
Abstract
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease for which there are limited therapeutic strategies. Protein phosphatase 2A (PP2A) activity is decreased in AD brains, which promotes the hyperphosphorylation of Tau and APP, thus participate in the formation of neurofibrillary tangles (NFTs) and β-amyloid (Aβ) overproduction. In this study, the effect of synthetic tricyclic sulfonamide PP2A activators (aka SMAPs) on reducing AD-like pathogenesis was evaluated in AD cell models and AD-like hyperhomocysteinemia (HHcy) rat models. SMAPs effectively increased PP2A activity, and decreased tau phosphorylation and Aβ40/42 levels in AD cell models. In HHcy-AD rat models, cognitive impairments induced by HHcy were rescued by SMAP administration. HHcy-induced tau hyperphosphorylation and Aβ overproduction were ameliorated through increasing PP2A activity on compound treatment. Importantly, SMAP therapy also prevented neuronal cell spine loss and neuronal synapse impairment in the hippocampus of HHcy-AD rats. In summary, our data reveal that pharmacological PP2A reactivation may be a novel therapeutic strategy for AD treatment, and that the tricyclic sulfonamides constitute a novel candidate class of AD therapeutic.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is one of the most widely prevalent neurodegenerative diseases, and despite the significant and growing disease burden, there are no treatments capable of curing or even slowing AD progression. AD is characterized by two core pathologies: first extracellular deposits of β-amyloid (Aβ) plaques, and second, hyperphosphorylation of tau, a microtubule-associated protein that leads to intracellular formation of neurofibrillary tangles (NFTs) in the brain [1, 2]. Phosphorylation of tau is part of a normal intracellular trafficking to remove tau from microtubules, which is followed by dephosphorylation to allow tau to return to microtubules in normal cell function [3]. However, in AD and other tauopathies, tau is abnormally hyperphosphorylated, giving soluble phospho-tau species; these then aggregate into paired helical filaments (PHFs) and form NFTs [4]. Aβ peptides are produced by the cleavage of amyloid precursor protein (APP) via β-site APP cleaving enzyme 1 (BACE1) and γ-secretase, released and deposited into extracellular senile plaques [5]. Tau phosphorylation and dephosphorylation is modulated by protein kinases (PKs) and protein phosphatases (PPs), respectively. The functional imbalance between PKs and PPs contributes to tau hyperphosphorylation and AD pathogenesis [6, 7]. Also APP pathogenicity is regulated by phosphorylation. APP phosphorylation at Thr668 facilitates β-cleavage of APP [8, 9].
Protein phosphatase 2A (PP2A) is a ubiquitously expressed serine/threonine phosphatase and is a central factor in the regulation of diverse cellular processes including proliferation, growth, metabolism, cell motility, and apoptosis [10, 11]. The holoenzyme of PP2A is composed of a scaffolding subunit (A), a regulatory subunit (B), and a catalytic subunit (C). Both A and C subunits have two isoforms, α and β, and together form a catalytically competent AC heterodimer. Four families of regulatory, B subunit combine to form PP2A ABC holoenzymes, and the B subunit determines the substrate specificity and subcellular localization of PP2A heterotrimers [12, 13]. A second level of PP2A regulation is via endogenous cellular inhibitor proteins, prominently SET and CIP2A; they are overexpressed in multiple cancers [14], chronic obstructive pulmonary disease (COPD) [15], and AD [16] where they are associated with reduced PP2A activity.
PP2A phosphatase activities are decreased in postmortem brains of AD patients [17]. Recently, we demonstrated that overexpression of PP2A inhibitor protein CIP2A in normal mouse brain induced both Tau and APP phosphorylation, and full spectrum of AD characteristics including memory loss [16]. An evaluation of the dephosphorylation ability of different phosphatases suggested that PP2A makes up for about 71% of the total tau phosphatase activity in human brain [18]. In addition, PP2A inhibition enhances APP phosphorylation at Thr668, which increases the generation of Aβ in most studies [8, 16]. Therefore, abnormal suppression of PP2A activity is critical to both key AD-related pathogenesis: tau hyperphosphorylation and Aβ overproduction. Thus, PP2A reactivation may be an effective therapeutic strategy for the treatment of AD [19]. Some drugs, chemicals and peptides such as metformin [20], FTY720 [21], sodium selenate [22], COG1410 [23], and EHT [24] have been tested in AD models as PP2A activators, albeit by indirect mechanisms.
Homocysteine (Hcy) is an intermediate product of the methionine cycle. Hyperhomocysteinemia has been proposed to be an independent risk factor of AD [25, 26]. One of the neurotoxic effects of Hcy is to suppress the methylation of proteins, such as PP2A; reduced C-terminal methylation at Leu309 suppresses PP2A activity through interfering with B subunit loading to form PP2A ABC holoenzymes [27, 28]. Thus, HHcy-induced AD-like pathological features may be partially attributed to suppression of PP2A activity and a direct PP2A activator may reverse the associated pathology.
Recently, we reported characterization of a class of synthetic tricyclic sulfonamides which function as direct small-molecule activators of PP2A (SMAPs). Based on photoaffinity labeling studies, radiolytic footprinting, and mutagenesis of the binding site, the target of SMAPs has been identified as the interface of the A and C subunits in the PP2A complex [29]. Importantly, in cancer models, the effects of SMAPs can be reversed by either mutation of the drug-binding site on PP2A A-subunit, or overexpression of PP2A inhibitor small-T polyoma viral [29,29,30,32]. These studies clearly verify PP2A as a main target for SMAPs. Relevant to the current study, the SMAPs cross the blood-brain barrier after oral administration, and are active in an intracranial mouse model of invasive glioblastoma [33]. Given the above, and the significance of diminished PP2A activity in AD, we decided to investigate PP2A reactivation by SMAPs in neurons undergoing AD-like pathogenesis in vitro and in vivo. As a result, we found that two SMAPs (DBK-1154 and DBK-1160) decrease tau phosphorylation and Aβ40/42 generation in the HEK293/tau and N2a/APP cells. In HHcy-AD rat models, DBK-1160 effectively relieved the HHcy-induced cognitive deficits, and reduced the HHcy-induced tau hyperphosphorylation and Aβ overproduction through recovering PP2A activity. Moreover, DBK-1160 could simultaneously preserve the spine loss and synapse impairment.
Materials and Methods
Reagents
The primary antibodies used in this study and their properties are shown in Table 1. Secondary antibodies for Western blotting were purchased from Amersham Pharmacia Biotech (Little Chalfort, Buckinghamshire, UK). dl-Homocysteine (Hcy) was from Sigma (St. Louis, MO, USA). The chemical structures of DBK-1154 and DBK-1160 are shown in supplementary Fig 1 and their synthesis has been described previously [29]. Positive control compound SCR1693, a novel tacrine-dihydropyridine hybrid which has been proved to efficiently reduce tau pathology and amyloidosis [34, 35], was synthesized by Simovay (Purity: 99.3%; Jiangsu Simovay Pharmaceutical Co., Ltd. Nanjing, China).
Cell Culture and Treatment
HEK293/tau (HEK293 cells were stably transfected with human Tau 40) cell line was established in our laboratory. N2a/APP (N2a cells were stably transfected with human APP) cell line was gifted by Dr. Jia-zan Ni (Shenzhen University, China). Cells were seeded in 6-well plates in high-glucose DMEM with 200 mg/ml G418 and 10% fetal bovine serum in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. The cells were cultured for at least 24 h after plating. When the cell density reached to 70–80%, to minimize stress responses induced by serum deprivation, cells were switched to 0.5% fetal bovine serum media for 24 h, kept in serum- and G418-free high-glucose DMEM media for 2 h. Then the cells were treated with DBK-1154 or DBK-1160 (dissolved in DMSO) in different concentrations for 24 h. At the end of incubation, cells were collected and used for following tests.
LDH Cytotoxicity Assay
For LDH cytotoxicity assay, cells were cultured in 96-well plates (100 μl, 5 × 103 per well). Pierce LDH Cytotoxicity Assay Kit 88953 (Thermo scientific, USA) was used to detect the compounds’ (DBK-1154 and DBK-1160) toxic effect on HEK293/tau and N2a/APP cells according to the manufacturer’s instruction.
Animals and Drug Administration
Forty-nine male Sprague-Dawley rats (8 weeks old, 250 ± 25 g) were supplied by the Experimental Animal Central of Disease Control and Prevention Center of Hubei Province. These rats were housed five ones per cage with free access to food and water under a 12:12-h reversed light-dark cycle, relative humidity (55 ± 15%), and a constant room temperature (22 ± 2 °C). All experiments in this study were approved by the Animal Care and Use Committee of Huazhong University of Science and Technology, and performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Rats were divided into five groups including groups of control (control group), homocysteine injection (Hcy group), SMAP preventive (Hcy-P group), SMAP treatment (Hcy-1160 group), and positive control (Hcy-SCR group) (Table 2). Hcy was dissolved in normal saline (0.9% NaCl) to a final concentration of 400 μg/ml immediately before injection. The rats were injected via the vena caudalis with Hcy (400 μg/kg/day [36]) or normal saline (Ctrl) from 9:00 am to 2:00 pm for 14 days as previously reported [36]. DBK-1160 was solubilized in DMA, and then was added with Solutol–HS® (BASF; Lot 58169868EO, Germany) to obtain a clear organic mix. The mixture was then heated gently with addition of water to obtain a clear homogeneous solution. SCR-1693 was dissolved in normal saline (0.9%NaCl) to a final concentration of 0.25 mg/ml.
On day 1 to day 14, rats in the control group were injected with normal saline and the rats in groups of Hcy, Hcy-1160, and Hcy-SCR were injected with Hcy (400 μg/kg/day) via the vena caudalis. At the same time, rats in Hcy-P group were administered with DBK-1160 (twice a day and 35 mg/kg, oral gavage, based on the dosage used in mice, unpublished data). On day 15 to day 28, rats in the control, Hcy, and Hcy-P groups were only administered with normal saline (oral gavage). Rats in the Hcy-1160 and Hcy-SCR groups were administrated with DBK-1160 (twice a day and 35 mg/kg, oral gavage) or SCR-1693 (1 mg/kg/day, oral gavage) respectively (Table 2 and Fig. 3a).
Animal Behavior Tests
Open-Field Test
Locomotion activity was explored by open-field test (OFT). Before the test, rats were placed in the testing room for 3 h of habituation. The test was performed as previously reported [37]. Rats were placed inside a novel box, and movements inside the box were tracked over the period of 5 min (AniLab software and Instruments Co., Ltd., Chengdu, China). The total distance, average speed, center duration%, and center distance% were used to assess the general locomotor function of the rats.
Morris Water Maze Test
Spatial learning memory was detected by the Morris water maze (MWM) test. Three days after OFT, the MWM test was performed as previously described [38]. Rats were handled to swim freely for 60 s in the pool with the platform absent before the test. For spatial learning, rats were trained to be able to find the submerged platform within 60 s in each trial. If the rats failed to find the hidden platform within 60 s, they were manually guided to the platform and stayed there for 30 s. Each rat was subjected to 15 training trials: 3 trials per day for 5 training days. For spatial memory test on day 6, the platform was removed and escape latency, platform crossings times, duration in platform quadrant, and nonplatform quadrants were monitored by Noldus video tracking system (Ethovision, Noldus Information Technology, Holland).
Fear Conditioning Test
Seven days after MWM test, the cognitive function of the rats was further detected by fear conditioning test. Rats were placed into a square chamber (40 cm × 40 cm × 50 cm) with white board walls, a transparent front door, and grid floor. The equipment was cleaned with 75% ethanol and wiped with paper towels before each trial. On the first day, each rat was habituated to the chamber for 3 min, then followed by two foot shocks with a 2-min interval (0.5 mA, 2 s for each foot shock). The rat was then kept in the same chamber box for another 1 min and returned back to the cage. Twenty-four hours later, rats were sent into the same chamber for 3 min without foot shock for fear memory test. Total freezing time was recorded and analyzed.
Immunoprecipitation
The rat brain hippocampal tissues and cells were washed twice by PBS (4 °C, pH 7.5) and lysed with RIPA buffer (10,000 cells:1 μl, 1 mg:10 μl, Beyotime Biotechnology, Shanghai, China) with addition of 100 mg/ml phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor cocktail (EDTA-free, 1:100, Yeasen Biotech, Shanghai, China), then sonicated for 15 s on ice. The extracts (about 200 μg total proteins) were incubated with antibodies at 4 °C overnight, followed with addition of protein G agarose and incubated for 4 h at 4 °C. The agarose beads were collected, washed twice by PBS, and resuspended in 40 μl of loading buffer containing 50 mM Tris-HCl, pH 7.6, 2% sodium dodecyl sulfate (SDS), 10% glycerol, 10 mMDTT, and then added 0.2% bromophenol blue, boiled for 10 min for Western blotting.
Western Blotting
Cells were washed twice by PBS (4 °C, pH 7.5) and lysed with RIPA buffer (10,000 cells: 1 μl), 100 mg/ml PMSF, and protease inhibitor cocktail (EDTA-free, 1:100), with further addition of one volume of 2 × loading buffer containing Tris-Cl (pH 7.6) 100 mmol/L, 4% SDS, 20% glycerol, and DTT 200 mmol/L, boiled for 10 min and sonicated for 15 s.
Hippocampus tissues were separated from the rat brain and washed twice by PBS (4 °C, pH 7.5) and homogenated with RIPA buffer (1 mg tissue:10 μl), 100 mg/ml PMSF, and protease inhibitor cocktail (1:100). One volume of 2 ×loading buffer was added after homogenization, followed by 10-min boiling in water bathing and sonication for 15 s. Protein (cell and tissue extracts) concentration was measured by BCA kit (Pierce, Rockford, IL, USA), followed by electrophoresing via SDS-polyacrylamide gel (10% gel), and then transferred to nitrocellulose membranes (Amersham Biosciences, USA). After blocking in 5% non-fat milk for 1 h at room temperature, the membranes were incubated with primary antibody at 4 °C overnight. Then after washing in TBST, the membranes were incubated with secondary antibodies at room temperature for 1 h and visualized by Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, United States). Image J software (Rawak Software, Inc. Germany) was used to analyze the protein bands.
PP2A Activity Assay
The Serine/Threonine Phosphatase Assay System kit V2460 was used to detect PP2A activity in the HEK293/tau, N2a/APP cell extracts, and brain homogenates according to the manufacturer’s procedure (Promega, Madison, USA). In brief, cells or brain tissues were homogenized at 4 °C for 30 min using phosphatase free storage buffer (10,000 cells: 1 μl or 1 g tissue: 3 ml). Then cell lysates or tissue extracts were loaded into a Spin Column, centrifuged at 600×g at 4 °C for 5 min to remove the free phosphates. Protein concentrations of the phosphate-free samples were then assayed by using BCA kit (Pierce, Rockford, IL, USA). Five micrograms of protein samples in triplicates was incubated with a chemically synthesized phosphopeptide (RRA(pT) VA), an optimal substrate for PP2A, PP2B, and PP2C, but not for PP1 in a buffer optimized for PP2A activity while cation-dependent PP2B and PP2C were inhibited (protocol provided by the manufacturer) [39] for 30 min at 32 °C. Phosphate released from the substrate was detected by measuring the absorbance of a molybdate-malachite green-phosphate complex at 630 nm. PP2A activity was evaluated by the release of phosphate per microgram of protein and per minute (pmol/μg/min).
BACE Activity Assay
β-Secretase Activity Fluorometric Assay kit (Catalog no. K360-100) was applied to detect BACE activity in the N2a/APP cell extracts and brain homogenates according to the manufacturer’s procedure (BioVision, CA, USA).
Enzyme-Linked Immune-Sorbent Assay
The levels of Aβ40 and Aβ42 in cells and brain tissues were quantified by using a sandwich ELISA kit (Elab-science Biotechnology, Wuhan, China). N2a/APP cells or hippocampal tissues were homogenized by PBS (150–200 μl: 1 × 106 cells or 9 ml: 1 g tissue) with addition of protease inhibitor PMSF (100 mg/ml) and crushed by freeze/thaw. Then samples were centrifuged for 10 min at 5000×g to obtain the supernatant and Aβ40 /Aβ42 levels were detected according to the manufacturer’s instructions.
Golgi Staining
FD Rapid Golgi Staining Kit PK401 (FD NEURO TECHNOLOGIES, INC, Columbia MO, USA) was used to detect the morphology of neuronal dendrites and dendritic spines. In brief, the rats were decapitated under deep anesthesia, and brains were removed immediately and washed by normal saline. Golgi staining was then performed according to the manufacturer’s instructions.
Nissl Staining
The brain after perfusion and fixation was sliced at 25-μm thickness and the sections were mounted on gelatin-coated slides. Nissl staining solution was used to stain the neurons according to the manufacturer’s procedure (Beyotime Biotechnology, Shanghai, China).
Statistical Analyses
All data were expressed as mean ± SD or mean ± SEM and analyzed by GraphPad Prism 6 statistical software (GraphPad Software, Inc., La Jolla, CA). The one-way analysis of variance (ANOVA) procedure was used to determine the differences among groups followed by LSD’s post hoc tests to determine the differences among two groups. P < 0.05 was considered statistically significant in all experiments. All results shown correspond to individual representative experiments.
Results
SMAPs Reduce Tau Phosphorylation Through Activating PP2A in HEK-293/Tau Cells
To explore the effect of SMAPs on tau phosphorylation, HEK293/tau cells were treated with SMAPs (DBK-1154 or DBK-1160) in different concentrations (0, 0.1, 1, 5, 10 μM) for 24 h. LDH assay results showed that 5 μM of DBK-1160 or DBK-1154 was the highest dosage with tolerable cytotoxicity for HEK293/tau cells (Fig. 1a). In concentration lower than 5 μM, PP2A activity showed dose-dependent increase, with the highest at 5 μM after administration of DBK-1154 and DBK-1160 for 24 h (Fig. 1b). Compared with DBK-1154, DBK-1160 was more efficient in activation of PP2A, with a lower cytotoxicity (Fig. 1a, b).

SMAP (DBK-1154 or DBK-1160) treatments decrease tau phosphorylation levels in HEK-293/tau cells. a LDH assay in HEK293/tau cells treated with increasing doses of DBK-1154 or DBK-1160 (0, 0.1, 1, 5, and 10 μM) at 24 h. b PP2A activity assay in HEK293/tau cells treated with different doses of DBK-1154 or DBK-1160 (0, 0.1, 1, and 5 μM) at 24 h. **P < 0.01 versus DBK-1154. c Western blots for tau phosphorylation levels at the sites of pS199, pT205, pT231, pS396, pS404, and total tau (Tau-5) in HEK-293/tau cells treated with DBK-1154 or DBK-1160 (5 μM) for 24 h. d–i Quantification of the relative protein expression levels (pS199, pT205, pT231, pS396, pS404, and Tau-5) after normalization to the GAPDH signal. Data represent mean ± SD, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 versus control. 1160: DBK-1160, 1154: DBK-1154
With DBK-1154 or DBK-1160 treatment, tau phosphorylation levels at several AD-related sites, including Ser199 (pS199), Thr205 (pT205), Thr231 (pT231), and Ser396 (pS396), were significantly decreased whereas no significant effects were observed in total tau levels (Tau-5) (Fig. 1c–i). All these above sites are dephosphorylation sites for PP2A [17, 18], supporting the conclusions that SMAP treatment alleviates tau phosphorylation through activating PP2A.
SMAPs Decrease Aβ Production Through Modulating APP Phosphorylation and Secretase Activity in N2a/APP Cells
To further investigate the effect of SMAPs on Aβ generation, we incubated N2a/APP cells with SMAPs (DBK-1154 or DBK-1160). Similar with that in HEK293/tau cells, LDH assay results revealed that 5 μM of DBK-1160 or DBK-1154 was the highest dosage with tolerable cytotoxicity for N2a/APP cells (Fig. 2a). At 5 μM, PP2A activity also reached the highest level, and DBK-1160 had a higher PP2A activation efficiency than DBK-1154(Fig. 2b). ELISA results showed that both intracellular Aβ40 and Aβ42 levels were decreased upon SMAPs treatment in a dose-dependent manner, with 5 μM having the most effect (Fig. 2c, d).

SMAPs (DBK-1154 or DBK-1160) treatments reduce Aβ generation in N2a/APP cells. a LDH assay in N2a/APP cells treated with increasing doses of DBK-1154 or DBK-1160 (0, 0.1, 1, 5, and 10 μM) at 24 h. b PP2A activity assay in N2a/APP cells treated with different doses of DBK-1154 or DBK-1160 (0, 0.1, 1, and 5 μM) at 24 h. c, d ELISA assay of intracellular Aβ40 and Aβ42 levels after treatment with SMAPs at different dosages (0, 0.1, 1, and 5 μM) for 24 h. e Western blots for APP, sAPPβ, pT668APP, BACE1, and PS1 in N2a/APP cells treated with DBK-1154 or DBK-1160 (5 μM) for 24 h. f–j Quantification of the relative protein expression level (APP, sAPPβ, pT668APP, BACE1, and PS1) after normalization to the GAPDH signal. k IP blots for BACE1 interacted with APP in N2a/APP cells treated with DBK-1154 or DBK-1160 (5 μM) for 24 h. l Quantitative analysis of the IP blots. m BACE activity assay in N2a/APP cells treated with DBK-1154 or DBK-1160 (5 μM) at 24 h. Data represent mean ± SD, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 versus control. 1160: DBK-1160, 1154: DBK-1154
APP is cleaved by BACE1 to generate sAPPβ, which is further cleaved by γ-secretase to release Aβ peptides [40]. In addition, phosphorylation of APP at Thr668 (pT668APP) may increase Aβ production [8]. Therefore, in order to explore the possible mechanisms underlying the inhibitory effect of SMAPs on Aβ production, we assessed the levels of sAPPβ, pT668APP, BACE1, and PS1 (key component of γ-secretase). We observed that sAPPβ, pT668APP, and PS1 were all reduced compared to control after SMAP treatments, except that the less potent SMAP DBK-1154 did not decrease pT668APP at statistically significant levels (Fig. 2e, g, h, j). The expression levels of APP and BACE1 had no significant difference among these groups (Fig. 2e, f, i). Total BACE activity also showed no change in SMAP treatment (Fig. 2m). APP phosphorylation at Thr668 facilitates its cleavage by BACE1 [41]. Thus, we detected the BACE1 interacted with APP through immunoprecipitation. The result showed that SMAPs dramatically decreased APP and BACE1 interaction (Fig. 2k, l).These data suggested that SMAPs attenuate Aβ production through inhibiting APP phosphorylation, modulating γ-secretase expression level and reducing interaction of APP with BACE1 without influencing total BACE activity in cell models.
SMAP Ameliorates Memory Deficits in HHcy Rats
Based on the in vitro results, established in a head-to-head comparison that DBK-1160 showed less cytotoxicity and a stronger ability to activate PP2A versus DBK-1154. Both DBK-1154 and DBK-1160 have been evaluated in mouse toxicology and neither compound showed problems up to very high in vivo doses (800 mg/kg, data not shown). DBK-1160 has somewhat higher microsomal stability shown in Supplementary Figure 1b (i.e., more stable to oxidative metabolism) and comparable systemic exposure to DBK-1154 in mouse after oral dose. All of the above, taken together, led us to examine DBK-1160 in our in vivo rat hyperhomocysteinemia (HHcy) model.
Hyperhomocysteinemia (HHcy) is a high-risk factor of AD in human [25, 42]. We and others have established a sporadic AD rat model through inducing hyperhomocysteinemia in rats. These rats exhibit typical AD-like pathogenesis such as tau hyperphosphorylation, Aβ overproduction, PP2A inhibition, and cognitive deficits [35, 36, 43,42,43,46]. To explore the effect of SMAPs in AD model in vivo, HHcy rat model was established after 14 days injection of Hcy (400 μg/kg/day) via the vena caudalis, together with (preventive) or followed with the treatment of DBK-1160 (35 mg/kg, twice a day) or SCR-1693(1 mg/kg/day, treatment positive control) (Table 2 and Fig. 3a). In the open-field test, total distance, average speed, center distance%, and center duration% showed no significance difference among these groups (Fig. 3b–e), indicating that the locomotion activity was not influenced by Hcy, DBK-1160, or SCR-1693 treatment.

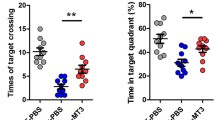
SMAP (DBK-1160) ameliorates memory deficits in HHcy rats. a Study design timeline performed in the study. b–e The total distance, average speed, center distance/total distance, and center duration/total duration were analyzed in the open-field test. f Escape latency in 6 days were analyzed in the Morris water maze (MWM) test, *P < 0.05, **P < 0.01, Hcy versus control. g Escape latency on test day 6 was analyzed in the MWM. h The platform crossing times on test day were analyzed in MWM test. i, j The duration in platform quadrant and nonplatform quadrants were analyzed in MWM test. k In the fear memory test, the total freezing time was analyzed on test day. Data represent mean ± SEM, control (treated with normal saline) n = 10, Hcy (treated with homocysteine) n = 11, Hcy-P (preventive group, treated with homocysteine and DBK-1160 simultaneously) n = 8, Hcy-1160 (treatment group, treated with DBK-1160 14 days after homocysteine injection) n = 9, Hcy-SCR (positive control group, treated with SCR-1693 14 days after homocysteine injection) n = 11, *P < 0.05, **P < 0.01, ***P < 0.001
Next, we explored the impact of DBK-1160 or SCR-1693 on learning and memory ability of the HHcy rats. In Morris water maze test, the escape latency was significantly increased in the Hcy group compared with the control group on days 2, 3, 4, 5, and 6, while supplementation with DBK-1160 simultaneously or after (preventive and treatment group) and SCR-1693 (positive control group) attenuated the HHcy-induced learning deficit (Fig. 3f). In the probe trial on day 6, rats in DBK-1160 preventive and treatment group spent less time to find the platform place (Fig. 3g), more time in the target quadrant (Fig. 3i), less time in the nonplatform quadrants (Fig. 3j), and crossed for more times above the platform (Fig. 3h) than the Hcy group. These data suggested that DBK-1160 effectively prevents the spatial learning and memory loss in HHcy rat model. We further performed fear conditioning test to assess the contextual fear memory. The results showed that the rats in the Hcy group exhibited significant shorter freezing time than the control group, indicating impaired fear memory. The deficit in contextual fear memory was almost completely rescued in DBK-1160 preventive, treatment, and positive control group (Fig. 3k).
SMAP Attenuates HHcy-Induced Tau Hyperphosphorylation Through Activation of PP2A
Tau hyperphosphorylation and accumulation contribute to learning and memory deficits in AD, and tau dephosphorylation is mainly regulated by PP2A [47]. Thereby, we measured the phosphorylated tau levels in the hippocampus of the rats between the control and DBK-1160 treated groups. The results indicated that tau phosphorylation level was increased at Thr205 (pT205), Ser396 (pS396), and Ser404 (pS404) with no significant change at Ser199 (pS199) and Thr231 (pT231) site (though there was a tendency) in the hippocampal extracts of the Hcy group comparing to the control group. Supplementation with DBK-1160 in preventive/treatment group or SCR-1693 in the positive control group effectively prevented tau hyperphosphorylation (Fig. 4a–f). Total tau level (Tau-5) remained comparable among these groups (Fig. 4a, g).

SMAP (DBK-1160) reduces tau phosphorylation levels and re-activates PP2A in HHcy rats. a Western blots for tau phosphorylation levels at the sites of pS199, pT205, pT231, pS396, pS404, and total tau (Tau-5) in rat hippocampus. b–g Quantification of the relative protein expression levels (pS199, pT205, pT231, pS396, pS404, and Tau-5) after normalization to the GAPDH signal. h–l Western blots and quantitatively analysis for the catalytic subunit of PP2A (PP2Ac), methylated PP2Ac (m-PP2Ac), demethylated PP2Ac (Dm-PP2Ac), and phosphorylated PP2Ac at Y307 (pY307-PP2Ac) in rat hippocampus. m PP2A activity assay in different groups in rat hippocampus. Data represent mean ± SD, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001
To confirm that DBK-1160 induce tau dephosphorylation through PP2A activation in HHcy rat model, we detected the active/inactive PP2A protein levels and measured the PP2A activity in the rat hippocampus in different groups. PP2A activity was significantly suppressed in the Hcy group compared with the control group and recovered after administration of DBK-1160. The effect of DBK-1160 on activating PP2A was comparable to that of SCR-1693 (Fig. 4m). In HHcy rat model, PP2A is inhibited partially through retarded methylation at Leu309 [36, 48, 49] and increased phosphorylation at Tyr307 [50, 51] of PP2Ac. Consistent with the previous report, we found that the methylation level at Leu309 (m-PP2Ac) was decreased in the Hcy group compared with the control group, and supplementation with DBK-1160 or SCR-1693 could partially restore it (Fig. 4h, j). In contrast, demethylation level at Leu309 (Dm-PP2Ac) was increased in the Hcy group compared with the control group and it returned to normal level after treatment with DBK-1160 or SCR-1693 (Fig. 4h, k). Furthermore, phosphorylation level at Tyr307 (pY307-PP2Ac) was increased in the Hcy group and reversed by DBK-1160 or SCR-1693 treatment (Fig. 4h, l). The total level of PP2Ac showed no significance difference among these five groups (Fig. 4h, i). These data suggested that DBK-1160 attenuated HHcy-induced tau hyperphosphorylation via reactivation of PP2A.
SMAP Relieves HHcy-Induced Aβ Overproduction
Next, we investigated the effect of SMAP on amyloidosis in HHcy-AD rat model. The ELISA results showed that DBK-1160 could decrease the HHcy-induced Aβ (Aβ40 and Aβ42) overproduction in preventive and treatment groups (Fig. 5j, k).

SMAP (DBK-1160) inhibits HHcy-induced Aβ overproduction in rats. a Western blots for APP, sAPPβ, pT668APP, BACE1, and PS1 in rat hippocampus. b–f Quantification of the relative protein levels (APP, sAPPβ, pT668APP, BACE1, and PS1) after normalization to the GAPDH signal. g, h IP blots and quantitatively analysis of BACE1 interacted with APP in rat hippocampus. P: preventive group, treated with homocysteine and DBK-1160 simultaneously; 1160: treatment group, treated with DBK-1160 14 days after homocysteine injection; SCR: positive control group, treated with SCR-1693 14 days after homocysteine injection. i BACE activity assay in different groups in rat hippocampus. j, k ELISA assay of the levels of Aβ40 and Aβ42 in the rat hippocampus. Data represent mean ± SD, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001
To further investigate the possible mechanisms underlying the effects of DBK-1160 on reducing Aβ generation, we detected the phosphorylation level of APP at Thr668 (pT668APP), and the levels of BACE1, sAPPβ, and PS1 (the catalytic subunit of γ-secretase). The results showed that the levels of pT668APP, sAPPβ, and PS1 were increased in Hcy group compared with the control group and were restored after DBK-1160 or SCR-1693 treatment (Fig. 5a, c, d, f). The level of total APP in the Hcy group did not differ from other groups (Fig. 5a, b). BACE1 level in the Hcy group only showed an upward trend without statistical significance (Fig. 5a, e). We further measured the BACE1 interaction with APP and found that the interaction between APP and BACE1 was enhanced in the Hcy group compared with the control group and attenuated after supplementation with DBK-1160 or SCR-1693 (Fig. 5g, h). Different with the results in N2a/APP cells, DBK-1160 treatment could reduce total BACE activity but SCR-1693 failed (Fig. 5i), indicating that DBK-1160 effectively inhibits abnormally activated BACE. These data suggested that DBK-1160 could suppress Aβ overproduction via decreasing β-cleavage of APP through inhibiting BACE activity and reducing APP interaction with BACE1, and decreasing γ-cleavage of APP through reducing expression of PS1, the catalytic subunit of γ-secretase. When DBK-1160 was used alone in normal rats without Hcy injection, no change of the behaviors, PP2A activity and modifications (methylation and phosphorylation), tau phosphorylation, and Aβ levels was observed (Supplementary Fig. 2–5), indicating that when used in vivo, DBK-1160 reactivates abnormally inhibited PP2A. This observation is consistent with pilot (non-GLP) toxicology studies on DBK-1160 , and that the compounds are well tolerated in multiple mouse models of cancer and COPD. Further it may indicate that mechanism related toxicity can be avoided in clinical studies for direct PP2A activators that normalize PP2A function in the context of chronic diseases such as AD and COPD.
SMAP Rescues HHcy-Induced Spine Loss and Synaptic Impairment
Synaptic impairment is an early event in AD development and underlies cognitive deficit [52]; therefore, we evaluated the synaptic impairment in rat brains. First, Nissl staining indicated that no neuronal loss was induced by HHcy (Fig. 6a, b). However, Hcy injection resulted in significant dendritic spine loss revealed in Golgi staining, and DBK-1160 or SCR-1693 effectively reversed the spine loss (Fig. 6c, d). Consistent with these findings, the levels of postsynaptic proteins such as PSD95, GluA1, GluA2, and GluN2B were reduced in the Hcy group and partially recovered after supplementation with DBK-1160 or SCR-1693 (Fig. 6e–i). Presynaptic proteins such as synapsin I and synaptophysin showed no significant change in these five groups (Fig. 6e, j, k). The results suggested that DBK-1160 prevented the synaptic impairment in HHcy rats through preserving dendritic spine density and postsynaptic protein expression levels.

SMAP (DBK-1160) reverses dendritic spine loss and recovers postsynaptic proteins. a, ) Representative Nissl staining images and the quantification of neuron number in the CA3 region of rat hippocampus, scale bar = 200 and 100 μm for low and high magnifications respectively. c, d Representative Golgi staining images and statistics analysis of spine number in the dentate gyrus region of rat hippocampus (scale bar, 10 μm). e–k Western blots and quantitatively analysis for synaptic proteins (PSD95, GluA1, GluA2, GluN2B, Synapsin I, and synaptophysin) levels in rat hippocampus. Data represent mean ± SD, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
The underlying pathogenesis of AD is poorly understood, and the current treatments do not reverse or prevent AD progression [53]. AD neuropathology is defined by the extracellular accumulation of amyloid-β (Aβ) peptide into amyloid plaques, and intraneuronal neurofibrillary tangles (NFTs) consisting of aggregated hyperphosphorylated tau protein [54, 55]. PP2A activity is suppressed in AD patients and it is involved in both tau hyperphosphorylation and Aβ overproduction [56, 57]. Therefore, pharmacologically improving or restoring PP2A activity is a promising strategy to inhibit tau hyperphosphorylation and Aβ generation. It was reported that antidiabetic drug metformin could activate PP2A directly and reduce tau phosphorylation at PP2A-dependent epitopes in AD models. Metformin disrupts microtubule associated PP2Ac-MID1-α4 protein complexs, which regulates the degradation of PP2Ac, and induces PP2A activity by freeing-up PP2Ac for holoenzyme assembly [20]. Another chemical, sodium selenate, can stabilize PP2A-tau complexes and enhance tau binding of PP2A in a dose-dependent manner [22]. Chronic treatment of sodium selenate in tau transgenic mouse strains can reduce tau hyperphosphorylation, improve contextual memory and motor performance, and prevent neurodegeneration. COG1410, an apoE-mimetic peptide, could specifically bind the PP2A inhibitor SET, disrupting SET-PP2A interaction and resulting in PP2A activity promotion [23]. It was reported that COG1410 could reduce AD-like brain pathologies in SwDI-APP/NOS2−/− (CVND-AD) mice. The synthetic sphingolipid drug, FTY-720 also acts by disrupting the interaction of SET with PP2A Ac and derepressing PP2A activity [58].
Small-molecule activators of PP2A (SMAPs) are efficacious in several in vivo cancer models [29, 59]. The compounds are well tolerated in in vivo models on oral administration and the molecular target of this class of compound has been identified as interface of the Aα and C subunits in the PP2A complex based on photoaffinity labeling studies, radiolytic footprinting, and mutagenesis of the binding site [29]. The SMAPs exhibit a robust PP2A reactivating phenotype across multiple cancer cell lines with dephosphorylation of direct PP2A targets such as ERK, AKT, FOXO1, MYC, and AR. In a mouse model of COPD, a smoking and age-related chronic degenerative lung disease, the compounds were effective in preventing loss of lung alveolar tissue over 2-month dosing by suppressing expression of cathepsin S, a protease also associated with AD progression [60, 61]. In this study, we tested the possibility of using SMAPs to re-activate PP2A and thereby reduce tau pathology and amyloidosis in cell and rat models. We demonstrate the potential of direct PP2A activation as a therapeutic strategy in AD and the viability of the tricyclic sulfonamides as leads for further development in neurodegenerative disease.
The compounds evaluated in our study, DBK-1154 and DBK-1160 (also known as SMAPs), are a pair of enantiomers (i.e., right and left hands of the same structure) and have the same polarity, lipophilicity, and comparable oral bioavailability. The compounds bind to the PP2A Aα scaffold subunit and drive PP2A conformational changes in the holoenzyme complex. In a cellular context PP2A catalytic C subunit is always associated with the A subunit (or a chaperone such as α4) and the two act cooperatively to modulate phosphatase activity [62,63,64]. It was identified that the compounds bind between HEAT repeats 5 and 6 of the A subunit, at a site close to the C-terminal tail of the C subunit, [29], SMAPs induce conformational change of A subunit and thus facilitate the catalytic activity of the C subunit.
Two cell lines stably overexpressed tau or APP are used to evaluate the effect of DBK-1154 and DBK-1160 on tau phosphorylation and Aβ generation and we found DBK-1160 to be a somewhat more effective PP2A activator versus the neuronal targets tested in vitro and DBK-1160 was used in an in vivo model. We used a rat model with hyperhomocysteinemia to induce AD-like pathology in vivo. Homocysteine (Hcy), a non-essential sulfur-containing amino acid, is a homolog of cysteine which is an intermediate form in the process of methionine metabolism [65, 66]. Hyperhomocysteinemia is an independent risk factor of AD [25, 26]. Elevated plasma level of Hcy was found in AD patients [67]. Hcy can lead to DNA breakage, oxidative stress, low methylation, apoptosis [68], and induction of microglial inflammatory markers and effectors [69]. Furthermore, Hcy can also increase phosphorylation of tau protein via inactivation of PP2A by interrupting its methylation [36, 70]. We and others have established the AD-like rat model through 14 days consecutive injection of Hcy (400 μg/kg/day) via the vena caudalis [36]. HHcy rats show typical AD-like spatial learning and memory deficits, synaptic impairment and loss, tau hyperphosphorylation, and Aβ overload, which make them suitable models in rapid drug efficacy evaluation [48, 50, 51]. SCR-1693 is a novel tacrine-dihydropyridine hybrid which has been proved to efficiently reduce tau pathology and amyloidosis, and ameliorate cognitive impairment both in AD cell and in rat models, with comparable or better effect than donepezil and nilvadipine [34, 35]. Thus, in our study, we used SCR-1693 as the positive control.
In cell experiments, both DBK-1154 and DBK-1160 showed potent effect on activating PP2A, and strong effect on reducing tau phosphorylation levels and Aβ levels. PP2A is the major tau phosphatase [71]; we conclude that DBK-1154 and DBK-1160 decrease tau phosphorylation via increasing PP2A activity. In N2a/APP cells, DBK-1154 or DBK-1160 treatment decreased the Aβ40 and Aβ42 levels in a dose-dependent manner. This effect may be also mediated by PP2A activation, because APP phosphorylation at Thr668 is regulated by PP2A [8]. SMAP treatment resulted in decreased level of pT668APP, indicating that APP is dephosphorylated upon PP2A activation. Under these conditions, interaction of APP with BACE1 is reduced, which was identified by immunoprecipitation. As a result, β-cleavage is attenuated, which was identified by decreased sAPPβ level in the two treatment groups. In addition, DBK-1154 or DBK-1160 could decrease the expression of PS1, the catalytic subunit of γ-secretase, indicating that SMAPs may further inhibit γ-cleavage of APP. These findings strongly suggest that direct activation of PP2A is a novel, effective therapeutic strategy for AD and the tricyclic sulfonamides are viable drug leads for AD therapeutics.
In HHcy rats, DBK-1160 did not influence the motor ability and anxiety evaluated in open-field test. However, DBK-1160 supplementation at the same time of Hcy injection (prevention) or after Hcy injection (treatment) rescued the rats from cognitive impairments in Morris water maze and fear conditioning test. The effect was comparable with that of SCR-1693. Similar with the results in cell models, DBK-1160 effectively activated PP2A in rat brains. This effect seems to be partially through modification of PP2Ac methylation. Hcy metabolism plays an important role in protein methylation. PP2Ac methylation at C-terminal Leu309 facilitates the B subunit binding thus improves PP2A activity towards certain substrates [28]. HHcy rats showed reduced PP2Ac-Leu309 methylation, which underlies PP2A inhibition. DBK-1160 could partially recover the PP2A methylation level, indicating that by directly interacting with PP2A at the AC subunit interface, SMAPs have the ability to facilitate PP2Ac methylation. Mechanistic details will be the subject of future reports.
With the PP2A activation in HHcy rats treated with DBK-1160, tau hyperphosphorylation, Aβ overproduction, and APP-Thr668 phosphorylation were all reduced. Consistent with the results in cell experiment, DBK-1160 decreased APP-BACE1 interaction and β-cleavage of APP. An inhibiting effect of DBK-1160 on BACE activity in HHcy rats indicated this chemical has the ability to repress abnormally activated BACE, which was not observed in the positive control SCR-1693 group. Increased PS1 expression was observed in HHcy rats, which was, once again, reversed by DBK-1160. These data suggest that SMAPs can efficiently reverse AD-like tau pathology and amyloidosis in rat brains.
Both tau and Aβ pathologies induce damage or loss of synapses, which is the foundation of cognitive deficits [72]. Loss of synapse is also an early event in AD development, which underlies learning and memory deficits in the patients and animal models [73]. Thus, the recovery of synaptic function may be a link between cognition rescuing and tau/Aβ pathologies alleviation. In all experimental groups, there was no neuronal loss in rat hippocampus. However, dendritic spine numbers were significantly reduced in the HHcy group. Consistent with this result, postsynaptic proteins, such as PSD95 and glutamate receptors, were decreased. All these synaptic impairments were rescued by DBK-1160 in a prevention or treatment dosing regime, indicating that SMAPs have a powerful restoring effect on synaptic function.
In summary, we demonstrated that SMAPs could efficiently ameliorate tau hyperphosphorylation and Aβ overproduction in AD-like cell and animal models through increasing PP2A activity. Furthermore, it can effectively attenuate HHcy-induced memory deficits and restore the number of dendritic spines and expression level of postsynaptic proteins. An unanticipated finding is that SMAPs may decrease BACE activity and PS1 expression and thus may inhibit APP β- and γ-cleavage. Thus, the tricyclic sulfonamide PP2A activators, originally described as tumor suppressor activators in cancer, have a multifaceted effect on drivers of AD neuropathology in vitro and in vivo. It suggests direct PP2A activation is an effective therapeutic option for AD treatment and that the tricyclic sulfonamides are viable leads as disease-modifying AD therapeutics. Results with next-generation analogs of DBK-1160 with improved metabolic stability and oral bioavailability will be reported in due course.
References
Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett 1986;65(3):351–5.
Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984;120(3):885–90.
Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev 2004;84(2):361–84.
Wang JZ, Wang ZH, Tian Q. Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer’s disease. Neurosci Bull 2014;30(2):359–66.
Zhang S, Wang Z, Cai F, et al. BACE1 Cleavage Site Selection Critical for Amyloidogenesis and Alzheimer’s Pathogenesis. J Neurosci 2017;37(29):6915–25.
Gong CX, Shaikh S, Wang JZ, et al. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem 1995;65(2):732–8.
Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett 1998;436(1):28–34.
Sontag E, Nunbhakdi-Craig V, Sontag JM, et al. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci 2007;27(11):2751–9.
Lee MS, Kao SC, Lemere CA, et al. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol 2003;163(1):83–95.
Shi Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell. 2009;139(3):468–84.
Apostolidis SA, Rodriguez-Rodriguez N, Suarez-Fueyo A, et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat Immunol 2016;17(5):556–64.
Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445(7123):53–7.
O’Connor CM, Perl A, Leonard D, Sangodkar J, Narla G. Therapeutic targeting of PP2A. Int J Biochem Cell Biol 2018;96:182–93.
Kauko O, Westermarck J. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int J Biochem Cell Biol 2018;96:157–64.
Nath S, Ohlmeyer M, Salathe MA, et al. Chronic Cigarette Smoke Exposure Subdues PP2A Activity by Enhancing Expression of the Oncogene CIP2A. Am J Respir Cell Mol Biol 2018;59(6):695–705.
Shentu YP, Huo Y, Feng XL, et al. CIP2A Causes Tau/APP Phosphorylation, Synaptopathy, and Memory Deficits in Alzheimer's Disease. Cell Rep 2018;24(3):713–23.
Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem 1993;61(3):921–7.
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 2005;22(8):1942–50.
Voronkov M, Braithwaite SP, Stock JB. Phosphoprotein phosphatase 2A: a novel druggable target for Alzheimer's disease. Future Med Chem 2011;3(7):821–33.
Kickstein E, Krauss S, Thornhill P, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A 2010;107(50):21830–5.
Asle-Rousta M, Kolahdooz Z, Oryan S, Ahmadiani A, Dargahi L. FTY720 (fingolimod) attenuates beta-amyloid peptide (Abeta42)-induced impairment of spatial learning and memory in rats. J Mol Neuroscience 2013; 50(3):524–532
van Eersel J, Ke YD, Liu X, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A 2010;107(31):13888–93.
Vitek MP, Christensen DJ, Wilcock D, et al. APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegener Dis 2012;10(1–4):122–6.
Basurto-Islas G, Blanchard J, Tung YC, . Fernandez JR, Voronkov M, Stock M, Zhang S, Stock JB, Iqbal K, (2014) Therapeutic benefits of a component of coffee in a rat model of Alzheimer's disease. Neurobiology of Aging 35 (12):2701–2712
Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett 2006;580(13):2994–3005.
Seshadri S. Elevated plasma homocysteine levels: risk factor or risk marker for the development of dementia and Alzheimer’s disease? J Alzheimers Dis 2006;9(4):393–8.
Longin S, Zwaenepoel K, Louis JV, et al. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J Biol Chem 2007;282(37):26971–80.
Wu J, Tolstykh T, Lee J, et al. Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. EMBO J 2000;19(21):5672–81.
Sangodkar J, Perl A, Tohme R, et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest 2017;127(6):2081–90.
Chen Y, Xu Y, Bao Q, et al. Structural and biochemical insights into the regulation of protein phosphatase 2A by small t antigen of SV40. Nat Struct Mol Biol 2007;14(6):527–34.
Cho US, Morrone S, Sablina AA, et al. Structural basis of PP2A inhibition by small t antigen. PLoS Biol 2007;5(8):e202.
Sablina AA, Hahn WC. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev 2008;27(2):137–46.
Merisaari J, Denisova OV, Doroszko M, Joncour V Le, Johansson P, Leenders WPJ, Kastrinsky DB, Zaware N, Narla G, Laakkonen P, Nelander S, Ohlmeyer M, Westermarck J (2020) Monotherapy efficacy of blood–brain barrier permeable small molecule reactivators of protein phosphatase 2A in glioblastoma. Brain Communications 2 (1)
Wang X-L, Xiong Y, Yang Y, et al. A novel tacrine-dihydropyridine hybrid (-)SCR1693 induces tau dephosphorylation and inhibits Aβ generation in cells. Eur J Pharmacol 2015;754:134–9.
Xia Y, Liu R, Chen R, et al. Novel Multipotent AChEI-CCB Attenuates Hyperhomocysteinemia-Induced Memory Deficits and Neuropathologies in Rats. J Alzheimers Dis 2014;42(3):1029–39.
Zhang CE, Tian Q, Wei W, et al. Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2A in rat hippocampus. Neurobiol Aging 2008;29(11):1654–65.
Seibenhener ML, Wooten MC. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp 2015(96):e52434.
Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984;11(1):47–60.
Liu R, Zhou XW, Tanila H, et al. Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J Cell Mol Med 2008;12(1):241–57.
Viola KL, Klein WL. Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 2015;129(2):183–206.
Triaca V, Sposato V, Bolasco G, et al. NGF controls APP cleavage by downregulating APP phosphorylation at Thr668: relevance for Alzheimer’s disease. Aging Cell 2016;15(4):661–72.
Jerneren F, Cederholm T, Refsum H, et al. Homocysteine Status Modifies the Treatment Effect of Omega-3 Fatty Acids on Cognition in a Randomized Clinical Trial in Mild to Moderate Alzheimer’s Disease: The OmegAD Study. J Alzheimers Dis 2019;69(1):189–97.
Chai GS, Jiang X, Ni ZF, et al. Betaine attenuates Alzheimer-like pathological changes and memory deficits induced by homocysteine. J Neurochem 2013;124(3):388–96.
Wei W, Liu YH, Zhang CE, et al. Folate/vitamin-B12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J Alzheimers Dis 2011;27(3):639–50.
Salissou MTM, Mahaman YAR, Zhu F, et al. Methanolic extract of Tamarix Gallica attenuates hyperhomocysteinemia induced AD-like pathology and cognitive impairments in rats. Aging. 2018;10(11):3229–48.
Guo J, Ni S, Li Q, Wang JZ, Yang Y. Folate/Vitamin B Alleviates Hyperhomocysteinemia-Induced Alzheimer-Like Pathologies in Rat Retina. Neurosci Bull 2019;35(2):325–35.
Martin L, Latypova X, Wilson CM, et al. Tau protein phosphatases in Alzheimer’s disease: the leading role of PP2A. Ageing Res Rev 2013;12(1):39–49.
Mahaman YAR, Huang F, Wu M, et al. Moringa Oleifera Alleviates Homocysteine-Induced Alzheimer’s Disease-Like Pathology and Cognitive Impairments. J Alzheimers Dis 2018;63(3):1141–59.
Guo J, Xu C, Ni S, et al. Elevation of pS262-Tau and Demethylated PP2A in Retina Occurs Earlier than in Hippocampus During Hyperhomocysteinemia. J Alzheimers Dis 2019;68(1):367–81.
Zeng P, Shi Y, Wang XM, et al. Emodin Rescued Hyperhomocysteinemia-Induced Dementia and Alzheimer’s Disease-Like Features in Rats. Int J Neuropsychopharmacol 2019;22(1):57–70.
Zhang Y, Xie JZ, Xu XY, et al. Liraglutide Ameliorates Hyperhomocysteinemia-Induced Alzheimer-Like Pathology and Memory Deficits in Rats via Multi-molecular Targeting. Neurosci Bull 2019;35(4):724–34.
Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci 2017;40(6):347–57.
Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol 2018;25(1):59–70.
Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 1986;83(13):4913–7.
Tian Q, Wang J. Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neuro-Signals. 2002;11(5):262–9.
Xiong Y, Luo DJ, Wang XL, et al. Zinc binds to and directly inhibits protein phosphatase 2A in vitro. Neurosci Bull 2015;31(3):331–7.
Liu R, Wang J-Z. Protein phosphatase 2A in Alzheimer’s disease. Pathophysiology. 2009;16(4):273–7.
De Palma RM, Parnham SR, Li Y, Oaks JJ, Peterson YK, Szulc ZM, Roth BM, Xing Y, Ogretmen B (2019) The NMR-based characterization of the FTY720-SET complex reveals an alternative mechanism for the attenuation of the inhibitory SET-PP2A interaction. The FASEB Journal 33 (6):7647–7666
Tohme R, Izadmehr S, Gandhe S, et al. Direct activation of PP2A for the treatment of tyrosine kinase inhibitor-resistant lung adenocarcinoma. JCI Insight 2019;4(4).
Doherty DF, Nath S, Poon J, et al. Protein Phosphatase 2A Reduces Cigarette Smoke-induced Cathepsin S and Loss of Lung Function. Am J Respir Crit Care Med 2019;200(1):51–62.
Lowry JR, Klegeris A. Emerging roles of microglial cathepsins in neurodegenerative disease. Brain Res Bull 2018;139:144–56.
Grinthal A, Adamovic I, Weiner B, Karplus M, Kleckner N (2010) PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proceedings of the National Academy of Sciences 107 (6):2467–2472
Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci U S A 2002;99(7):4221–6.
Wu CG, Chen H, Guo F, et al. PP2A-B' holoenzyme substrate recognition, regulation and role in cytokinesis. Cell Discov 2017;3:17027.
Forti P, Maioli F, Arnone G, et al. Homocysteinemia and early outcome of acute ischemic stroke in elderly patients. Brain Behav 2016;6(5):e00460.
Shirafuji N, Hamano T, Yen SH, et al. Homocysteine Increases Tau Phosphorylation, Truncation and Oligomerization. Int J Mol Sci 2018;19(3).
Clarke R, Smith AD, Jobst KA, et al. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol 1998;55(11):1449–55.
Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 2003;26(3):137–46.
da Cunha AA, Ferreira AG, Wyse AT. Increased inflammatory markers in brain and blood of rats subjected to acute homocysteine administration. Metab Brain Dis 2010;25(2):199–206.
Li JG, Chu J, Barrero C, Merali S, Pratico D. Homocysteine exacerbates beta-amyloid pathology, tau pathology, and cognitive deficit in a mouse model of Alzheimer disease with plaques and tangles. Ann Neurol 2014;75(6):851–63.
Ma RH, Zhang Y, Hong XY, et al. Role of microtubule-associated protein tau phosphorylation in Alzheimer’s disease. J Huazhong Univ Sci Technol Med Sci. 2017;37(3):307–12.
Rajmohan R, Reddy PH. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J Alzheimers Dis 2017;57(4):975–99.
Kamat PK, Kalani A, Rai S, et al. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer's Disease: Understanding the Therapeutics Strategies. Mol Neurobiol 2016;53(1):648–61.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 31970964, 31771189) (RL), the Natural Science Foundation of Hubei Province, China (grant number 2017CFA065) (RL), the Fundamental Research Funds for the Central Universities (2019kfyXKJC079) (RL), and the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST (JZW). DBK-1160 synthesis was funded by PFNYC Bioaccelerate award (MO)
Author information
Authors and Affiliations
Contributions
HW, HLZ, YZ, and DDA performed animal experiments and established HHcy AD model; HW, HLZ, LW, and JZX cultured the cells and HW performed cell experiments; HW, MO, and RL wrote the manuscript draft. XCW, JZW, YML, and RL helped to design the experiments. XJS and JW provided expert advice and contributed to the manuscript writing. MO supplied the SMAP compounds. RL, JW and MO conceived the project, designed the experiments, and finalized the manuscript.
Corresponding authors
Ethics declarations
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Conflict of Interest
Mount Sinai School of Medicine has filed patents on DBK-1154, DBK-1160 and related PP2A activating compounds. Other authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wei, H., Zhang, Hl., Wang, Xc. et al. Direct Activation of Protein Phosphatase 2A (PP2A) by Tricyclic Sulfonamides Ameliorates Alzheimer’s Disease Pathogenesis in Cell and Animal Models. Neurotherapeutics 17, 1087–1103 (2020). https://doi.org/10.1007/s13311-020-00841-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00841-6