparison. **P < 0.01, ***P < 0.001
Methylselenocysteine and Selenocystamine but not Selenocystine Are Protective Against MCAO-Induced Ischemic Injury in Mice
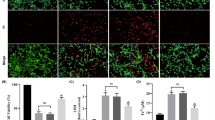
Based on our in vitro data, we selected selenocystine, selenocystamine, and methylselenocysteine as candidate drugs to test rescue against ferroptosis in vivo because they were organic compounds, exhibited weaker and stronger anti-ferroptotic activity, respectively (in order to try to achieve a preliminary study of structure–activity relationship), while only moderately increasing intracellular selenium (Table 2), and have reported a favourable toxicity profile over other selenocompounds in preclinical animal models [23, 24]. We chose MCAO-induced focal cerebral ischemia–reperfusion, a previously characterized model in mice, for inducing neuronal death following ischemic injury [19]. Previously, we validated the involvement of ferroptosis in neuronal death induced by MCAO in 3-month-old C57/BL6 mice by rescuing ischemia–reperfusion injury, and consequent neurological deficit, using intranasal administration of anti-ferroptotic compounds, liproxstatin-1 (antioxidant/radical trapping agent), ferrostatin-1 (antioxidant/radical trapping agent), and ML351 (15-lipoxygenase-1 inhibitor) [19]. We further confirmed the involvement of ferroptosis in our animal model and showed that GPX4 levels significantly decreased in the ipsilateral hippocampus brain region (Fig. 4a, b), and that GPX activity was reduced in the ipsilateral hippocampus and cortex regions (Fig. 4c) following ischemia–reperfusion injury. In contrast to the classical intranasal administration, which is preferred for the current anti-ferroptotic drugs due to their low penetrability in the brain, mice in this study received intraperitoneal injections (0.5 mg/kg) or vehicle (saline) for 5 days prior to MCAO. The dose of 0.5 mg/kg body weight is much lower than the reported LD50 for methylselenocysteine (LD50: 9.26 to 12.60 mg/kg body weight) [24].

Selenocompounds prevent ischemia/reperfusion neuronal damage in the MCAO mouse model. a Representative Western blot of GPX4 expression in hippocampus from the ipsilateral and contralateral brain regions in MCAO mice after 24-h reperfusion following ischemia, and the corresponding GPX4 expression levels normalized to β-actin are provided in b. c GPX activity measurement in the ipsilateral and contralateral hippocampus and cortex regions in MCAO mice after 24-h reperfusion following ischemia. d Pretreatment with either selenocystamine or methylselenocysteine but not selenocystine (0.5 mg/kg body weight for 5 days) attenuated progressive neurological deficits in the MCAO mouse model and reached significance 24 h post-reperfusion. e Representative images of stained brain sections showing the change of infarct area caused by MCAO 24 h after reperfusion in selenium-treated animals (scale bar 10 mm). f Quantification of the infarct volume in the MCAO model after 24-h reperfusion in control and selenium-pretreated animals. Data are means ± SEM, n = 5 (a–c), n = 4 (d–f). P values were calculated using unpaired t-test (b) and two-way ANOVA corrected for multiple comparison (c, d, and f). *P < 0.05, **P < 0.01, ***P < 0.001
We found that selenocystamine and methylselenocysteine were both effective in attenuating MCAO-induced neurological deficits 6 h post-reperfusion (selenocystamine—18%; methylselenocysteine—22%) and reached statistical significance 24 h post-reperfusion (selenocystamine—27%, P = 0.012; methylselenocysteine—31%, P = 0.0034; compared with lesioned control mice, two-way ANOVA corrected for multiple comparison with Dunnett; Fig. 4d). Furthermore, both selenocompounds markedly reduced infarct volume 24 h post-reperfusion with methylselenocysteine being the most potent compound (selenocystamine—42%, P = 0.0030; methylselenocysteine—58%, P = 0.0005, two-way ANOVA corrected for multiple comparison with Dunnett; Fig. 4e, f). In contrast, selenocystine failed to attenuate neurological deficits or infarct volume (Fig. 4d–f). A possible reason for this result is the low expression of xCT in vivo, which is the transporter that delivers selenocystine to the cell, while the expression of this cystine importer is known to be highly induced in cells cultured in vitro [25]. Taken together, this indicated that treatment with either selenocystamine or methylselenocysteine prior to ischemia–reperfusion injury prevents the loss of neurons and functional impairment, consistent with the in vitro activities of the compounds for inhibiting ferroptosis.
Discussion
Here, we report the pharmacology of different organic, inorganic, and synthetic selenocompounds against ferroptosis in vitro, and demonstrate that selenocystamine and methylselenocysteine are protective in the MCAO model, where ferroptosis is implicated. The translational potential of selenium for stroke is evidenced by a recent study that demonstrated that combined use of selenite with another anti-ferroptotic compound, N-acetylcysteine, improved multiple outcomes in a clinical trial of 123 patients with intracerebral haemorrhage [26]. This clinical trial supports prior findings in animal models that use selenium for hemorrhagic and ischemic stroke.
Recently, the potential benefit of systemic application of selenium in preventing neuronal ischemia–reperfusion injury was demonstrated in vivo using a selenopeptide, Tat Sel-Pep, a fusion of HIV-Tat protein, and C-terminal domain of SelP (23). However, small molecule selenocompounds such as sodium selenate were also shown to penetrate the brain to attenuate neuronal damage in other animal studies [27]. A comparison between different selenocompounds against ferroptosis, selenium delivery, and functional incorporation into selenoproteins has not previously been investigated. We found that both inorganic and organic selenocompounds have varying potencies against ferroptosis that are unlikely to be merely related to the extent of selenium uptake. Similarly, the degree of upregulation of different selenoenzymes did not explain the differences in potencies. This is perhaps not surprising, because elevating GPX4 may benefit a cell challenged by RSL3 (GPX4 inhibitor) or erastin (depleting the GSH substrate required for GPX4 activity). Therefore, these data might be consistent with a GPX4-independent mechanism of protection by selenium, such as activating the selenome gene transcription cassette [22].
Ebselen is used as an antioxidant that mimics GPX, but does not release its selenium for incorporation into selenoproteins [28, 29]. Ebselen has been previously reported to protect against ferroptosis in some in vitro studies [14, 30], but it has not been approved as a drug after it showed insufficient efficacy in clinical trials of cerebral ischemia [31]. We have confirmed that ebselen at pharmaco-therapeutically useful concentrations (e.g. 10 μM) does protect against ferroptosis using our neuronal cell model and does not release its selenium to be incorporated into GPX proteins in this system, in contrast to all other selenocompounds, which were effective in increasing both GPX expression and activity. Ebselen only showed anti-ferroptotic activity at micromolar concentrations that also induced some toxicity. In contrast, all other selenocompounds showed a robust anti-ferroptotic activity with selenocystine and selenite emerging as the most potent candidate drugs with EC50 values within the low nanomolar range comparable to the best-in-class current anti-ferroptotic compound, liproxstatin-1. Interestingly, we observed a tenfold increase in selenium upon treatment with selenomethionine but this increase did not translate into an increase in GPX protein expression or activity. This indicates that selenomethionine may not be readily transformed by the cells into the selenocysteine that comprises the GPX active site.
We tested the translatability of selenium supplementation in vivo in the mouse cerebral ischemia–reperfusion model, finding that methylselenocysteine and selenocystamine mitigated infarct volume and neurological deficits. The relative preventive potency matched the relative potencies of these compounds in preventing ferroptotic death in neuronal cells. The mechanism of preventing ferroptosis might be via upregulation of GPX activity or protective transcriptional changes prior to ischemia–reperfusion injury. Elevated GPX4 activity may arise from an enhanced selenium availability for de novo generation of GPX4 and/or from transcriptional changes driven by selenium [22]. These data therefore demonstrate the proof of principle that brain selenium confers protection against ischemic stroke, possibly via inhibition of ferroptosis. Thus, suggesting the use of ferroptosis inhibitors for treatment and select selenium compounds for prevention of neuronal damage in ischemic stroke. In summary, our data indicate that select selenocompounds afford robust protection against ferroptotic death in neuronal cells in vitro and in vivo, providing the rationale for evaluating their clinical efficacy as selective anti-ferroptotic drugs capable of crossing the blood–brain barrier and preventing neuronal death in ischemic stroke, with potential for other neurovascular and neurogenerative diseases where ferroptosis is implicated such as Alzheimer’s disease and Parkinson’s disease.
Methods
Cell Culture and Reagents
N27 cells, derived from E12 rat mesencephalic tissue (Cat# SCC048, Millipore), were cultured in RPMI 1640 medium (Cat# 72,400,120, Life Technologies Australia Pty., Ltd.) supplemented with 10% FBS (Bovogen Biologicals), penicillin (100 U/mL), and streptomycin (100 μg/mL) in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were utilized between passages 11 to 20. Erastin (Cat#S7242) and RSL3 (Cat#S8155) were purchased from Selleckchem. Liproxstatin-1 (Cat#SML1414) and MTT (Cat# M2128) and selenium compounds were purchased from Sigma-Aldrich.
Cell Treatments
Western blot analysis, metal measurement, and GPX activity assay were all evaluated from the same treated cells. All selenocompounds were dissolved in water. For each compound, cells were seeded in three 25-cm2 culture dish and incubated in growth media for 16 h before they were treated with the corresponding selenocompound or control (water) in growth media for a subsequent 24 h. Cells were harvested in phosphate-buffered saline (PBS), split into four tubes, and centrifuged, and pellets were stored at –20 °C for subsequent analyses. One pellet was used to determine protein concentration using the Pierce™ BCA protein assay (Thermo Scientific).
Selenium Measurement
Cell pellets were dissolved in concentrated nitric acid + hydrogen peroxide, and selenium content was measured by inductively coupled mass spectrometry (ICP-MS) with an Ultramass 700 (Varian, Victoria, Australia) as previously described [32] and selenium concentration was normalized to protein concentration.
GPX Activity Assay
Cell pellets were resuspended in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100 containing protease inhibitors (Roche)). Resultant cell lysate was centrifuged at 10,000 g for 10 min at 4 °C to remove insoluble cell debris, and supernatant was transferred to fresh micro tubes. In a 96-well plate, 20 μL of cell supernatant (30 μg total protein) was added to GPX assay buffer (final concentrations: Tris–HCl (pH 8.0, 50 mM), NADPH (0.5 mM), GSH (0.25 mM), glutathione reductase (0.5U/mL)) and mixed. Tert-butyl hydroperoxide (t-Bu-OOH) substrate was added to a final concentration of 250 μM to initiate the reaction. NADPH was measured kinetically by reading absorbance at 340 nm every 15 s over 30 min. GPX activity was normalized to protein concentration and determined as micromolars per minute per milligram of protein (units/mg protein).
Western Blot Analysis
Proteins were extracted from cell pellets using RIPA buffer as described above. Fifteen micrograms of protein was separated by SDS-PAGE on 4–20% Bis–Tris protein gels (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membrane using iBlot (Invitrogen). Primary antibodies used were rabbit anti-GPX1 (Cat#ab22604, Abcam), rabbit anti-GPX4 (Cat#ab125066, Abcam), and mouse anti-SelP (Cat#sc-376858, Santa Cruz Biotechnology). Mouse anti-β-actin (Cat#A5441, Sigma) was used as a loading control. Membranes were probed with horseradish peroxidase-conjugated secondary antibodies and signal was detected with ECL reagent (GE Healthcare Life Science) and a LAS-4000 Luminescence Imaging Analyzer (GE Healthcare Life Science). Densitometry analyses were carried out using ImageJ software, and quantitation was normalized to β-actin levels.
Cell Viability Assay
Cell viability was evaluated using the MTT colorimetric assay as previously described [21]. Briefly, cells were cultured in 96-well plates at a density of 20,000 cells/well in growth media for 8 h. Cells were subsequently incubated with selenocompounds in growth medium for 16 h prior to treatment with ferroptosis toxins (erastin/RSL3) and additional compounds (selenocompounds/liproxstatin-1) in growth medium for an additional 24 h. MTT was then added to the plate and absorbance was measured at 570 nm using a microplate reader (BioTek). Cell viability was expressed as a percentage of control cells.
Animal Studies
All mice were purchased from Beijing Huafukang Bioscience Company (Beijing, China). Adult male C57BL/6 mice (25–30 g) were housed under standard conditions of temperature and humidity, and a 12-h light/dark cycle (lights on at 08:00), with free access to food and water before use. Adequate measures were taken to minimize pain or discomfort during surgeries. Experiments were carried out following the Institutional Guidelines of the Animal Care and Use Committee (K2018071, Sichuan University, China). Mice were chosen randomly for treatment. Selenocystine (0.5 mg/kg/day, i.p., S1650, Sigma-Aldrich), selenocystamine (0.5 mg/kg/day, i.p., S0520, Sigma-Aldrich), methylselenocysteine (0.5 mg/kg/day, i.p., M6680, Sigma-Aldrich), or saline (0.9% NaCl) were administered for 5 days before the induction of focal cerebral ischemia in C57BL/6 mice.
Focal Cerebral Ischemia Model
All surgeries were conducted under aseptic conditions by a skilled animal surgeon. Transient acute focal cerebral ischemia was induced by reversible intraluminal MCAO, as described previously [19]. Mice were anesthetized with isoflurane (5% induction, and 1% maintenance). A 2-cm incision was opened in the middle of the anterior neck. Left unilateral MCAO was accomplished by inserting a silicon rubber-coated nylon monofilament (Guangzhou Jialing Biotechnology Co., Ltd., China) into the internal artery via the common carotid artery, advanced 9–10 mm past the carotid bifurcation until a slight resistance was felt. Adequacy of MCAO was confirmed by monitoring cortical blood flow using a PeriCam PSI System (Perimed, Järfälla, Sweden). Animals were excluded if mean ipsilateral laser speckle signal was > 30% of the pre-ischemic ipsilateral hemisphere baseline. Body temperature was controlled at 36.5 ± 0.5 °C throughout MCAO surgery with a heating pad. After 60 min of occlusion, the occluding filament was withdrawn to allow for reperfusion and the incision was closed with 4–0 surgical sutures (Jinhuan, Co., Ltd.). In the sham-operated animals, the occluding filament was inserted only 5 mm above the carotid bifurcation. The surgeon was blinded to treatment groups.
Western Blot and GPX Activity in Tissue Extract
Mice were deeply anesthetized using chloral hydrate (BBI Life Sciences) and transcardially perfused with PBS before the brains were removed. The cerebral cortex and hippocampus were dissected and homogenized in ice-cold lysis buffer containing 50 mM Tris–HCl (pH = 7.6), 150 mM NaCl, 1%(v/v) Triton X-100, protease inhibitor cocktail (1:50, Roche), and phosphatase inhibitors II and III (1:1000). After clearing debris by centrifuging at 14,000 × g at 4 °C, protein concentration in the extracts was determined by BCA Protein Assay Kit (Beyotime). Western blot analysis and GPX activity were measured following the protocols described above.
Neurological Assessment
The neurological assessment post-surgery was performed by an investigator blinded to the experimental groups and confirmed by a second investigator blinded to the experimental groups. After 0, 6, and 24 h of MCAO/reperfusion, the neurological deficit of each mouse was evaluated by a 5-point scale as described previously [19]: 0, no observable deficit; 1, right forelimb flexion; 2, decreased resistance to left lateral push (and right forelimb flexion) without circling; 3, the same behaviour as grade 2, with circling to right; 4, severe rotation progressing into barreling, loss of walking, or righting reflex.
Infarct Volume Analysis
The individual performing the infarct volume analysis was blinded to the treatment group. At 24 h of reperfusion, mice were euthanized, and brains were removed rapidly and placed at –20 °C for 20 min. Coronal slices were made at 2-mm intervals from the frontal poles, and the 2-mm brain sections were incubated in 1% 2,3,5-triphenyl tetrazolium chloride (TTC, T8877, Sigma-Aldrich) in phosphate-buffered saline (PBS) for 15 min at 37 °C, and then fixed in 10% formalin for 24 h. Infarction volume was measured using digital imaging, and images were analyzed using ImageJ (1.49 m, NIH) by an investigator blinded to the experimental groups. The area of infarct (white, unstained), the area of the ipsilateral hemisphere (white, unstained, plus red brick, stained), and the area of the contralateral hemisphere (red brick, stained) were measured for each section by a blinded operator. The volume was calculated by summing the representative areas in all sections and multiplying by the slice thickness, then correcting for edema, as previously described [19]: corrected infarct volume (CIV) = contralateral hemisphere volume − (ipsilateral hemisphere volume − infarct volume).
Data and Statistical Analyses
All quantitative data were analyzed using Prism 9.0.1 software (GraphPad). Replicates within experiments and three independent experiments were used to ensure reliability of the reported values. Data were expressed as mean ± standard error of the mean (SEM) from independent experiments. Unpaired t-test was used to compare between two groups, and one-way and two-way ANOVAs were used to compare between three or more groups and calculate the P-value as indicated in each figure legend. P-value < 0.05 was considered statistically significant. For the cell viability data, nonlinear regression analysis with a variable slope model was used to fit a curve to dose response and calculate EC50 with 95% confidence interval.
References
Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci. 2014;39(3):112-20.
Pillai R, Uyehara-Lock JH, Bellinger FP. Selenium and selenoprotein function in brain disorders. IUBMB Life. 2014;66(4):229-39.
Prohaska JR, Ganther HE. Selenium and glutathione peroxidase in developing rat brain. J Neurochem. 1976;27(6):1379-87.
Akbaraly TN, Hininger-Favier I, Carriere I, Arnaud J, Gourlet V, Roussel AM, et al. Plasma selenium over time and cognitive decline in the elderly. Epidemiology. 2007;18(1):52-8.
Gao S, Jin Y, Hall KS, Liang C, Unverzagt FW, Ji R, et al. Selenium level and cognitive function in rural elderly Chinese. Am J Epidemiol. 2007;165(8):955-65.
Shahar A, Patel KV, Semba RD, Bandinelli S, Shahar DR, Ferrucci L, et al. Plasma selenium is positively related to performance in neurological tasks assessing coordination and motor speed. Movement disorders : official journal of the Movement Disorder Society. 2010;25(12):1909-15.
Clark LC, Combs GF, Jr., Turnbull BW, Slate EH, Chalker DK, Chow J, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276(24):1957–63.
Hatfield DL, Gladyshev VN. The Outcome of Selenium and Vitamin E Cancer Prevention Trial (SELECT) reveals the need for better understanding of selenium biology. Mol Interv. 2009;9(1):18-21.
Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39-51.
Rayman MP. Selenium and human health. Lancet. 2012;379(9822):1256-68.
Nicastro HL, Dunn BK. Selenium and prostate cancer prevention: insights from the selenium and vitamin E cancer prevention trial (SELECT). Nutrients. 2013;5(4):1122-48.
Krol MB, Gromadzinska J, Wasowicz W. SeP, ApoER2 and megalin as necessary factors to maintain Se homeostasis in mammals. J Trace Elem Med Biol. 2012;26(4):262-6.
Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, et al. Characterization of mammalian selenoproteomes. Science. 2003;300(5624):1439-43.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-72.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180-91.
Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell. 2018;172(3):409–22 e21.
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6(1):49.
Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med. 2019;133:221-33.
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Molecular psychiatry. 2017;22(11):1520-30.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273-85.
Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020;177(3):656-67.
Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell. 2019;177(5):1262–79 e25.
Cao S, Durrani FA, Toth K, Rustum YM. Se-methylselenocysteine offers selective protection against toxicity and potentiates the antitumour activity of anticancer drugs in preclinical animal models. Br J Cancer. 2014;110(7):1733-43.
Yang H, Jia X. Safety evaluation of Se-methylselenocysteine as nutritional selenium supplement: acute toxicity, genotoxicity and subchronic toxicity. Regul Toxicol Pharmacol. 2014;70(3):720-7.
Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants & redox signaling. 2013;18(5):522-55.
Kim M, Byun J, Chung Y, Lee SU, Park JE, Park W, et al. Reactive Oxygen Species Scavenger in Acute Intracerebral Hemorrhage Patients: A Multicenter, Randomized Controlled Trial. Stroke. 2021;52(4):1172-81.
Sanchez-Elexpuru G, Serratosa JM, Sanchez MP. Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of Lafora disease. Epilepsia. 2017;58(3):467-75.
Muller A, Cadenas E, Graf P, Sies H. A novel biologically active seleno-organic compound--I. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem Pharmacol. 1984;33(20):3235–9.
Wendel A, Fausel M, Safayhi H, Tiegs G, Otter R. A novel biologically active seleno-organic compound--II. Activity of PZ 51 in relation to glutathione peroxidase. Biochem Pharmacol. 1984;33(20):3241–5.
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555-68.
Parnham MJ, Sies H. The early research and development of ebselen. Biochem Pharmacol. 2013;86(9):1248-53.
Belaidi AA, Gunn AP, Wong BX, Ayton S, Appukuttan AT, Roberts BR, et al. Marked Age-Related Changes in Brain Iron Homeostasis in Amyloid Protein Precursor Knockout Mice. Neurotherapeutics. 2018;15(4):1055-62.
Funding
This study received funds from the Ministry of Science and Technology of China (2018YFC1312300), the National Natural Science Foundation of China (82071191, 81801182, 81722016), the National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Z2018B03), Sichuan University postdoctoral interdisciplinary Innovation Fund, the Post-Doctor Research Project, West China Hospital, Sichuan University (2018HXBH044), and the Alzheimer’s Association (AARFD-16–442821).
Author information
Authors and Affiliations
Contributions
Disclosure forms provided by the authors are available with the online version of this article.
Corresponding authors
Ethics declarations
Conflict of Interest
AIB is a shareholder in Alterity Ltd., Cogstate Ltd., and Mesoblast Ltd. He is a paid consultant for, and has a profit share interest in, Collaborative Medicinal Development Pty Ltd.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tuo, QZ., Masaldan, S., Southon, A. et al. Characterization of Selenium Compounds for Anti-ferroptotic Activity in Neuronal Cells and After Cerebral Ischemia–Reperfusion Injury. Neurotherapeutics 18, 2682–2691 (2021). https://doi.org/10.1007/s13311-021-01111-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01111-9