
Abstract
The major forms of autoimmune myopathies include dermatomyositis (DM), polymyositis (PM), myositis associated with antisynthetase syndrome (ASS), immune-mediated necrotizing myopathy (IMNM), and inclusion body myositis (IBM). While each of these conditions has unique clinical and histopathological features, they all share an immune-mediated component. These conditions can occur in isolation or can be associated with systemic malignancies or connective tissue disorders (overlap syndromes). As more has been learned about these conditions, it has become clear that traditional classification schemes do not adequately group patients according to shared clinical features and prognosis. Newer classifications are now utilizing myositis-specific autoantibodies which correlate with clinical and histopathological phenotypes and risk of malignancy, and help in offering prognostic information with regard to treatment response. Based on observational data and expert opinion, corticosteroids are considered first-line therapy for DM, PM, ASS, and IMNM, although intravenous immunoglobulin (IVIG) is increasingly being used as initial therapy in IMNM related to statin use. Second-line agents are often required, but further prospective investigation is required regarding the optimal choice and timing of these agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction and Classification
The autoimmune myopathies consist of five main conditions, namely dermatomyositis (DM), polymyositis (PM), myositis associated with antisynthetase syndrome (ASS), immune-mediated necrotizing myopathy (IMNM), and inclusion body myositis (IBM). While each of these conditions has unique clinical and histopathological features, they all share an immune-mediated component (Table 1). These conditions can occur in isolation or can be associated with systemic malignancies or connective tissue disorders (overlap syndromes) and all are often associated with autoantibodies.
Accurate incidence and prevalence of the autoimmune myopathies are difficult to obtain due to use of different diagnostic criteria employed across epidemiological studies. The Bohan and Peter 1975 criteria, employed by many older studies, do not require a muscle biopsy, tend to overestimate the incidence of PM, and preceded the discovery of IBM or IMNM as unique entities [2, 3]. Various revised diagnostic criteria for the autoimmune myopathies have since been proposed which take into account clinical features, autoantibodies, and histopathology [4,5,6,7,8]. One included a weighted scoring system estimating probability of an inflammatory myopathy [7], although this was not externally validated using controls and only included individuals with a disease duration of more than 6 months, limiting generalizability to individuals with a more acute presentation.
Clinical Features
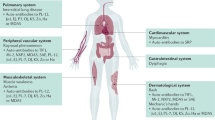
DM can present at any age. The incidence is higher in females compared to males, similar to most autoimmune conditions [9]. Weakness can present relatively acutely over days to weeks, or it can present more slowly over a few months. The proximal arms and legs are typically affected, along with a tendency for early involvement of the neck flexors. In up to 30% of individuals, weakness of oropharyngeal and esophageal muscles results in dysphagia [10]. Dysarthria and facial muscle weakness can occur but are uncommon [11]. The characteristic rash associated with DM includes a periorbital purplish discoloration (heliotrope rash), a papular erythematous rash over the knuckles (Gottron papules), and an erythematous macular rash on the face, neck, and anterior chest (V-sign) and on the shoulders and upper back (shawl sign), as well as on the extensor surfaces of elbows, knuckles, and knees (Gottron sign). The rash can develop months after disease onset, but more typically it accompanies or predates the onset of muscle weakness. In up to 20% of individuals, the characteristic rash occurs in the absence of weakness, a form of DM known as amyopathic DM or DM sine myositis [12, 13]. On the contrary, some individuals present with muscle weakness and classic histopathological features on muscle biopsy but never develop the characteristic rash, a form of DM referred to as adermatopathic DM or DM sine dermatitis [14]. Calcinosis cutis (subcutaneous calcium deposits) can occur infrequently in children and adults with DM [15]. The presentation of DM in children is similar to that in adults.
DM is also associated with multiple systemic complications, including cardiac, pulmonary, gastrointestinal, and rheumatological involvement, as well as systemic malignancies (in adult-onset DM). Potential cardiac complications include conduction system abnormalities and arrhythmias, pericarditis, myocarditis, coronary artery disease, and congestive heart failure/diastolic dysfunction [16,17,18,19,20]. Interstitial lung disease (ILD) can occur in 15 to 20% of DM patients, typically presenting with a dry cough, shortness of breath, fine inspiratory bibasal crackles on lung examination, and a restrictive pattern on pulmonary function testing [21, 22]. Bronchiolitis obliterans with organizing pneumonia is a much rarer pulmonary complication. Involvement of ventilatory and oropharyngeal muscles in DM can also result in significant weakness and risk of aspiration pneumonia. Gastrointestinal complications include difficulties with swallowing, as noted above, aspiration of gastric contents, and delayed gastric emptying, thought to be due to a reduction in gastric peristalsis [23,24,25,26]. Rheumatological complications include arthralgias, arthritis, and joint contractures.
The risk of malignancy is increased in adult patients with DM, to approximately 10 to 15% within 2 to 3 years of initial presentation and with the majority of cases occurring in individuals over the age of 40 years [27, 28]. Juvenile-onset DM is not associated with cancer. The most common cancers associated with adult-onset DM include hematological and lymphatic cancers (particularly non-Hodgkin lymphoma, leukemia, and multiple myeloma), followed by solid organ adenocarcinomas of the lung, colon, bladder, breast, ovary, cervix, pancreas, and esophagus [27, 29]. Successfully treating the underlying malignancy can result in improved muscle strength [30]. Individuals with DM should undergo comprehensive malignancy screening, which should include a detailed history and physical examination including breast, pelvic, testicular, and prostate examinations, as appropriate. Basic workup should include a complete blood count, electrolytes and renal function, serum protein electrophoresis with immunofixation and serum free light chains, urinalysis, computerized tomography of the chest, abdomen, and pelvis, pelvic ultrasound and mammography in women, and colonoscopy for patients aged over 50 or those have symptoms concerning for a gastrointestinal malignancy, including fluctuating bowel habits, tenesmus, bleeding per rectum, melena, and/or weight loss. Positron emission tomography is useful in individuals where there is a high clinical suspicion for an underlying malignancy despite negative initial cancer screening.
PM also occurs more frequently in women compared to men [31]. The true incidence of PM is unknown, due to the previously described limitations of diagnostic criteria employed in many epidemiological studies to date. Patients with PM tend to present over the age of 20 years with symmetric weakness in a proximal distribution in the upper and lower extremities, although involvement of distal muscles can also be seen to a lesser degree. Muscle tenderness and myalgia are also reported, as well as difficulties with swallowing. Cardiac manifestations including conduction system abnormalities and heart failure are reported by up to 30% of patients. Like DM, PM is also associated with an increased risk of malignancy [28]. The frequency of pulmonary complications, including ILD, is similar to DM. Interestingly, ILD tends to occur less frequently in cases of PM or DM which are associated with malignancy [32].
Myositis overlap syndromes occur when an autoimmune myopathy (DM or PM) occurs in association with other connective tissue diseases, typically mixed connective tissue disease, systemic lupus erythematosus, Sjögren syndrome, scleroderma, or rheumatoid arthritis [33]. In up to 15% of patients at initial presentation, clinical features of a connective tissue disease are not present although overlap antibodies are present. In such patients, clinical features of a connective tissue disorder typically occur on follow-up [33].
Myositis associated with ASS is a category of myositis associated with anti-aminoacyl-tRNA synthetase (ARS) antibodies, most commonly anti-Jo1 antibodies. These patients typically manifest with ILD, constitutional symptoms including fevers and weight loss, nonerosive arthritis, Raynaud’s phenomenon, and skin changes known as “mechanic’s hands” [34,35,36,37]. The co-occurrence of an erythematous rash can result in many patients being misdiagnosed with DM. ASS is important to recognize because of the high prevalence of ILD (50–60%) that can be very difficult to treat.
IMNM accounts for up to 20% of all autoimmune myopathies and can present acutely or more insidiously with proximal upper and lower extremity weakness which progresses over time, as well as with facial muscle weakness [38,39,40]. IMNM has been associated with antibodies against 3-hydoxy-3-methylglutaryl-coenzyme A reductase (HMGCR) and against the signal recognition particle (SRP), and there are likely others yet to be discovered. The frequency of anti-HMGCR antibodies in IMNM has been reported to range from 22 to 61% [41, 42]. Anti-HMGCR antibodies have been found in up to 60% of statin-exposed patients with IMNM [39]. It is important to distinguish anti-HMGCR myopathy from the more common toxic myopathy associated with statin use. The later improves with discontinuation of the statin, while anti-HMGCR myopathy requires immunotherapy for improvement (discussed below). Anti-HMGCR antibodies have also been reported in children and adults unexposed to statin therapy and with underlying cancer [43]. Of note, IMNM, particularly anti-HMGCR myopathy, can manifest in children and mimic a limb girdle muscular dystrophy. Therefore, it is important to assess for anti-HMGCR antibodies in children and adults with suspected limb girdle muscular dystrophy in whom no mutation is found, as the IMNM is treatable [42, 44]. Although most anti-HMGCR myopathies have histopathological features of IMNM, occasionally there is endomysial inflammation suggestive of PM. Anti-SRP antibodies have been reported in up to 16% of patients with IMNM [41] and are associated with severe and aggressive disease that can be difficult to adequately control [41, 45, 46]. Anti-SRP myopathy tends to occur more commonly in women compared to men [47]. Some studies have suggested there may be an increased risk of cardiac complications in younger patients with anti-SRP antibodies [48]. Anti-SRP myopathy tends to be associated with more severe weakness and a higher risk of pulmonary involvement or dysphagia compared to anti-HMGCR myopathy [40, 49]. Axial muscle weakness, e.g., manifesting as head drop, can also occur [47, 50].
IMMN can also be seen with connective tissue diseases (e.g., mixed connective tissue disease and scleroderma) and as a paraneoplastic complication (most commonly lung cancer or gastrointestinal adenocarcinomas), or it can be idiopathic, occurring in the absence of myositis-specific or myositis-associated antibodies or cancer [50, 51]. A workup for malignancy is required for all patients, similar to DM and PM. Of note, the more recent cancer immunotherapies, namely the programmed death-1 (PD-1) inhibitors (pembrolizumab and nivolumab), have also been associated with numerous neuromuscular complications, including a severe necrotizing immune-mediated myopathy which can be difficult to treat, despite aggressive immunotherapy [52,53,54,55].
IBM, the most common myopathy in individuals over the age of 50 years, occurs more frequently in men compared to women. It typically presents in individuals aged 40 or over with a slow onset of progressive, asymmetric, proximal, and distal atrophy and weakness, with a predilection for the quadriceps femoris, wrist, and finger flexors and ankle dorsiflexors (Fig. 1) [56]. An estimated 35 to 50% of affected individuals require a wheelchair within 14 years [56, 57]. Dysphagia occurs in at least 60% of patients and can be the presenting complaint, preceding the onset of extremity weakness by up to 7 years [58, 59]. A generalized sensory polyneuropathy has been noted in up to 20% of patients [60]. IBM has also been associated with sarcoidosis as well as with viral infections, including hepatitis C and HIV [61,62,63,64]. IBM is not known to be associated with pulmonary complications, cardiac manifestations, or an increased risk of systemic malignancies, with perhaps the exception of granulocytic leukemia (see below) [65].

Characteristic distribution of weakness and atrophy in inclusion body myositis. (a) Quadriceps atrophy. (b) Asymmetric atrophy of flexor muscles in the forearm, with asymmetric weakness of the deep finger flexors (including the flexor pollicis longus) > superficial finger flexors (the patient was asked to curl his fingers and thumbs)
Diagnostic Evaluation
Laboratory Features Including Myositis Antibodies
Serum creatine kinase (CK) activity is elevated in approximately 70% of patients with DM [16, 66]. In approximately 10% of cases with normal CK, serum aldolase levels are elevated [67, 68]. CK is always elevated in PM, myositis associated with ASS and IMNM. In IBM, serum CKs are normal or only mildly elevated, usually less that 10× the upper limit of normal. Notably, serum CK does not correlate with the severity of clinical weakness in any of the myositides.
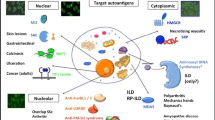
Myositis antibodies include both myositis-specific antibodies (MSA) and myositis-associated antibodies (MAA). MSA are primarily present solely in the presence of an autoimmune myopathy whereas MAA are associated with myositis but are also found in other autoimmune rheumatological conditions (myositis overlap syndromes).
The utility of MSA in autoimmune myopathies is increasingly being recognized, including 1) diagnosing and characterizing autoimmune myopathies, 2) guiding additional workup and screening, 3) predicting response to treatment, and 4) indicating prognosis (Table 1). MSA include ARS antibodies, SRP antibodies, and DM-specific antibodies (namely anti-Mi-2, anti-MDA5, anti-TIF1-ϒ, and anti-NXP2). Of the ARS antibodies, anti Jo-1 antibodies (against histidyl-tRNA synthetase) are the most common and are strongly associated with ASS, including ILD (66% of patients), arthralgias (56%), fever (27%), Raynaud’s phenomenon (40%), and mechanic’s hands (31%) [34]. Less commonly occurring ARS antibodies include anti-PL-12 (anti-alanyl), anti-PL-7 (anti-threonyl), anti-EJ (anti-glycyl), anti-OJ (anti-isoleucyl), anti-KS (anti-asparaginyl), anti-Zo (anti-phenylalanyl), anti-Ha (or anti-YRS, anti-tyrosyl), anti-SC (anti-lysyl), anti-JS (anti-glutaminyl), anti-tryptophanyl, and anti-SAE (anti-small ubiquitin-like modifier activating enzyme) [35].
DM-specific antibodies are found in 60 to 70% of patients with DM [69]. Anti-Mi-2 antibodies are found in 7 to 30% of patients with DM and are typically associated with acute onset of classic DM, particularly severe skin manifestations at initial presentation, good response to therapy, a favorable prognosis, and a reduced risk of malignancy, compared to other patients with DM [70,71,72,73]. Antibodies directed against melanoma differentiation-association protein 5 (MDA5) occur in approximately 15% of patients with DM [74, 75]. Anti-MDA5 antibodies are usually associated with minimal muscle involvement (amyopathic or hypomyopathic DM), severe vasculopathy often with digital ulcerations, and rapidly progressive ILD [74,75,76,77]. Antibodies directed against transcriptional intermediary factor 1-ϒ (TIF1-ϒ), also known as anti-p155/140 antibody, have a prevalence of 14 to 31% in DM and strongly predict risk of malignancy, showing 89% specificity and 78% sensitivity for malignancy diagnosis, with a positive predictive value of 58% and a negative predictive value of 95% [78,79,80]. In addition, these antibodies are associated with particularly severe skin manifestations including palmar hyperkeratotic papules, psoriatic type lesions, and hypopigmented and telangiectatic skin patches [81]. Antibodies against nuclear matrix protein 2 (NXP-2), another anti-p140 antibody and also known as anti-MJ antibody, are found in up to 30% of patients with DM and are associated with a younger age of onset, severe muscle weakness, calcinosis, good response to treatment, and an increased risk of malignancy [82,83,84,85,86]. Antibodies to SRP and HMGCR are discussed above.
The MAA include anti-Ro, anti-La, anti-Sm, anti-PM-Scl, or anti-U1-ribonucleoprotein (RNP) antibodies. As mentioned above, the occurrence of MAA in a patient with myositis is suggestive of a myositis overlap syndrome.
Antibodies targeting cytosolic 5′ nucleotidase 1A (cN-1A) are found in approximately 70% of individuals with IBM and can be of assistance in distinguishing IBM from other autoimmune myopathies. This antibody has a reported sensitivity of 70% and specificity of 92% for the diagnosis of IBM [87, 88]. However, anti-cN-1A antibodies are also detectable in up to 20% of patients with Sjögren syndrome and systemic lupus erythematosus in the absence of a myopathy, and have also been found in patients with DM or PM as well as healthy volunteers [89, 90]. There is also an association between IBM and certain autoimmune conditions, including Sjögren syndrome, sarcoidosis, and lymphoproliferative disorders (chronic lymphocytic leukemia/lymphocytosis) [63, 65], and less specific autoimmune antibodies, including anti-Ro, anti-La, antinuclear antibody, anti-rheumatoid factor, anti-Smith, and anti-RNP antibodies, have also been reported in up to 20% of patients with IBM [60, 91, 92].
In a recent study looking at the association between IBM and lymphocytic leukemia/lymphocytosis, almost 60% of patients with IBM were found to have aberrant clonal populations of large granular T lymphocytes meeting criteria for T cell large granular lymphocytic leukemia, compared to 14% of age-matched patients with DM, PM, or IMNM, and 0% of age-matched controls [65]. A reduced ratio of CD4 to CD8 cells, an elevated peripheral CD8 cell count, and a lymphocyte predominant blood count correlated highly with the flow cytometry expression of large granular lymphocytes. Muscle histopathology revealed large granular lymphocytic invasion of muscle in all affected IBM patients, with the extent of the CD8+ cell invasion on muscle biopsy directly correlating with the size of the peripheral blood population of large granular lymphocytes. Cells invading myocytes were found to express CD57, which is a marker of T call aggressiveness. This study suggested that autoimmune T cell expansion in IBM might evolve into a neoplastic-like disorder, which may explain the refractoriness of this disorder to standard immune therapies (treatment of IBM is discussed below).
Electrophysiological Features
Characteristic electrophysiological findings in patients with inflammatory myopathies include 1) increased spontaneous activity as evidenced by fibrillation potentials and positive sharp waves; 2) low-amplitude, short-duration, polyphasic motor unit action potentials; and 3) early recruitment. In chronic disease, high-amplitude, long-duration motor unit potentials may be observed if there is fiber splitting with regeneration, as well as reduced recruitment of fast firing motor unit potentials if there is prominent muscle fiber atrophy, which can mimic a neurogenic process. In up to one third of patients with IBM, a mixed picture of tall, long-duration, polyphasic motor unit potentials along with small amplitude, short-duration, polyphasic motor units with early recruitment is seen, which can lead to an erroneous diagnosis of motor neuron disease [60, 93, 94]. In addition, nerve conduction studies can show evidence of a mild axonal sensory neuropathy in up to 20% of IBM patients [60].
Muscle Imaging
Magnetic resonance imaging (MRI) of skeletal muscle can reveal signs of edema (felt to indicate active inflammation) that is best appreciated on short tau inversion recovery (STIR) imaging sequences. However, these features are often nonspecific for inflammatory myopathies and can also be seen in toxic myopathies, rhabdomyolysis, muscular dystrophies, and occasionally in acute neurogenic disease [95, 96]. A particular pattern of fibro-fatty infiltration and atrophy of the vastus intermedius and vastus medialis muscles in a distal to proximal gradient (with relative sparing more proximally) has been noted on thigh MRI in some patients with IBM and was noted to occur in 10 out of 17 patients with definite IBM in one study [97]. MRI can also offer utility, in addition to the clinical exam, in identifying the optimal muscle to biopsy [98, 99].
Muscle Biopsy
Muscle biopsy should be considered in the diagnostic workup of all patients with autoimmune myopathy. In patients with characteristic clinical features of DM, a skin biopsy confirming the diagnosis of DM may be sufficient for the diagnosis. In patients with MSA and characteristic clinical features of DM or ASS, a muscle biopsy may not be required. Similarly, in IBM, the presence of anti-cN-1A antibodies are very specific for the disease, particularly in the presence of characteristic clinical features, and so an experienced clinician may forego muscle biopsy in such cases.
Choosing the optimal muscle to biopsy is based on the clinical examination, ideally selecting a muscle that is moderately weak (grade 4 on the Medical Research Council scale) but not severely weak, due to the limited diagnostic yield with biopsies of very weak muscles which tend to show nonspecific end-stage fibro-fatty changes. EMG or MRI can be particularly useful in choosing a good muscle biopsy site in cases where patients are weak in muscles that are less routinely biopsied.
Histopathology
The pathognomonic histological feature of DM is perifascicular atrophy; however, this tends to be seen in only 50% of adult cases as it is often absent in those biopsied at an early stage in the disease. Perivascular and perimysial inflammatory infiltrate consisting of macrophages, B cells, CD4+ T cells, and dendritic cells is seen. The majority of the CD4+ cells are plasmacytoid dendritic cells overexpressing interferon-1, which is thought to be toxic to perifascicular muscle fibers and neighboring capillaries [100]. Immunohistochemistry staining reveals major histocompatibility complex-1 (MHC-1) and interferon-alpha/beta-inducible protein myxovirus resistance A (MxA) expression on muscle fibers, particularly in the perifascicular regions [100]. One recent study reported that MxA sarcoplasmic expression had a sensitivity of 71% and specificity of 98% for diagnosis of DM, i.e., a more sensitive marker compared to perifascicular atrophy or MAC deposition on capillaries (sensitivity of 47% and specificity of 98%, sensitivity of 35% and specificity of 93%, respectively) [101]. In contrast to PM and IBM, in DM, there is typically no invasion of nonnecrotic muscle fibers. On electron microscopy, tubuloreticular inclusions can be seen in endothelial cells.
PM tends to be characterized histologically by variability in fiber size and shape, mixed areas of necrotic and regenerating muscle fibers, and endomysial and perivascular inflammation, consisting primarily of CD8+ T cells and macrophages surrounding and sometimes invading nonnecrotic muscle fibers expressing major MHC-1 [102, 103].
In ASS-associated myositis, typical histopathological findings include deposits of plasmacytoid dendritic cells and macrophages in the perimysial and perivascular regions, deposition of MAC on capillaries, and evidence of perimysial connective tissue fragmentation and damage with positive staining for alkaline phosphatase [104]. Similar to DM, perifascicular atrophy is seen, although compared to DM, there tends to be greater perifascicular myocyte necrosis and deposition of MHC-1 and MAC. Myofiber HLA-DR expression has been reported in over 80% of patients with ASS-associated myositis compared to 24% of patients with DM, with perifascicular expression uniquely observed in ASS myositis, suggesting a role for HLA-DR detection in differentiating these diagnoses [105].
IMNM is typically characterized histologically by the presence of scattered necrotic muscle fibers undergoing myophagocytosis and scant inflammation localized to necrotic fibers [106]. However, in up to 20% of patients with anti-SRP antibody-associated myositis, a prominent lymphocytic infiltrate can be seen [47]. In addition, in up to 30% of patients with anti-HMGCR myopathy, prominent lymphocytic collections can be seen, with scattered endomysial CD4+ and CD8+ cells in up to 50% of patients [38, 106, 107]. Diffuse expression of MAC and MHC-1 on the sarcolemma of nonnecrotic muscle fibers (more commonly seen in patients with anti-HMGCR antibodies compared to those with anti-SRP antibodies) and deposition of MAC on small blood vessels can be seen [40, 46]. Thickening of capillary basement membranes (pipestem capillaries) has also been noted [108]. The MHC class II human leukocyte antigen (HLA) allele DRB1 11:01 is a strong immunogenetic risk factor for developing anti-HMGCR autoantibodies while B 5001 and DQA1 0104 are associated with an increased risk of developing anti-SRP autoantibodies [109,110,111].
Histologically, IBM is characterized by endomysial inflammation consisting of CD8+ T cells and macrophages invading nonnecrotic muscle fibers expressing MHC-1, similar to what is seen in PM. What helps distinguish IBM from PM is the presence of rimmed vacuoles and cytoplasmic inclusions within muscle fibers; however, these are not demonstrated in 20 to 30% of any given IBM muscle biopsies. Mitochondrial pathology, as evidence by an increased number of ragged red fibers and COX-negative muscle fibers compared to patients with DM, PM, or age-matched controls, can also be seen, as well as small deposits of amyloid in rare (< 1%) muscle fibers using Congo red stain. Immunohistochemistry staining for Tar DNA-binding protein-43 (TDP-43) is very specific for IBM, while staining for p62 (demonstrating cytoplasmic aggregates) is very sensitive for IBM [112]; p62 is typically employed more in most myopathology laboratories. On electron microscopy, intranuclear and cytoplasmic tubulofilaments can be observed in rare muscle fibers, although these can be difficult to find [60].
Treatment
The goal of treatment in a patient with myositis should be to return a patient to normal strength and function and minimize extra-muscular involvement while avoiding side effects of treatment. There are few randomized placebo-controlled trials of patients with myositis. A 2012 Cochrane systematic review, for example, identified only 10 randomized controlled trials encompassing 258 total patients suitable for inclusion, none of which demonstrated benefit of second-line oral immunosuppressive therapies over placebo [113]. Accordingly, treatment recommendations are largely guided by small uncontrolled trials, retrospective case series, and expert opinion.
Immunotherapy is the mainstay of treatment for DM, PM, myositis associated with ASS, and IMNM; IBM, by contrast, does not typically respond to immunotherapy (Table 2). The approach to therapy is similar among PM, DM, ASS, and IMNM, with corticosteroids used as first-line treatment. The major exception is anti-HMGCR myopathy, in which patients may respond to IVIG monotherapy as a first-line treatment [114]. Many patients will require the addition of a second-line agent due to either incomplete response to corticosteroids or intolerable side effects. Occasionally, patients who have relative contraindications to corticosteroids (e.g., patients with poorly controlled diabetes, osteoporosis) may be treated with a traditional second-line agent (discussed below). Particularly refractory patients may require third-line agents, at times utilizing three medications concurrently (e.g., corticosteroids, an oral steroid-sparing agent, and IVIG or rituximab).
Initiating Therapy
Although no randomized placebo-controlled trials have assessed their effectiveness in myositis patients, corticosteroids are considered effective first-line treatment [8, 115, 116]. We start patients on high-dose daily prednisone (0.7 to 1 mg/kg/day, up to a maximum of 60 mg daily). In patients with severe weakness (e.g., inability to walk or dysphagia) or multisystem involvement (e.g., ILD, myocarditis, severe skin rash), a 3- to 5-day course of intravenous methylprednisolone (1 g/daily) can be given before starting oral prednisone.
Alternative strategies exist for patients with an increased risk of steroid-induced side effects—those with diabetes, osteoporosis (or those at increased risk, e.g., postmenopausal women), obesity, or neuropsychiatric disorders, for example. We prefer to still use high-dose prednisone, but to also start a second-line agent immediately to facilitate a more rapid prednisone taper (see below). Patients with mild disease and favorable prognostic factors may instead be started on a low dose of prednisone, gradually increasing the dose until response is evident.
Emerging evidence from observational studies supports the role of IVIG as first-line monotherapy in patients with IMNM and contraindication to steroids. One case series of three patients with IMNM associated with statin exposure and positive HMGCR antibodies who refused steroids and were treated with IVIG (2 g/kg per month) as monotherapy [114]. After 2 to 3 cycles, two of the three patients became asymptomatic, with full muscle strength on exam, and the third improved but had ongoing mild hip flexor weakness. CK decreased in all patients. Another four patients with IMNM (autoantibodies unspecified) have been reported to respond to IVIG as first-line monotherapy [8]. Additional prospective studies are warranted to confirm these findings.
In some cases, prednisone monotherapy is sufficient for adequate disease control. The starting dose of prednisone should be maintained without taper until strength normalizes or plateaus and the CK returns to the normal range. This typically requires a patient to remain on high-dose prednisone for 2 to 4 months. Once strength has normalized or plateaued, we typically taper the daily dose of prednisone by 10 mg every 4 weeks until 20 mg/day, then by 5 mg every 4 weeks until 10 mg/day. If possible, we taper by 1 to 2.5 mg every 4 weeks thereafter. In our experience, tapering more rapidly often leads to relapse and an inevitable re-escalation of the dose.
Changes in the steroid dose should be guided primarily by the patient’s strength on clinical exam. Most patients will require an ongoing small dose of prednisone to maintain long-term control—the goal is to find the lowest possible dose that achieves adequate disease control. A rising serum CK alone does not require escalation of treatment but may herald impending worsening weakness. In such cases, our practice is to hold the dose of prednisone steady temporarily and follow the patient closely for changes in exam. Patients with IMNM commonly have persistently elevated CK. In one cohort, even among patients achieving full strength on treatment, 55% continued to have CK higher than 500 IU/L [43]. In patients with worsening weakness after initial improvement, EMG can be helpful in addition to monitoring CK. Patients with active myositis usually have a rising CK and increased insertional activity or spontaneous activity evident on needle EMG. Patients with type 2 fiber atrophy related to corticosteroid use have normal or even decreased insertional activity on EMG.
What if Treatment Is Ineffective or Only Partially Effective?
If there is no response to corticosteroids, the first step is to reconsider the diagnosis. Muscular dystrophy may sometimes be confused for myositis based on a similar clinical presentation and the presence of inflammation on muscle biopsy. Dysferlinopathy, calpainopathy, and facioscapulohumeral muscular dystrophy (FSHD) often have inflammatory cellular infiltrates evident on biopsy [117]. In one cohort, for example, 25% of dysferlinopathy patients were initially misdiagnosed with polymyositis due to inflammatory muscle biopsies and rapid clinical progression or pain [118]. Similarly, IBM patients are often first diagnosed as having PM given the overlapping histopathologic features described above. Assessing for cytochrome oxidase-negative fibers, MHC1 expression on the sarcolemma of fibers, and p62 inclusions, as well as testing serum for anti-cN-1A antibodies, is of particular utility in such cases.
The more common situation, however, is the patient in whom response to treatment is only partial. Normalization of strength may not be possible in those patients with chronic untreated or aggressive disease in which there has already been significant fatty replacement of muscle. Initiation of treatment soon after disease onset is therefore critical, before irreversible damage can accumulate. In a retrospective analysis of 113 patients with inflammatory myopathy, no patient with treatment initiation more than 18 months after symptom onset had complete response to treatment (i.e., return to normal strength and reduce in CK to normal range) [119]. Anti-SRP myopathy is particularly aggressive; atrophy and fatty replacement of muscle are more often evident at disease onset than in other patients [120]. In one cohort of 100 anti-SRP myopathy patients, the degree of weakness and muscle atrophy evident at treatment initiation (not the preceding duration of symptoms) predicted response to treatment after 2 years [47].
When faced with a patient with partial response to treatment, a distinction must be made between ongoing disease activity that is amenable to treatment and irreversible muscle damage that will not respond to treatment. Serum CK and EMG may help, as described above. MRI of the thigh muscles can also aid in the distinction, as T1 sequences can demonstrate fatty infiltration. In a small study of patients with anti-SRP antibodies, the presence of prominent fatty infiltration on thigh MRI correlated with a poor response to therapy [121]. As discussed above (see “Muscle Imaging”), increased STIR signal can identify myoedema, which suggests ongoing inflammation that may be amenable to therapy [122].
Starting a Second-Line Agent
In one study, 55% of patients had poor response or severe side effects with corticosteroid monotherapy [123]. Many patients will accordingly require treatment with an additional agent, but there is uncertainty regarding the optimal time to start second-line therapy. We add a second-line medication if there has been incomplete response to high-dose prednisone after 2 to 4 months or if the patient relapses during prednisone taper.
In some patients, it may be appropriate to start a second-line agent at disease onset together with prednisone. We do this in patients with severe weakness at onset, multisystem involvement (e.g., ILD, myocarditis), or patients with contraindications to corticosteroids. Additionally, there is increasing evidence from observational studies that testing for autoantibodies can help guide expectations about a patient’s clinical course and response to therapy (see Table 2). Compared to patients with other autoantibodies or seronegative patients, patients with anti-Mi-2 antibodies may require the lowest dose of prednisone for adequate disease control, infrequently require a second-line agent, and rarely experience relapse during tapering of therapy [124]. By contrast, anti-Jo-1 antibodies have been associated with a poor response to prednisone alone and an increased need for second-line agents [125, 126]. In a cohort of 40 patients with various ARS antibodies, 83% required additional immunotherapy beyond prednisone over a mean 40 months of follow-up [37]. Patients with ASS and ILD are especially likely to be refractory to corticosteroid monotherapy. In one single-center retrospective study, all 17 patients with anti-Jo-1 antibodies and ILD either failed to respond to prednisone monotherapy or had disease relapse within 12 months of initiation of therapy [127].
Prednisone monotherapy is rarely sufficient in patients with IMNM. In one cohort, 90% of patients required addition of a second-line agent within 6 months of treatment initiation [50]. In fact, 56% of these patients required the concurrent use of three medications to control disease activity. In another cohort, patients with anti-SRP myopathy were significantly less likely to be successfully managed with corticosteroid monotherapy than those with anti-HMGCR myopathy (8% of SRP patients vs 31% of anti-HMGCR patients, p = 0.0048) [40], similar to the rates reported in a French cohort [8]. In other observational studies, rates of steroid failure in anti-HMGCR patients were as high as 92 to 100% [42, 128, 129]. Such findings have led some experts to argue that second-line agents should be used more systematically in these patients. In 2016, the European Neuromuscular Centre (ENMC) convened a workshop on the definition and treatment of IMNM. The assembled panel of experts reached consensus that patients with IMNM should be treated both with corticosteroids as well as a second-line agent either immediately or within 1 month of presentation, depending on severity of disease and response to steroid treatment [8]. Additionally, they recommended that IVIG should be added for all anti-HMGCR myositis patients within 6 months of presentation and that rituximab should be added for all anti-SRP myositis patients within 6 months if other measures are ineffective (see below).
Choice of Second-Line Agents
Based on expert consensus and observational studies, methotrexate, azathioprine, and mycophenolate mofetil are considered preferred second-line agents [8, 116, 130,131,132,133]. We use methotrexate as our second-line agent of choice. In the authors’ experience, methotrexate has a faster time to therapeutic effect than either azathioprine or mycophenolate, with response evident within 4 weeks in some patients (Table 2). We tend to avoid methotrexate in patients with ILD or ARS antibodies who are at high risk for developing ILD. The pulmonary toxicity associated with methotrexate can mimic ILD, a diagnostic dilemma and therapeutic dilemma we find best avoided. We use mycophenolate as our treatment of choice in patients with ASS or DM/PM patients with ILD. Retrospective studies suggest mycophenolate to be a safe and effective treatment for ILD and small series support its use specifically in patients with ILD related to autoimmune myopathy [134, 135].
A small randomized, factorial-design, double-blinded, placebo-controlled trial in adult myositis patients failed to demonstrate any benefit of the addition of methotrexate over prednisone monotherapy, either alone or in combination with cyclosporine [136]. In one retrospective single-center cohort study of 160 patients with PM or DM, patients treated with methotrexate (at any point during their illness) had a significantly better 10-year survival in comparison to patients treated with azathioprine [137]. Given this was a retrospective observational study, imbalances in known and unknown confounders may have accounted for some of the difference between patients treated with azathioprine compared to methotrexate; however, the authors did adjust for baseline CK, presence of ILD, or presence of cancer. Other studies have suggested similar efficacy between the two agents [119, 122].
IVIG is one of the few treatments with randomized placebo-controlled trial data to support its use in patients with autoimmune myopathy [138]. In one study, 15 patients with dermatomyositis were randomized to receive IVIG 2 g/kg/month or placebo for 3 months, followed by an optional cross-over phase for another 3 months. Patients treated with IVIG had a significant increase in muscle strength after 3 months. Including crossovers, 9/12 IVIG-treated patients met prespecified criteria for major improvement in strength compared to 0/11 receiving placebo. In a review of seven open-label studies of IVIG in myositis patients, it was estimated that 71 to 100% of patients had a reported response [139]. An ongoing international multicenter phase III randomized controlled trial of Octagam (NCT02728752) may soon provide additional evidence.
Because of the aggressive nature of the disease, there has been increasing interest in the use of IVIG in patients with IMNM. In addition to the reported success of IVIG as up-front monotherapy as discussed above, treatment with IVIG within the first 3 months was associated with a better clinical response at 6 months (p = 0.047) in a single-center cohort of IMNM patients studied retrospectively [50]. ENMC consensus recommendations suggest that IVIG can be used instead of or an addition to methotrexate as second-line therapy [8]. For patients that respond to IVIG and remain dependent on it, clinical stability can usually be achieved with a maintenance dose of 1 g/kg/month after the initial 3 months [140].
Case series have suggested a high rate of response to rituximab among patients with autoimmune myopathies—an estimated 80% of 88 PM and DM patients and 40 ASS patients improved after rituximab administration [141]. The Rituximab in Myositis (RIM) trial—the largest randomized, prospective, double-blind trial of myositis patients to date—failed to demonstrate a benefit of rituximab based on its primary outcome [142]. The trial utilized a randomized, cross-over design in which all patients received rituximab—half at study onset, and half after a delay of 8 weeks. Flaws of study design have been postulated to be the reason for statistical failure of the trial, including an underestimation of the placebo effect and a longer time to onset of rituximab effect than expected [143].
Some experts have argued for the specific utility of rituximab in patients with anti-SRP-antibodies. Recent work has supported the pathogenic role of anti-SRP and anti-HMGCR antibodies in IMNM [144, 145], and disease severity has been shown to correlate with antibody titers in anti-SRP myopathy patients [146]. Although a case series suggested that there is a significant reduction in anti-SRP titers after rituximab [147], post hoc analysis from the RIM trial failed to show a significant decrease after rituximab [148]. Small case series of anti-SRP myopathy patients suggest frequent but not universal response [49, 147]. ENMC consensus recommendations suggest that rituximab can be used instead of or in addition to methotrexate as second-line therapy [8].
Rituximab rarely induces long-term remission. Some clinicians redose rituximab routinely at 6-month intervals, but we wait until early signs of relapse emerge as some patients may derive clinical benefit for a year or more before a repeat dose is required. Retrospective data suggests patients can have sustained response from a single cycle from less than 1 year to more than 2 years [149].
Cyclosporine [127, 150,151,152,153] and tacrolimus [152, 154, 155] are typically reserved as third-line agents, mainly because of their side effect profiles. Treated patients reported in the literature typically have had either myositis refractory to multiple other immunosuppressants or ILD. Cyclosporine and tacrolimus have similar side effect profiles, with nephrotoxicity and worsening hypertension being the most common side effects requiring dose adjustment or cessation of the drug. Due to the risk of severe side effects including infertility and later secondary malignancy, as well as the burden of monitoring required during therapy, cyclophosphamide is typically reserved as a last option, usually in patients with severe ILD or systemic vasculitis [156, 157].
Minimizing Deleterious Effects of Treatment
While assuring adequate therapeutic effect, it is equally important to mitigate the risk associated with immunosuppression and prolonged corticosteroid use. Together with the patient’s primary care physician, we ensure fasting blood glucose, serum potassium levels, and blood pressure are monitored regularly during corticosteroid therapy. We also recommend annual eye exams. To limit weight gain, we advise patients to follow a low-sodium, low-carbohydrate diet. Because of the risk of incipient osteoporosis, we obtain a baseline bone density scan (DEXA) and repeat annually while steroids are continued. Lower baseline bone density and higher daily dose of corticosteroids are predictive of vertebral fracture, but even those patients with normal bone density are at an increased risk [158, 159]. We also prescribe calcium (500 to 600 mg BID or TID) and vitamin D (1000 IU/daily) to all patients. Bisphosphonates may be considered in patients with concerning DEXA results or those at higher risk for osteoporosis such as postmenopausal women. Patients on second-line agents associated with an increased risk of skin cancer should be advised to wear sunscreen and avoid UV exposure. In patients with high-risk ARS antibodies, we perform pulmonary function testing annually.
Immunosuppressive therapy increases the risk of infection. Over 15 years of follow-up in one cohort of PM/DM patients, 37% were admitted to the hospital for severe infection. Aspiration pneumonia was most common, but severe opportunistic infection occurred in 11.8% of the patients [160]. Accordingly, we recommend pneumococcal vaccine and yearly influenza vaccination. Influenza vaccination has been demonstrated to be safe and to generate adequate immunogenicity in patients with myositis on immunosuppressive therapy [161]. Before starting second-line immunosuppressive therapies, we also screen for tuberculosis and hepatitis B and C. There are no consensus guidelines regarding prophylaxis for pneumocystis pneumonia (PCP) in patients with myositis, but some authors have suggested thresholds of total lymphocyte count < 800/μL or CD4 counts < 200/μL [162]. Our practice is to recommend prophylaxis to patients receiving a daily dose of prednisone greater than 20 mg/day together with a second immunosuppressive agent. Finally, progressive multifocal leukoencephalopathy (PML) due to reactivation of the JC virus has been reported in myositis patients during treatment with rituximab, azathioprine, mycophenolate, cyclosporine, and cyclophosphamide and IVIG [163,164,165]. The incremental risk associated with any one agent remains uncertain in this population, as many cases occurred in patients treated with multiple agents and PML has also been reported in myositis patients on prednisone monotherapy and even without immunosuppressive therapy [163].
Treatment of IBM
Unfortunately, immunotherapies to date have generally been ineffective for IBM. Treated patients will often have a decreased serum CK and even reduced inflammation on repeat muscle biopsy, but this does not correlate with a meaningful clinical improvement [166]. Moreover, an observational study found that patients treated with immunosuppressive therapies were more severely disabled at last follow-up and had more rapid progression toward handicap with walking [56]. Because of the lack of objective evidence of improvement and this potential for harm, we do not recommend immunosuppressive therapy for patients with IBM.
Myostatin and related ligands in the TGFβ family inhibit differentiation and growth of skeletal muscle. Bimagrumab is an antagonistic antibody against activin receptors which mediate signaling downstream from myostatin. Despite a promising proof of concept trial that demonstrated increased thigh muscle volume after intramuscular injection of the drug [167], a phase II/III trial of intravenous use failed to meet its primary efficacy outcome (NCT01925209). Follistatin is an endogenous molecule that binds activins and blocks binding to their receptors, thus inhibiting the myostatin pathway in a different way. Gene therapy using a rAAV1 vector to deliver an alternatively spliced cDNA isoform of follistatin into muscle was recently reported to be safe in an open-label study of six patients with IBM, with no adverse events attributed to gene therapy [168].
Finally, a phase 2b trial of rapamycin was recently completed. Rapamycin is an mTOR inhibitor and not only helps regulate cell survival, protein synthesis, and autophagy, but also inhibits IL-2. Although the primary outcome—quadriceps strength by quantitative muscle testing—was negative, significantly less fatty replacement of muscle was seen in treated patients after 1 year and other clinical measures improved. An open-label extension study is reportedly planned [169].
Nonpharmacologic Management
A multidisciplinary approach to treatment—including physical therapy, occupational therapy, speech/swallowing therapy, and other consultants such as rheumatology, dermatology, and pulmonology—is recommended to limit patient disability and to monitor for and manage extramuscular involvement. Stretching and passive range of motion exercises are important for severely weak patients to prevent joint contractures. Patients with difficulty walking may benefit from assist devices (such as ankle-foot orthotics [AFO] or knee-ankle-foot orthotic [KAFO]) and aids like a cane or walker. Rehabilitation programs can also teach patients compensatory actions that reduce fall risk. Multiple small studies have demonstrated that many forms of aerobic exercise and moderate intensity strength training are safe in myositis patients, and help maintain or even improve strength [170, 171]. A small, open-label study of a home exercise program for patients with IBM demonstrated increased muscle strength after 16 weeks [172]. Occupational therapy is helpful for those with functional impairment from upper extremity weakness; splinting and assist devices can be recommended. Patients with dysphagia should be referred to a speech-language pathologist. Dynamic imaging can demonstrate the anatomic and physiologic nature of the dysphagia [173] and compensatory swallowing maneuvers and dietary modifications can be taught to improve swallowing safety and maintain nutrition. Some IBM patients may benefit from botulinum toxin injection, esophageal dilation, or cricopharyngeal myotomy depending on the nature of the dysphagia [174].
Long-Term Prognosis and Mortality
Observational studies suggest that 20 to 40% of patients with DM and PM will achieve a long-term remission (improved and stable strength, normal serum muscle enzymes, and absence of other organ system involvement) off pharmacologic therapy [175,176,177]. For the remainder of patients, the disease course will either be chronic continuous or polycyclic, with relapse(s) after periods of quiescence. Relapse during tapering of therapy is common, but recurrence of symptoms can also occur many years after discontinuation of therapy so ongoing follow-up is important. Many patients with a chronic continuous course can return to and maintain normal strength, but require ongoing treatment. Even among those patients with normal muscle strength, ongoing perception of disability is common and patients frequently report reduced quality of life [178, 37]. Patients with IBM will have slow but continuous progression of weakness throughout their disease course, leading to the use of a cane on average 10 years and the use of a wheelchair on average 15 years after disease onset [57, 179]. Patients with disease onset after age 55 to 60 years have been shown to require gait aids after a significantly shorter period of time than younger patients [179, 180].
Antibodies may better predict a patient’s prognosis than the histology seen on muscle pathology, further highlighting the limitations of the historical classification scheme of inflammatory myopathy. As an example, 16 of 100 patients with anti-SRP myopathy in one cohort had an inflammatory muscle biopsy rather than one typical of necrotizing myopathy; these differences on biopsy were not predictive of clinical outcome [47]. Anti-Jo-1 antibodies have been associated with reduced risk of remission off pharmacologic therapy [175]. Patients with SRP or HMGCR antibodies have been shown to have low rates of complete response to therapy. In a Japanese cohort of patients with either SRP or HMGCR antibodies, less than 20% of patients had become free of symptoms or disability at 2 years [40]. In a cohort of patients from the USA, 48% of anti-SRP myopathy patients reached full or near-full strength after 4 years of treatment and 44% of 50 anti-HMGCR myopathy patients reached full strength after 2 years of treatment [43, 149]. Both anti-SRP and anti-HMGCR myopathy patients with a younger age of onset have been shown to be significantly less likely to return to full strength than older patients [43, 47, 149]. Statin-exposed anti-HMGCR myopathy patients, by contrast, have been shown to be more treatment responsive [181]. Up to 12% of reported anti-SRP and anti-HMGCR myopathy patients with longitudinal follow-up achieved remission off immunosuppressive entirely [42, 43, 50, 128, 181], leading some experts to recommend that second-line agents should not tapered or stopped until patients have had at least 2 years of well-controlled disease or minimal or no steroids [8, 182].
Although survival has improved over time for myositis patients, studies have estimated a mortality risk two- to threefold higher than that of the general population [182, 183]. Cancer, cardiac involvement, lung complications, and infections are cited as the most common causes of death [184]. In a cross-sectional cohort of 831 patients with PM (53%), DM (43%), and amyopathic DM (4%) over median follow-up of 4.5 years, the risk of death was significantly higher in patients with ILD (HR 2.13, CI 1.06–4.25, p = 0.03) [185]. In those without ILD, the survival rates at 1, 5, and 10 years were 99, 95, and 90%, respectively. In those with ILD, the survival rates at 1, 5, and 10 years were 97, 91, and 81%, respectively. Interestingly, a 2014 meta-analysis including 27 studies of 3487 patients failed to identify significant differences in mortality between any autoantibodies even despite their often-distinct clinical presentations [34]. Patients with IBM have been shown to have normal life expectancy, but complications of the disease—dysphagia leading to aspiration and pneumonia and severe weakness leading to falls, for example—can contribute to death in some patients [57, 186].
Future Directions
Given that much of the available evidence comes from observational studies, there is a need for more prospective trials in patients with autoimmune myopathies. Recent work has proven that large clinical trials are possible despite the rarity of these diseases if multicenter, multidisciplinary designs are utilized [142, 187]. There is increasing evidence that type-1 interferon (IFN)-mediated mechanisms are important in the pathogenesis of DM. Type-1 IFN-induced genes are the highest differentially upregulated genes in DM muscle, and studies have demonstrated intracellular aggregation of IFN-upregulated proteins, with IFN-β playing a particularly important role [188,189,190]. In 2013, a phase 1b study of sifalimumab—an anti-interferon-α monoclonal antibody—in patients with PM and DM was reported. Measurements of IFN-gene signature (IFNGS) were suppressed in both blood and muscle after administration of the drug, and patients that improved by > 15% on MMT had a greater degree of IFNGS suppression [191]. A phase II trial of sifalimumab was also completed for patients with SLE. The pharmaceutical company has since abandoned development in favor of anifrolumab, an anti-interferon-α/β receptor monoclonal antibody, but there are no myositis trials for this drug at the time of this writing. A phase 2 trial of an IFN-Kinoid vaccine that induces production of anti-IFN-α antibodies is currently enrolling patients (clinicaltrials.gov, NCT02980198).
Janus kinase (JAK) inhibitors, which have a suppressive effect on interferon signaling, have recently been studied for use in autoimmune disorders. Interferon-β in particular relies on these molecules as downstream signal transducers. Tofacitinib is an oral JAK 1/3 inhibitor that has been approved in the USA for treatment of rheumatoid arthritis and psoriatic arthritis. After case reports suggested impressive responses to the medication in patients with DM refractory to multiple immunosuppressive therapies [192, 193], a phase 1 trial of patients with IIM has been initiated (clinicaltrials.gov, NCT300264).
Recent work has attempted to identify subsets of patients that may be more likely to benefit from B cell-depleting therapy. In post hoc analysis of patients in the RIM trial, an increased signature of IFN-regulated chemokines (IFNCK) in either blood [194] or muscle [195] correlated with certain measures of clinical response. Further work has demonstrated that IFNCK scores differ among patients with different autoantibodies both at baseline and after rituximab treatment [196]. For example, IFNCK scores were high at baseline and decreased after rituximab in patients with ASS or anti-Mi-2 antibodies, whereas IFNCK scores were high at baseline but did not decrease after rituximab in patients with TIF-γ. By contrast, patients with SRP antibodies had among the lowest IFNCK scores at baseline and there was no significant change after rituximab. This underscores that the pathogenesis of the autoimmune myopathies is not uniform and that the mechanism by which various immunosuppressive therapies affect the disease process may not be uniform, either. Finally, belimumab, a B cell-depleting agent that inhibits B cell-activating factor (BAFF), is also being studied in autoimmune myopathy patients (NCT02347891). BAFF mRNA expression has been shown to correlate with measures of disease activity [197].
References
AA Amato, JA Russell (eds): Neuromuscular Disorders, 2nd ed. New York, McGraw-Hill Education; 2016, Table 33-1, p. 828.
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344–347.
Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403–407.
Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscular disorders : NMD. 2004;14:337–345.
Dalakas MC. Inflammatory muscle diseases. N Engl J Med. 2015;372:1734–1747.
Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011, Naarden, The Netherlands. Neuromuscular disorders : NMD. 2013;23:1044–1055.
Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Annals of the rheumatic diseases. 2017;76:1955–1964.
Allenbach Y, Mammen AL, Benveniste O, Stenzel W. 224th ENMC International Workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscular disorders : NMD. 2018;28:87–99.
Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26–30.
Marie I, Hatron PY, Levesque H, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore). 1999;78:139–147.
Amato AA, Gronseth GS, Jackson CE, et al. Inclusion body myositis: clinical and pathological boundaries. Ann Neurol. 1996;40:581–586.
Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54:597–613.
Stonecipher MR, Jorizzo JL, White WL, Walker FO, Prichard E. Cutaneous changes of dermatomyositis in patients with normal muscle enzymes: dermatomyositis sine myositis? J Am Acad Dermatol. 1993;28:951–956.
Mammen AL. Autoimmune Myopathies. Continuum (Minneapolis, Minn). 2016;22:1852–1870.
Nielsen AO, Johnson E, Hentzer B, Kobayasi T. Dermatomyositis with universal calcinosis. A histopathological and electron optic study. Journal of cutaneous pathology. 1979;6:486–491.
Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255–286.
Lundberg IE. The heart in dermatomyositis and polymyositis. Rheumatology (Oxford, England). 2006;45 Suppl 4:iv18–21.
Rai SK, Choi HK, Sayre EC, Avina-Zubieta JA. Risk of myocardial infarction and ischaemic stroke in adults with polymyositis and dermatomyositis: a general population-based study. Rheumatology (Oxford, England). 2016;55:461–469.
Lai YT, Dai YS, Yen MF, et al. Dermatomyositis is associated with an increased risk of cardiovascular and cerebrovascular events: a Taiwanese population-based longitudinal follow-up study. Br J Dermatol. 2013;168:1054–1059.
Diederichsen LP, Simonsen JA, Diederichsen AC, et al. Cardiac Abnormalities in Adult Patients With Polymyositis or Dermatomyositis as Assessed by Noninvasive Modalities. Arthritis care & research. 2016;68:1012–1020.
Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Annals of the rheumatic diseases. 2004;63:297–301.
Marie I, Hachulla E, Chérin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis care & research. 2002;47:614–622.
de Merieux P, Verity MA, Clements PJ, Paulus HE. Esophageal abnormalities and dysphagia in polymyositis and dermatomyositis. Arthritis and rheumatism. 1983;26:961–968.
Mugii N, Hasegawa M, Matsushita T, et al. Oropharyngeal Dysphagia in Dermatomyositis: Associations with Clinical and Laboratory Features Including Autoantibodies. PLoS One. 2016;11:e0154746.
Laskin BL, Choyke P, Keenan GF, Miller FW, Rider LG. Novel gastrointestinal tract manifestations in juvenile dermatomyositis. The Journal of Pediatrics. 1999;135:371–374.
Horowitz M, McNeil JD, Maddern GJ, Collins PJ, Shearman DJC. Abnormalities of gastric and esophageal emptying in polymyositis and dermatomyositis. Gastroenterology. 1986;90:434–439.
Olazagasti JM, Baez PJ, Wetter DA, Ernste FC. Cancer risk in dermatomyositis: a meta-analysis of cohort studies. American journal of clinical dermatology. 2015;16:89–98.
Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282–291.
Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96–100.
Luu X, Leonard S, Joseph K-A. Dermatomyositis presenting as a paraneoplastic syndrome with resolution of symptoms following surgical management of underlying breast malignancy. Journal of Surgical Case Reports. 2015;2015:rjv075.
Bernatsky S, Joseph L, Pineau CA, et al. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Annals of the rheumatic diseases. 2009;68:1192–1196.
Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. American journal of respiratory and critical care medicine. 2001;164:1182–1185.
Troyanov Y, Targoff IN, Tremblay JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005;84:231–249.
Lega JC, Fabien N, Reynaud Q, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmunity reviews. 2014;13:883–891.
Witt LJ, Curran JJ, Strek ME. The Diagnosis and Treatment of Antisynthetase Syndrome. Clinical pulmonary medicine. 2016;23:218–226.
Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Current rheumatology reports. 2011;13:175–81.
Noguchi E, Uruha A, Suzuki S, et al. Skeletal Muscle Involvement in Antisynthetase Syndrome. JAMA neurology. 2017;74:992–999.
Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis and rheumatism. 2011;63:713–721.
Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle & nerve. 2013;48:477–483.
Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. Journal of Neurology, Neurosurgery and Psychiatry. 2016;87:1038–1044.
Christopher-Stine L, Casciola-Rosen LA, Hong G, et al. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis and rheumatism. 2010;62:2757–2766.
Allenbach Y, Drouot L, Rigolet A, et al. Anti-HMGCR autoantibodies in european patients with autoimmune necrotizing myopathies: Inconstant exposure to statin. Medicine (United States). 2014;93:150–157.
Tiniakou E, Pinal-Fernandez I, Lloyd TE, et al. More severe disease and slower recovery in younger patients with anti-3-hydroxy-3-methylglutarylcoenzyme A reductase-associated autoimmune myopathy. Rheumatology (United Kingdom). 2017;56:787–794.
Liang WC, Uruha A, Suzuki S, et al. Pediatric necrotizing myopathy associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies. Rheumatology (Oxford, England). 2017;56:287–293.
Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry. 2002;73:420–428.
Hengstman GJ, ter Laak HJ, Vree Egberts WT, et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Annals of the rheumatic diseases. 2006;65:1635–1638.
Suzuki S, Nishikawa A, Kuwana M, et al. Inflammatory myopathy with anti-signal recognition particle antibodies: case series of 100 patients. Orphanet journal of rare diseases. 2015;10:61.
Rider LG, Shah M, Mamyrova G, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013;92:223–243.
Pinal-Fernandez I, Parks C, Werner JL, et al. Longitudinal Course of Disease in a Large Cohort of Myositis Patients With Autoantibodies Recognizing the Signal Recognition Particle. Arthritis care & research. 2017;69:263–270.
Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA neurology. 2015;72:996–1003.
Allenbach Y, Keraen J, Bouvier AM, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain. 2016;139:2131–2135.
Liewluck T, Kao JC, Mauermann ML. PD-1 Inhibitor-associated Myopathies: Emerging Immune-mediated Myopathies. Journal of immunotherapy (Hagerstown, Md : 1997). 2017.
Kao JC, Liao B, Markovic SN, et al. Neurological Complications Associated With Anti-Programmed Death 1 (PD-1) Antibodies. JAMA neurology. 2017;74:1216–1222.
Haddox CL, Shenoy N, Shah KK, et al. Pembrolizumab induced bulbar myopathy and respiratory failure with necrotizing myositis of the diaphragm. Annals of oncology : official journal of the European Society for Medical Oncology. 2017;28:673–675.
Vallet H, Gaillet A, Weiss N, et al. Pembrolizumab-induced necrotic myositis in a patient with metastatic melanoma. Annals of oncology : official journal of the European Society for Medical Oncology. 2016;27:1352–1353.
Benveniste O, Guiguet M, Freebody J, et al. Long-term observational study of sporadic inclusion body myositis. Brain. 2011;134:3176–3184.
Cox FM, Titulaer MJ, Sont JK, et al. A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain. 2011;134:3167–3175.
Verma A, Bradley WG, Adesina AM, Sofferman R, Pendlebury WW. Inclusion body myositis with cricopharyngeus muscle involvement and severe dysphagia. Muscle & nerve. 1991;14:470–3.
Riminton DS, Chambers ST, Parkin PJ, Pollock M, Donaldson IM. Inclusion body myositis presenting solely as dysphagia. Neurology. 1993;43:1241–1243.
Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis. Observations in 40 patients. Brain. 1989;112 ( Pt 3):727–747.
Uruha A, Noguchi S, Hayashi YK, et al. Hepatitis C virus infection in inclusion body myositis: A case-control study. Neurology. 2016;86:211–217.
Lloyd TE, Pinal-Fernandez I, Michelle EH, et al. Overlapping features of polymyositis and inclusion body myositis in HIV-infected patients. Neurology. 2017;88:1454–1460.
Bouillot S, Coquet M, Ferrer X, et al. [Inclusion body myositis associated with sacroidosis: a report of 3 cases]. Annales de pathologie. 2001;21:334–336.
Sanmaneechai O, Swenson A, Gerke AK, Moore SA, Shy ME. Inclusion body myositis and sarcoid myopathy: coincidental occurrence or associated diseases. Neuromuscular disorders : NMD. 2015;25:297–300.
Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM. Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain. 2016;139:1348–1360.
Tymms KE, Webb J. Dermatopolymyositis and other connective tissue diseases: a review of 105 cases. J Rheumatol. 1985;12:1140–1148.
Carter JD, Kanik KS, Vasey FB, Valeriano-Marcet J. Dermatomyositis with normal creatine kinase and elevated aldolase levels. J Rheumatol. 2001;28:2366–2367.
Nozaki K, Pestronk A. High aldolase with normal creatine kinase in serum predicts a myopathy with perimysial pathology. J Neurol Neurosurg Psychiatry. 2009;80:904–908.
Pinal-Fernandez I, Casciola-Rosen LA, Christopher-Stine L, Corse AM, Mammen AL. The Prevalence of Individual Histopathologic Features Varies according to Autoantibody Status in Muscle Biopsies from Patients with Dermatomyositis. J Rheumatol. 2015;42:1448–1454.
Roux S, Seelig HP, Meyer O. Significance of Mi-2 autoantibodies in polymyositis and dermatomyositis. J Rheumatol. 1998;25:395–396.
Shamim EA, Rider LG, Pandey JP, et al. Differences in idiopathic inflammatory myopathy phenotypes and genotypes between Mesoamerican Mestizos and North American Caucasians: ethnogeographic influences in the genetics and clinical expression of myositis. Arthritis and rheumatism. 2002;46:1885–1893.
Hamaguchi Y, Kuwana M, Hoshino K, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147:391–398.
Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70:360–374.
Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis and rheumatism. 2009;60:2193–2200.
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25–34.
Koga T, Fujikawa K, Horai Y, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford, England). 2012;51:1278–1284.
Gil B, Merav L, Pnina L, Chagai G. Diagnosis and treatment of clinically amyopathic dermatomyositis (CADM): a case series and literature review. Clinical rheumatology. 2016;35:2125–2130.
Trallero-Araguas E, Rodrigo-Pendas JA, Selva-O'Callaghan A, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis and rheumatism. 2012;64:523–532.
Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis and rheumatism. 2006;54:3682–3689.
Fujimoto M, Hamaguchi Y, Kaji K, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis and rheumatism. 2012;64:513–522.
Fiorentino DF, Kuo K, Chung L, et al. Distinctive cutaneous and systemic features associated with antitranscriptional intermediary factor-1gamma antibodies in adults with dermatomyositis. J Am Acad Dermatol. 2015;72:449–455.
Gunawardena H, Wedderburn LR, Chinoy H, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis and rheumatism. 2009;60:1807–1814.
Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis and rheumatism. 2013;65:2954–2962.
Valenzuela A, Chung L, Casciola-Rosen L, Fiorentino D. Identification of clinical features and autoantibodies associated with calcinosis in dermatomyositis. JAMA dermatology. 2014;150:724–729.
Tansley SL, Betteridge ZE, Shaddick G, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford, England). 2014;53:2204–2208.
Ceribelli A, Fredi M, Taraborelli M, et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis research & therapy. 2012;14:R97.
Greenberg SA. Cytoplasmic 5′-nucleotidase autoantibodies in inclusion body myositis: Isotypes and diagnostic utility. Muscle & nerve. 2014;50:488–492.
Larman HB, Salajegheh M, Nazareno R, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. 2013;73:408–418.
Lloyd TE, Christopher-Stine L, Pinal-Fernandez I, et al. Cytosolic 5'-Nucleotidase 1A As a Target of Circulating Autoantibodies in Autoimmune Diseases. Arthritis care & research. 2016;68:66–71.
Herbert MK, Stammen-Vogelzangs J, Verbeek MM, et al. Disease specificity of autoantibodies to cytosolic 5′-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Annals of the rheumatic diseases. 2016;75:696–701.
Koffman BM, Rugiero M, Dalakas MC. Immune-mediated conditions and antibodies associated with sporadic inclusion body myositis. Muscle & nerve. 1998;21:115–117.
Rojana-Udomsart A, Bundell C, James I, et al. Frequency of autoantibodies and correlation with HLA-DRB1 genotype in sporadic inclusion body myositis (s-IBM): a population control study. Journal of neuroimmunology. 2012;249:66–70.
Eisen A, Berry K, Gibson G. Inclusion body myositis (IBM): myopathy or neuropathy? Neurology. 1983;33:1109–1114.
Joy JL, Oh SJ, Baysal AI. Electrophysiological spectrum of inclusion body myositis. Muscle & nerve. 1990;13:949–951.
Finanger EL, Russman B, Forbes SC, et al. Use of skeletal muscle MRI in diagnosis and monitoring disease progression in Duchenne muscular dystrophy. Physical medicine and rehabilitation clinics of North America. 2012;23:1–10, ix.
Charlot-Lambrecht I, Brochot P, Noblet H, Varoquier C, Eschard JP. Neurogenic muscle hypertrophy. Joint, bone, spine : revue du rhumatisme. 2009;76:401–403.
Tasca G, Monforte M, De Fino C, et al. Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle & nerve. 2015;52:956–962.
Tomasova Studynkova J, Charvat F, Jarosova K, Vencovsky J. The role of MRI in the assessment of polymyositis and dermatomyositis. Rheumatology (Oxford, England). 2007;46:1174–1179.
Pitt AM, Fleckenstein JL, Greenlee RG, Jr., et al. MRI-guided biopsy in inflammatory myopathy: initial results. Magnetic resonance imaging. 1993;11:1093–1099.
Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–678.
Uruha A, Nishikawa A, Tsuburaya RS, et al. Sarcoplasmic MxA expression: A valuable marker of dermatomyositis. Neurology. 2017;88:493–500.
Greenberg SA. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology. 2007;69:2008–2019.
Engel AG, Arahata K. Monoclonal antibody analysis of mononuclear cells in myopathies. II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol. 1984;16:209–215.
Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry. 2000;68:472–478.
Aouizerate J, De Antonio M, Bassez G, et al. Myofiber HLA-DR expression is a distinctive biomarker for antisynthetase-associated myopathy. Acta neuropathologica communications. 2014;2:154.
Chung T, Christopher-Stine L, Paik JJ, Corse A, Mammen AL. The composition of cellular infiltrates in anti-HMG-CoA reductase-associated myopathy. Muscle & nerve. 2015;52:189–195.
Alshehri A, Choksi R, Bucelli R, Pestronk A. Myopathy with anti-HMGCR antibodies: Perimysium and myofiber pathology. Neurology(R) neuroimmunology & neuroinflammation. 2015;2:e124.
Emslie-Smith AM, Engel AG. Necrotizing myopathy with pipestem capillaries, microvascular deposition of the complement membrane attack complex (MAC), and minimal cellular infiltration. Neurology. 1991;41:936–939.
O'Hanlon TP, Carrick DM, Targoff IN, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine (Baltimore). 2006;85:111–127.
Mammen AL, Gaudet D, Brisson D, et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis care & research. 2012;64:1233–1237.
Limaye V, Bundell C, Hollingsworth P, et al. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle & nerve. 2015;52:196–203.
Hiniker A, Daniels BH, Lee HS, Margeta M. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta neuropathologica communications. 2013;1:29.
Gordon Pa, Winer JB, Hoogendijk JE, Choy EHS. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database of Systematic Reviews. 2012;8:CD003643.
Mammen AL, Tiniakou E. Intravenous Immune Globulin for Statin-Triggered Autoimmune Myopathy. NEJM. 2015;373:1680–1682.
Dimachkie MM, Barohn RJ, Amato A. Idiopathic Inflammatory Myopathies. Neurol Clin. 2014;32:595–628.
Mammen A. Autoimmune muscle disease. Handbook of clinical neurology. 2016;133:467–484.
Mammen AL. Which nonautoimmune myopathies are most frequently misdiagnosed as myositis? Current Opinion in Rheumatology. 2017;29:618–622.
Nguyen K, Bassez G, Krahn M, et al. Phenotypic Study in 40 Patients With Dysferlin Gene Mutations. Archives of Neurology. 2007;64:1176.
Joffe MM, Love La, Ph D Leff RL. Drug Therapy of the Idiopathic Inflammatory Myopathies : Predictors of Response to Prednisone , Azathioprine , and. The American Journal of Medicine. 1993;94:379–387.
Pinal-Fernandez I, Casal-Dominguez M, Carrino JA, et al. Thigh muscle MRI in immune-mediated necrotising myopathy: Extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Annals of the rheumatic diseases. 2017;76:681–687.
Zheng Y, Liu L, Wang L, et al. Magnetic resonance imaging changes of thigh muscles in myopathy with antibodies to signal recognition particle. Rheumatology (United Kingdom). 2015;54:1017–1024.
Miller FW. New approaches to the assessment and treatment of the idiopathic inflammatory myopathies. Annals of the rheumatic diseases. 2012;71:i82-i85.
van de Vlekkert J, Hoogendijk JE, de Haan RJ, et al. Oral dexamethasone pulse therapy versus daily prednisolone in sub-acute onset myositis, a randomised clinical trial. Neuromuscular Disorders. 2010;20:382–389.
Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositis-specific autoantibodies define useful homoegeneous patient groups. Medicine (Baltimore). 1991;70:360–374.
Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senécal J-L. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis research & therapy. 2007;9:R78.
Stanciu R, Guiguet M, Musset L, et al. Antisynthetase syndrome with anti-Jo1 antibodies in 48 patients: Pulmonary involvement predicts disease-modifying antirheumatic drug use. Journal of Rheumatology. 2012;39:1835–1839.
Cavagna L, Caporali R, Abdì-Alì L, et al. Cyclosporine in anti-Jo1-positive patients with corticosteroid-refractory interstitial lung disease. Journal of Rheumatology. 2013;40:484–492.
Grable-Esposito P, Katzberg HD, Greenberg SA, et al. Immune-mediated necrotizing myopathy associated with statins. Muscle and Nerve. 2010;41:185–190.
Ramanathan S, Langguth D, Hardy TA, et al. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurology: Neuroimmunology and NeuroInflammation. 2015;2:e96.
Distad BJ, Amato AA, Weiss MD. Inflammatory myopathies. Curr Treat Options Neurol. 2011;13:119–130.
Giannini M, Callen JP. Treatment of Dermatomyositis with Methotrexate and Prednisone. Archives of Dermatology. 1979;115:1251–1252.
Bunch TW. Prednisone and azathioprine for polymyositis. Long-term followup. Arthritis & Rheumatism. 1981;24:45–48.
Majithia V, Harisdangkul V. Mycophenolate mofetil (CellCept): An alternative therapy for autoimmune inflammatory myopathy. Rheumatology. 2005;44:386–389.
Fischer A, Brown KK, Du Bois RM, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. Journal of Rheumatology. 2013;40:640–646.
Morganroth PA, Kreider ME, Weth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis care & research. 2010;62:1496–1501.
Ibrahim F, Choy E, Gordon P, et al. Second-line agents in myositis: 1-year factorial trial of additional immunosuppression in patients who have partially responded to steroids. Rheumatology (United Kingdom). 2015;54:1050–1055.
Schiopu E, Phillips K, MacDonald PM, Crofford LJ, Somers EC. Predictors of survival in a cohort of patients with polymyositis and dermatomyositis: Effect of corticosteroids, methotrexate and azathioprine. Arthritis Research and Therapy. 2012;14:1–9.
Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000.
Anh-Tu Hoa S, Hudson M. Critical review of the role of intravenous immunoglobulins in idiopathic inflammatory myopathies. Seminars in arthritis and rheumatism. 2017;46:488–508.
Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: An open study with thirty-five adult patients. Arthritis and rheumatism. 2002;46:467–474.
Nalotto L, Iaccarino L, Zen M, et al. Rituximab in refractory idiopathic inflammatory myopathies and antisynthetase syndrome: Personal experience and review of the literature. Immunologic Research. 2013;56:362–370.
Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyosistis and adult polymyositis: A randomized placebo phase trial. Arthritis and rheumatism. 2013;65:314–324.
De Visser M. The efficacy of rituximab in refractory myositis: The jury is still out. Arthritis and rheumatism. 2013;65:303–306.
Arouche-Delaperche L, Allenbach Y, Amelin D, et al. Pathogenic role of anti–signal recognition protein and anti–3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Annals of Neurology. 2017;81:538–548.
Allenbach Y, Arouche-Delaperche L, Preusse C, et al. Necrosis in anti-SRP and anti-HMGCR myopathies. Neurology. 2018;90:e507-e517.
Benveniste O, Drouot L, Jouen F, et al. Correlation of anti-signal recognition particle autoantibody levels with creatine kinase activity in patients with necrotizing myopathy. Arthritis and rheumatism. 2011;63:1961–1971.
Valiyil R, Casciola-Rosen L, Hong G, Mammen A, Christopher-Stine L. Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: A case series. Arthritis Care and Research. 2010;62:1328–1334.
Aggarwal R, Oddis CV, Goudeau D, et al. Autoantibody levels in myositis patients correlate with clinical response during B cell depletion with rituximab. Rheumatology (United Kingdom). 2016;55:991–999.
Pinal-fernandez I, Parks C, Werner JL, et al. Longitudinal course of disease in a large cohort of patients with autoantibodies recognizing the signal recognition particle. Arthritis care & research. 2017;69:263–270.
Danieli M, Malcangi G, Palmieri C, et al. Cyclosporin A and intravenous immunoglobulin treatment in polymyositis / dermat ... Annals of the rheumatic diseases. 2002;61:37–41.
Qushmaq KA, Chalmers A, Esdaile JM. Cyclosporin A in the treatment of refractory adult polymyositis/dermatomyositis: population based experience in 6 patients and literature review. J Rheumatol. 2000;27:2855–2859.
Takada K, Nagasaka K, Miyasaka N. Polymyositis/dermatomyositis and interstitial lung disease: A new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity. 2005;38:383–392.
Kotani T, Takeuchi T, Makino S, et al. Combination with corticosteroids and cyclosporin-A improves pulmonary function test results and chest HRCT findings in dermatomyositis patients with acute/subacute interstitial pneumonia. Clinical rheumatology. 2011;30:1021–1028.
Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis and rheumatism. 2005;52:2439–2446.
Oddis CV, Sciurba FC, Elmagd KA, Starzl TE. Tacrolimus in refractory polymyositis with interstitial lung disease. Lancet. 1999;353:1762–1763.
Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology. 2007;46:124–130.
Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. Journal of Rheumatology. 2005;32:1719–1726.
Naganathan V, Jones G, Nash P, et al. Vertebral Fracture Risk With Long-term Corticosteroid Therapy: Prevalence and relation to age, bone density, and corticosteroid use. Archives of Internal Medicine. 2000;160:2917–2922.
Van Staa TP, Laan RF, Barton IP, et al. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis and rheumatism. 2003;48:3224–3229.
Marie I, Ménard JF, Hachulla E, et al. Infectious Complications in Polymyositis and Dermatomyositis: A Series of 279 Patients. Seminars in arthritis and rheumatism. 2011;41:48–60.
Shinjo SK, de Moraes JCB, Levy-Neto M, et al. Pandemic unadjuvanted influenza A (H1N1) vaccine in dermatomyositis and polymyositis: Immunogenicity independent of therapy and no harmful effect in disease. Vaccine. 2012;31:202–206.
Efthimiou P, Pokharna H, Kukar M, Hennessey K. PCP chemoprophylaxis is essential for lymphopenic dermatomyositis patients treated with immunomodulators. Muscle & nerve. 2011;43:918–919.
Dastmalchi M, Laki J, Lundberg IE, Iacobaeus E. The Journal of Rheumatology Progressive Multifocal Leukoencephalopathy in a Patient with Polymyositis : Case Report and Literature Review The Journal of Rheumatology is a monthly international serial edited by Earl D . Silverman featuring research article. The Journal of Rheumatology. 2012;39.
Belhassen-Garcia M, Rabano-Gutierrez A, Velasco-Tirado V, et al. Atypical Progressive Multifocal Leukoencephalopathy in a Patient with Antisynthetase Syndrome. Internal Medicine. 2015;54:519–524.
Marie I, Guegan-Massardier E, Levesque H. Progressive multifocal leukoencephalopathy in refractory polymyositis treated with rituximab. European Journal of Internal Medicine. 2011;22:e13-e14.
Barohn RJ, Amato AA, Sahenk Z, Kissel JT, Mendell JR. Inclusion body myositis: explanation for poor response to immunosuppressive therapy. Neurology. 1995;45:1302–1304.
Amato AA, Sivakumar K, Goyal N, et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology. 2014;83:2239–2246.
Mendell JR, Sahenk Z, Al-Zaidy S, et al. Follistatin Gene Therapy for Sporadic Inclusion Body Myositis Improves Functional Outcomes. Molecular Therapy. 2017;25:870–879.
Lilleker JB, Bukhari M, Chinoy H. Rapamycin for inclusion body myositis: targeting non-inflammatory mechanisms. Rheumatology. 2018:1–2.
Habers GEA, Takken T. Safety and efficacy of exercise training in patients with an idiopathic inflammatory myopathy—a systematic review. Rheumatology. 2011;50:2113–2124.
Alemo Munters L, Dastmalchi M, Andgren V, et al. Improvement in health and possible reduction in disease activity using endurance exercise in patients with established polymyositis and dermatomyositis: A multicenter randomized controlled trial with a 1-year open extension followup. Arthritis Care and Research. 2013;65:1959–1968.
Johnson LG, Edwards DJ, Walters S, Thickbroom GW, Mastaglia FL. The Effectiveness of an Individualized, Home-Based Functional Exercise Program for Patients With Sporadic Inclusion Body Myositis. Journal of clinical neuromuscular disease. 2007;8:187–194.
Olthoff A, Carstens PO, Zhang S, et al. Evaluation of dysphagia by novel real-time MRI. Neurology. 2016;87:2132–2138.
Darrow DH, Hoffman HT, Barnes GJ, Wiley CA. Management of dysphagia in inclusion body myositis. Archives of otolaryngology--head & neck surgery. 1992;118:313–317.
Bronner IM, van der Meulen MFG, de Visser M, et al. Long-term outcome in polymyositis and dermatomyositis. Annals of the rheumatic diseases. 2006;65:1456–1461.
Marie I HE, Hatron PY, Hellot MF,Levesque H, Devulder B. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors. J Rheumatol. 2001;28:22–30.
van de Vlekkert J, Hoogendijk JE, de Visser M. Long-term follow-up of 62 patients with myositis. J Neurol. 2014;261:992–998.
Ponyi A, Borgulya G, Constantin T, et al. Functional outcome and quality of life in adult patients with idiopathic inflammatory myositis. Rheumatology. 2005;44:83–88.
Cortese A, Machado P, Morrow J, et al. Longitudinal observational study of sporadic inclusion body myositis: Implications for clinical trials. Neuromuscular Disorders. 2013;23:404–412.
Peng A, Koffman BM, Malley JD, Dalakas MC. Disease progression in sporadic inclusion body myositis: Observations in 78 patients. Neurology. 2000;55:296–298.
Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody Levels Correlate with Creatine Kinase Levels and Strength in Anti-HMG-CoA Reductase-Associated Autoimmune Myopathy. Arthritis Rhem. 2012;64:4087–4093.
Limaye V, Hakendorf P, Woodman RJ, Blumbergs P, Roberts-Thomson P. Mortality and its predominant causes in a large cohort of patients with biopsy-determined inflammatory myositis. Intern Med J. 2012;42:191–198.
Airio A, Kautiainen H, Hakala M. Prognosis and mortality of polymyositis and dermatomyositis patients. Clinical rheumatology. 2006;25:234–239.
Marie I. Morbidity and mortality in adult polymyositis and dermatomyositis. Current rheumatology reports. 2012;14:275–285.
Johnson C, Pinal-Fernandez I, Parikh R, et al. Assessment of Mortality in Autoimmune Myositis With and Without Associated Interstitial Lung Disease. Lung. 2016;194:733–737.
Price MA, Barghout V, Benveniste O, et al. Mortality and Causes of Death in Patients with Sporadic Inclusion Body Myositis: Survey Study Based on the Clinical Experience of Specialists in Australia, Europe and the USA. Journal of Neuromuscular Diseases. 2016;3:67–75.
Ruperto N, Pistorio A, Oliveira S, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: A randomised trial. The Lancet. 2016;387:671–678.
Suárez-Calvet X, Gallardo E, Nogales-Gadea G, et al. Altered RIG-I/DDX58-mediated innate immunity in dermatomyositis. Journal of Pathology. 2014;233:258–268.
Salajegheh M, Kong SW, Pinkus JL, et al. Interferon-Stimulated Gene 15 (ISG15) Conjugates Proteins in Dermatomyositis Muscle with Perifascicular Atrophy. Ann Neurol. 2010;67:53–63.
Liao AP, Salajegheh M, Nazareno R, et al. Interferon β is associated with type 1 interferon-inducible gene expression in dermatomyositis. Annals of the rheumatic diseases. 2011;70:831–836.
Higgs BW, Zhu W, Morehouse C, et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-α monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Annals of the rheumatic diseases. 2014;73:256–262.
Kurtzman DJB, Wright NA, Lin J, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: An alternative treatment. JAMA dermatology. 2016;152:944–945.
Paik JJ, Christopher-Stine L. A case of refractory dermatomyositis responsive to tofacitinib. Seminars in arthritis and rheumatism. 2017;46:e19.
Lopez De Padilla CM, Crowson CS, Hein MS, et al. Interferon-regulated chemokine score associated with improvement in disease activity in refractory myositis patients treated with rituximab. Clinical and experimental rheumatology. 2015;33:655–663.
Nagaraju K, Ghimbovschi S, Rayavarapu S, et al. Muscle myeloid type I interferon gene expression may predict therapeutic responses to rituximab in myositis patients. Rheumatology (United Kingdom). 2016;55:1673–1680.
Reed AM, Crowson CS, Hein M, et al. Biologic predictors of clinical improvement in rituximab-treated refractory myositis. BMC musculoskeletal disorders. 2015;16:257.
López De Padilla CM, McNallan KT, Crowson CS, et al. BAFF expression correlates with idiopathic inflammatory myopathy disease activity measures and autoantibodies. Journal of Rheumatology. 2013;40:294–302.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Additional information
Emer R. McGrath and Christopher T. Doughty are joint first authors
Rights and permissions
About this article

Cite this article
McGrath, E.R., Doughty, C.T. & Amato, A.A. Autoimmune Myopathies: Updates on Evaluation and Treatment. Neurotherapeutics 15, 976–994 (2018). https://doi.org/10.1007/s13311-018-00676-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-018-00676-2