
Abstract
In neuro-oncology, there has been a movement towards personalized medicine, or tailoring treatment to the individual patient. Ideally, tumor and patient evaluations would lead to the selection of the best treatment (based on tumor characterization) and the right dosing schedule (based on patient characterization). The recent advances in the molecular analysis of glioblastoma have created optimism that personalized targeted therapy is within reach. Although our understanding of the molecular complexity of glioblastoma has increased over the years, the path to developing effective targeted therapeutic strategies is wrought with many challenges, as described in this review. These challenges include disease heterogeneity, clinical and genomic patient variability, limited number of effective treatments, clinical trial inefficiency, drug delivery, and clinical trial support and accrual. To confront these challenges, it will be imperative to devise innovative and adaptive clinical trials in order to accelerate our efforts in improving the outcomes for our patients who have been in desperate need.
Similar content being viewed by others


Introduction
In recent years, our understanding of glioma tumor biology has grown significantly as a result of large-scale molecular profiling efforts. Glioblastoma was selected as one of the first 3 cancers to be profiled by the National Institutes of Health’s The Cancer Genome Atlas (TCGA) and is now one of the most molecularly profiled of all human cancers. These molecular profiling efforts have resulted in the identification of molecular prognostic factors and molecular vulnerabilities that could be potentially targeted in the development of novel treatments in gliomas. TCGA has dramatically furthered our understanding regarding the genomic alterations driving gliomagenesis. Comprehensive molecular profiling has renewed a sense of optimism that personalizing therapy for glioblastoma may be possible. However, to date, our understanding of the molecular underpinnings of glioblastoma is far outpacing the development of new therapies.
Historically, the efficacy of molecularly based targeted therapy has been validated in the treatment of other cancers and is best exemplified by the development of imatinib. Chronic myeloid leukemia (CML), a myeloproliferative disorder, has a characteristic abnormality, the Philadelphia (Ph) chromosome, which results from a reciprocal translocation between the long arms of chromosomes 9 and 22. This translocation results in the generation of the fusion protein BCR–ABL which is a constitutively activated tyrosine kinase and necessary for its oncogenic activity in CML. This recognition of the essential role of BCR–ABL tyrosine kinase activity in CML was exploited in the development of imatinib, which acts as an inhibitor of the BCR–ABL tyrosine kinase [1]. Imatinib revolutionized the treatment of Ph (+) CML and has emerged as a paradigm for the development of targeted therapy in cancer therapeutics.
Although we are hopeful that molecularly based targeted therapy will be transformative in the management of gliomas, particularly glioblastoma, like it has been for other cancers, challenges are clearly evident in the actual clinical implementation, which will be described in this review. Many targets have been identified, but few therapies have demonstrated efficacy and none have been approved, underscoring the need for us to better understand mechanisms of resistance and why trials fail.
Glioblastoma
Glioblastoma is the most common malignant primary brain tumor in adults and invariably carries a poor prognosis. Despite optimized multimodality treatment that typically includes surgery, radiation, and cytotoxic chemotherapy, recent clinical trials have reported a median survival of only 14–16 months with a 26 to 33% 2-year survival rate [2, 3]. Recurrent disease is inevitable, emphasizing the pressing need for effective salvage therapies. Current cytotoxic therapies in recurrent glioblastoma are disappointing, with an average progression-free survival rate at 6 months of 9 to 15%, and an average survival of 3 to 9 months [4–7]. Since the establishment of the use of combined temozolomide with radiation (often referred to as the Stupp protocol), few therapeutic advances have been noteworthy. One negative clinical trial after another, including several large scale phase III studies, has plagued the field of neuro-oncology [3, 8–10].
Personalized Medicine for Gliomas
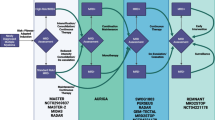
As part of our current clinical practice, most treatment decisions for gliomas, including glioblastoma, are largely made based on the patient’s age and performance status. This one-size-fits-all approach is becoming less and less appealing in the setting of our growing understanding regarding the extensive variability in the mutations and pathways driving tumorigenesis. Efforts have been directed in high-dimensional profiling studies in order to better understand the biology of glioblastoma. The recognition of the critical role genetic alterations have on patient outcome has led to the revision of the 2007 World Health Organization criteria of central nervous system tumors in order to integrate genomic and histopathologic data. In the wake of growing use of comprehensive molecular profiling, there has been a movement towards tailoring therapy to the unique features of the tumor. We are striving to move into an era of personalizing therapy in gliomas with molecularly targeted approaches, but embarking on this era has come with many challenges (Fig. 1).

Obtacles in the development of personalized, targeted therapeutics in glioblastoma
Challenges and Strategies
Challenge: Disease Heterogeneity
Glioblastoma is a molecularly heterogenous disease. The heterogeneity is both intratumoral and intertumoral, further complicated by continued molecular changes over time [11–15]. No single driver mutation explains tumorigenesis, and it is this molecular diversity that partially explains the therapeutic resistance seen. Genomic testing has afforded us the insight into the discovery of molecular alterations, some of which are drivers and some of which are passengers. Driver mutations enable the proliferation of a clonal population of cells within a tumor microenvironment. The detection of actionable driver mutations for which there is a targeted therapy can potentially open up treatment options for patients. In the clinical setting, this can have an extraordinary impact on patient care introducing options and clinical trial opportunities that otherwise would have not been known if the molecular testing had not been performed.
The first cancer studied by the TCGA was glioblastoma, and the efforts in comprehensively profiling the genomic characterization of glioblastoma have furthered our understanding of this biologically complex disease. This analysis has yielded the identification of important genetic events in glioblastoma, including: 1) dysregulation of growth factor signaling via amplification and mutational activation of receptor tyrosine kinase genes; 2) activation of the phosphatidylinositol-3OH-kinase pathway; and 3) inactivation of the p53 and retinoblastoma tumor suppressor pathways (Fig. 2) [11, 16]. Recognition of these key signaling pathways as obligatory events in most glioblastomas spurred the hope that patterns of mutations could inform the development of future therapies for patients who fit a specific molecular subgroup.

Pathway alterations in glioblastoma. Overall alteration rate summarized for canonical PI3K/mitogen-activated protein kinase (MAPK), p53, and RB regulatory pathways. Permission obtained from Elsevier Publishing Group. The Somatic Genomic Landscape of Glioblastoma. Cell 155, 462-477 (2013)
The vast differences in gene expression in glioblastoma have been described giving rise to 4 glioblastoma subtypes: classical, neural, proneural, and mesenchymal [15, 17, 18]. Of the 4 subgroups, the neural subtype is the most controversial, owing to the expression pattern being between the mesenchymal and proneural subtypes [19]. Aberrations and gene expression of EGFR, NF1, and PDGFRA/IDH1 characterize the classical, mesenchymal, and proneural subtypes, respectively. Despite a common morphologic diagnosis of glioblastoma, these subtypes harbor differential responses to aggressive therapy impacting survival, with the greatest benefit seen in the classical subgroup and the least benefit seen in the proneural subgroup [15]. The identification of these subtypes of glioblastoma has highlighted the challenge that different therapeutic approaches may be needed depending on the subtype. This challenge is further complicated by the known intratumoral cellular heterogeneity, even within subtype designations. Presently, there are no prospective clinical trials enrolling based on these subclassifications of glioblastoma. The challenge will be the identification of which therapies will work in a specified subgroup and how to enrich therapeutic groups. Although the subgroup classification system of glioblastoma has provided us a furthered understanding of the heterogeneity associated with the disease, the classification system plays no current role in clinical decision making as there are no treatments presently available that are distinguished by glioblastoma subtype.
Single-cell analyses have resulted in a growing understanding of intratumoral heterogeneity or the coexistence of different cell populations in the same glioblastoma. Intratumoral heterogeneity is becoming increasingly recognized as a determinant of treatment failure and disease recurrence [20]. Patel et al. [21] examined the genome sequence in samples of primary glioblastoma using single-cell RNA sequencing and found inherent variability in the expression of diverse transcription of factors related to oncogenic signaling, proliferation, complement/immune response, and hypoxia. Their work additionally demonstrated that the glioblastoma subtypes are variably expressed across individual cells within a tumor underscoring the therapeutic challenge of intratumoral heterogeneity. These findings convey critical implications regarding cell-to-cell variability expression of signaling molecules relevant to targeted therapy and the concern that targeting a specific tumor subtype will result in the regrowth of the suboptimally treated tumor cells from other subtypes.
The expansion of tumoral heterogeneity over time poses a tremendous challenge in the development of effective therapies. Exposure of a tumor to treatment results in the clonal selection of pre-existing passenger or acquired mutations suggesting that these cells diverted early from the primary tumor. It is necessary for us to understand the molecular profile of the residual population of tumor-initiating cells (TICs), also commonly referred to as cancer stem cells, left behind following treatment with surgery, radiation, and chemotherapy. TICs are those tumor cells that possess the ability to regrow the tumor [22]. In glioblastoma, TICs are a subpopulation of glioblastoma stem cells that are capable of self-renewal and apopotic resistance [23]. TICs are speculated to account for the aggressive nature of glioblastoma.
The molecular profile of the tumor at time of diagnosis compared with the profile at the time of recurrence or progression may share similarities or may be very different as a result of the acquisition of mutations in response to the selective pressures exerted by the therapy the patient was exposed to. The genomic road from primary to recurrent glioblastoma has been classified into linear or divergent recurrences. Linear recurrences share extensive genetic similarity with the primary tumor and can be directly traced to one of its specific sectors. In divergent recurrences, few genetic alterations are shared with the primary tumor originating from cells that branched off early during tumorigenesis [24]. In an analysis comparing primary and post-treatment glioblastoma whole-genome and multisector exome sequencing, deregulation of the p53 pathway has been found to be associated with an increased fraction of subclonal mutations at time of relapse, independent of age [24]. This analysis exemplifies divergent recurrence. Other interesting findings in this analysis included that the number of clonal but not subclonal mutations found at time of diagnosis increased with the age of the patient, and clonal mutations reflected the signature of the germline substitutions [24]. A high rate of molecular complexity may afford the tumor more routes to escape treatment, further complicating the therapeutic challenge of glioblastoma.
Strategy: Disease Heterogeneity
The 4 molecular subtypes that have been characterized in glioblastoma should be utilized to personalize treatment for each subgroup. Validation of prognostic characteristics of molecular subgroups and the prognostic impact of individual targets is critical to enhance our clinical trial design and development. Patient enrichment should be employed in all future glioblastoma trials in order to expedite the discovery of novel and effective therapies. Treatment impact in a molecularly defined subgroup will augment the efficiency of clinical trial execution in identifying a subset of patients most likely to benefit from treatment. This selection strategy to evaluate treatment impact on a biomarker-specific subgroup has been used in other solid tumors such as the investigation of trastuzumab in patients with breast cancer with human epidermal growth factor 2 overexpression. The addition of adjuvant trastuzumab significantly improved disease-free and overall survival among women with human epidermal growth factor 2-positive breast cancer [25], exemplifying that patient enrichment is key. For patients receiving trastuzumab in addition to standard therapy, the 5-year rate of disease-free survival was 84% compared with those patients receiving standard therapy alone (hazard ratio 0.64, p < 0.001) [25].
Overcoming the challenge of tumor heterogeneity may also require the development of novel polytherapeutic strategies. Combination therapy may be superior to monotherapy, and consideration will need to be given to rational combinatorial strategies utilizing immunotherapy, radiotherapy, targeted therapy, antiangiogenic therapy, and/or chemotherapy. Rational polytherapy strategies will need to be balanced with the likely risk and cost of increased toxicities, so-called “toxic synergy”.
The recognition of molecular changes over time underscores the need for smarter and adaptive clinical trial designs in order to better delineate those changes seen at the time of recurrence or progression. When feasible, recurrent glioblastoma trials should include biopsy or re-resection into the clinical trial design. In the setting of intratumoral heterogeneity and the recognized heterogeneity over time, sampling of multiple tumor regions at diagnosis and at time of recurrence may provide invaluable insight. This technique will likely only be feasible for tumors that are amenable for extensive resections. Paired tissue samples of the primary tumor compared with the recurrent disease would further our understanding of the molecular changes driving the disease initially versus those driving progression in one specific patient.
Challenge: Interpatient Heterogeneity
In addition to tumor heterogeneity, the heterogeneity from patient to patient further complicates the development of effective treatments in glioblastoma. The molecular intragroup heterogeneity among patients with glioblastoma is vast, resulting in very small numbers of patients with similar molecular aberrations. A myriad of genetic alterations have been described in glioblastoma, with many of these mutations seen only in a small percentage of patients. Even the most common mutations in glioblastoma, such as epidermal growth factor receptor (EGFR) amplification, are seen in less than half of all glioblastomas [16].
Germline (genomic) polymorphisms can play a role in predicting response and toxicity. For example, genetic polymorphisms of UGT1A1 can have a major influence on the pharmacokinetics and toxicity of irinotecan, with those patients having low UGT1A1 activity being at increased risk for irinotecan toxicity [26]. This knowledge of a differential risk for toxicity based on UGT1A1 genotyping has been used in clinical trial designs for patients with metastatic colorectal cancer, in order to identify those patients who could receive escalated doses of irinotecan without significant adverse events [27]. Preliminary analyses of clinical and genomic risk factors for temozolomide indicated that single nucleotide polymorphisms (SNPs) of O-6-methylguanine-DNA methyltransferase (MGMT; not related to promoter methylation) correlate with the risk of myelotoxicity, indicating that this normal variant of the protein is far less efficient in reversing the alkylation of DNA on normal bone marrow elements [28]. Patients carrying the G allele of MGMT (rs2308327) exhibited a 240% increase in risk of toxicity [95% confidence interval (CI) 0.99–5.84] [28].
SNP analysis has been evaluated in glioblastoma in predicting response to temozolomide. Promoter methylation of MGMT has been established as a predictive biomarker for response to temozolomide in glioblastoma [29]. A survival benefit was seen in patients with MGMT-methylated tumors compared with those with unmethylated MGMT tumors receiving concurrent radiation therapy and temozolomide (23.4 months vs 12.6 months median survival) [29]. However, 14.8% of the patients with unmethylated MGMT still survived at least 2 years, indicating that methylation testing alone may not be able to detect all temozolomide responders. In a study by Rapkins et al. [30], carriage of the T allele of the rs16906252 SNP was associated with both MGMT methylation and low MGMT protein expression predicting significantly longer survival in patients treated with temozolomide with both MGMT methylated and unmethylated glioblastoma [30]. The median survival for carriers of the T allele was 19.96 months (95% CI 14.07–25.85) compared with wild-type patients [12.26 months (95% CI 10.39–14.13, p = .010] [30]. The identification of this T carriage allele has the potential for significant translational implications in clinical practice through the detection of the SNP in blood DNA. This information could then be used to stratify response to temozolomide aiding in the personalization of treatment for glioblastoma.
Clinical factors additionally result in alterations in treatment efficacy, pharmacokinetics, and toxicity risk. For example, patients on hepatic enzyme-altering anticonvulsants may be at risk for enhanced drug clearance or be at increased risk for toxicity based on the alteration of cytochrome p450 activity of the anticonvulsant. Renal and/or hepatic impairment may also place patients at different risks for toxicity. Body surface area and the presence of comorbid medical conditions are additional clinical factors that have to be accounted for when evaluating the differences seen in treatment efficacy, pharmacokinetics, and toxicity.
Strategy: Interpatient Heterogeneity
Recognition of genomic factors that can influence outcomes affords us the opportunity to enhance our clinical trial design. The inclusion of SNP analysis from genomic DNA in clinical trials could potentially enrich our understanding of patients who respond versus do not respond to therapy and those who are at higher risk for developing toxicity. It is also possible that innovative informatics strategies to identify putative SNPs in early testing could then be validated in larger-scale trials. The clinical factors affecting the differences seen between patients must be recorded in a comprehensive fashion including the monitoring of concurrent medication(s) during early phase and subsequent efficacy testing. It will also be imperative to integrate toxicity data evaluating an agent under investigation. Ideally, in clinical trial designs in the future, we will be incorporating clinical and genomic factors to create paradigms for personalizing the dosing for our patients.
Challenge: Limited Efficacy With Single-Agent Targeted Therapy
Single-agent targeted therapy to date has been extremely disappointing owing to the inherent heterogeneity of the disease and to multiple driver mutations in various cell populations within a tumor. Redundant signaling pathways preclude single-agent targeted therapy from resulting in long-term responses [31, 32]. Many agents have been developed, but response rates continue to be poor. The redundancy and or “cross-talk” in pathways make the approach of attacking a single target ineffective.
EGFR is a drug target in glioblastoma that has been of interest given the high frequency of EGFR alterations seen in glioblastoma, with approximately 40% of glioblastomas harboring oncogenic EGFR alterations [16]. The success seen with EGFR inhibitors in EGFR-mutant lung cancer has only added to the appeal of this target [33]. Unfortunately, the EGFR kinase inhibitors, including erlotinib and gefitinib, have produced disappointing results in glioblastoma [34], with the lack of response ascribed to redundancy in signaling pathways and intratumoral heterogeneity [32, 35]. The most common oncogenic EGFR alterations in glioblastoma are relatively insensitive to erlotinib. Vivanco et al. [36] showed that EGFR mutant glioma and lung cell lines have differential sensitivity to irreversible EGFR inhibitors [36]. Mutations in glioblastoma are actually preferentially inhibited by EGFR inhibitors that can only be accommodated by the inactive conformation of the EGFR catalytic pocket as the result of their aniline substituents such as lapatinib [36, 37].
Strategy: Limited Efficacy With Single-Agent Targeted Therapy
Observations over time has furthered our understanding that glioblastoma growth is driven by signaling built on functional redundancy whereby blockade of a single pathway or node of signal transduction is insufficient to affect patient outcomes substantially. Although clear examples have not been described, it is likely that adaptation in the setting of targeted therapy occurs in the treatment of glioblastoma similar to that noted with B-Raf-mutated melanoma after treatment with B-Raf inhibitors, notable for upregulation of the mitogen-activated protein kinase/extracellular regulated kinase pathway. Future therapeutic strategies in clinical trials should employ combination therapies with targeted drugs to attack tumor cells harboring driver mutations while using treatments with less specificity to target cells with passenger and/or secondary mutations. Ideally, the pharmacologic agent should be tailored to target differences between patients and the clonal diversity within each patient’s tumor. The lack of effectiveness of single-agent therapies is likely attributable to the tumor’s capability to utilize multiple cellular pathways in a redundant fashion, underscoring the need to investigate the combination of targeted therapies with other agents both targeted and less targeted. Future trials with single-agent targeted therapy in glioblastoma should be, for the most part, abandoned unless a very specific driver mutation is clearly identified. Although the use of multiagent rational drug combinations may result in responses and less resistance to treatment, initiation of combination trials in the absence of testing each treatment individually will be challenging, and combination therapies will likely be associated with an increased risk for more toxicity.
Challenge: Brain Tumor Pharmacology
To date, targeted therapy developed in glioblastoma has also been hampered by limited drug delivery. Most new agents in glioblastoma have failed to demonstrate any efficacy, in large part because drug delivery to the tumor remains poor and subtherapeutic. The blood–brain barrier limits drug delivery and poor tumor perfusion secondary to tumor pressure, and aberrant vessel perfusion further complicates delivery of therapeutic concentrations of drug. If we continue to achieve only subtherapeutic concentrations, the desired effect on the tumor will never be achieved.
Lapatinib, one of the most selective adenosine triphosphate-site competitive kinase inhibitors [38], was evaluated in a window trial administered preoperatively at a dose of 750 mg twice a day. In this study, lapatinib failed to achieve sufficient intratumoral concentrations in glioblastoma. The concentrations found were below that required to induce cell death in EGFR-mutant glioblastoma cell lines [36]. In another study evaluating the combination of lapatinib and pazopanib, an oral multitargeted tyrosine kinase inhibitor of vascular endothelial growth factor receptors, the plasma levels of lapatinib were also found to be subtherapeutic suggesting that dosing strategies to achieve improved intratumoral lapatinib concentrations are needed, such as a pulsatile, intermittent schedule at higher dose levels [39]. It will be critical to incorporate pharmacokinetic analyses into the early development of therapeutic agents.
Strategy: Brain Tumor Pharmacology
In order to overcome the challenge of brain tumor pharmacology, it will be imperative that our preclinical models critically address blood–brain barrier delivery, pharmacokinetic and pharmacodynamics testing validation, and correlative makers (blood, hair, urine, etc.). Window trials where patients undergo biopsy or, ideally, a resection when feasible, followed by treatment with the investigational agent prior to a more definitive surgical procedure is a valuable trial design that affords the opportunity to confirm delivery of the drug to the tumor and its target activity. This model of trial design additionally allows for validation of correlative markers and quickly provides data to guide the decision to proceed with development or not. Although this model of trial design is ideal, the patient population that would be eligible for such a trial is often narrow. Some patients are not surgical candidates and other patients may not have the luxury of time owing to increasing symptom burden to be administered an investigational drug prior to surgery.
Challenge: Clinical Trial Inefficiency
The development of an investigational agent from the initial discovery stage through clinical testing and regulatory review is an expensive, complex, and inefficient process. Even though the Food and Drug Administration reviews oncology drugs, on average, 6 months faster than other drugs, the process is complicated by difficulties in recruiting patients and longer times required to establish efficacy (especially if survival is and endpoint). The median clinical phase times are 1.5 years longer for oncology drugs (7.8 vs 6.3 years) [40]. Oncology drugs additionally take 1 year longer to move from the start of clinical testing in the USA to regulatory marking approval [40]. The cost of oncology drug development is also 20% higher when compared with the average of other drugs ($1042 million vs $868 million) [40].
The standard procedure of the typical phase I trial leading to a phase II and then possibly a phase III study is entirely inefficient, especially in the setting of a rare disease such as glioblastoma. Given the rarity of the disease and the wide number of potential targets and agents, a more rapid, “screening” process is needed for molecularly profiling newly diagnosed and recurrent disease when patients do undergo re-resection. A centralized database to facilitate data sharing and knowledge is needed in the effort to facilitate and expedite the development of clinical trials. Molecular information that is readily accessible must then be used to enrich the patient population in order to allow for small, efficient trials capable of detecting early, efficacy signals.
Strategy: Clinical Trial Inefficiency
“Phase 0” trials are first-in-human studies designed in order to primarily evaluate the pharmacodynamics and/or pharmacokinetic properties of the investigational agent prior to traditional phase I testing [41]. Phase 0 trials involve limited exposure of a study agent administered at low doses and/or for a short period of time with one of the major objectives being to interrogate and refine a target or biomarker assay in a small number of patients prior to initiation of a larger trial involving more patients receiving toxic doses of the investigational agent [41]. Utilizing this type of trial design has the potential to improve the efficiency and success of subsequent molecularly targeted trials.
Innovative trial designs may also reduce clinical trial inefficiency. Several novel trial designs have emerged over the last several years, with the common thread of having a “targeted” strategy. The 2 critical aspects of these new trial designs operate on the components of screening and treatment. For the screening component, patients are evaluated systematically for the presence of the biomarkers of interest, while in the treatment component, patients are assigned to substudies with investigational agents targeting the biomarkers present in their tumors [42]. The 2 primary clinical trial designs that exemplify this model are umbrella and basket trials. Umbrella trials evaluate the impact of different drugs on different mutations in a single type of cancer. Examples of umbrella trials include BATTLE [43, 44], Lung-MAP [42], and FOCUS4 [45]. In contrast, basket trials test the effect of 1 or more drugs on 1 or more single mutation in a variety of cancer types. Examples of basket trials include NCI MATCH [46] and NCI IMPACT (NCT01827384). The design of umbrella and basket trials results in more efficient screening and facilitates the addition of new agents and biomarkers into the protocol on a rolling basis [42].
In glioblastoma, innovative adaptive trial designs are in development that provide for dynamic real-time changes in arm allocation or treatment arms based on outcomes (Fig. 3). The INSIGhT (INdividualized Screening trial of Innovative Glioblastoma Therapy) umbrella trial has been developed for newly diagnosed unmethylated glioblastoma. INSIGhT is biomarker agnostic and harbors the potential for adaptive designs with the ability to add or drop arms. Ineffective combinations are eliminated earlier and promising agents are identified. AGILE (Adaptive Global Innovative Learning Environment) is an international adaptive trial currently being conceptualized to include both patients with newly diagnosed and recurrent glioblastoma. At time of study entry, patients undergo biomarker testing and are stratified into groups according to biomarkers that are potentially predictive of response to therapy. Patients are subsequently adaptively randomized to a specific treatment arm. The AGILE effort is being spearheaded by leaders in neuro-oncology around the world.

Umbrella trials versus basket trials. NCI = National Cancer Institute
Challenge: Clinical Trial Support and Patient Accrual
Limitations in funding have compromised clinical trial designs and progress. Slow patient accrual to clinical trials and the delays in study results leads to diminished enthusiasm from stakeholders. Additionally, there is often a discrepancy in the number of clinical trials available at any one center, with some centers having a robust clinical trial portfolio, while other centers have few clinical trials. A balance in available clinical trials across centers is needed to remedy slow patient accrual. Protocol development and approval time is a long and arduous process as a result of a regulatory environment causing more costs, delays, and overhead. Access to pharmaceutical drug pipelines is another challenging aspect as collaborations between pharmaceutical companies can be difficult. In addition, a lack of collaboration moving forward amongst institutions and clinical research groups can result in redundancy of effort and a disharmonious, competitive environment.
The identification of and accrual of biomarker-selected patients to clinical trials is challenging. The rarity of specific molecular aberrations among patients with glioblastoma underscores the need for the molecular profiling of as many patients as possible. Screening requires substantial time in addition to tissue with often a low chance of enrollment. Although the number of patients with a selected molecular aberration will result in a small pool of eligible candidates for accrual in a clinical trial, small numbers of patients may suffice in demonstrating a clinical benefit if the patient population is appropriately selected.
Clinical Trial Support and Patient Accrual
Future success in the effort to personalize therapy for patients with glioblastoma will be contingent upon an interdisciplinary collaborative environment. It will also be critical for clinicians to inform and engage potential patients for clinical trials. The internet and social media has provided for increased opportunities to disseminate information about clinical trials, which generates enthusiasm. Partnering with philanthropy and patient advocate groups can additionally create excitement, ultimately fostering clinical trial support and patient accrual. Working guidelines with government agencies and pharma to speed agreements and protocol implementation will also aid in the process of clinical trial development overall by reducing the inefficiency of our current process. Collaborations on multiple levels will be a critical factor for success, as will data sharing. Institutional and clinical-derived data availability will make it possible to accelerate progress. Many institutions are now using electronic health record systems that enable better clinical annotation allowing data to be mined more easily for information, including natural history studies.
Concluding Remarks
Precision medicine is a valuable and important approach to therapy in glioblastoma in our efforts to improve the prognosis for these patients. The molecular characterization of glioblastoma has been markedly enhanced over the last several years, but translation to the development of improved targeted therapies yielding improved patient outcomes continues to lag behind. The challenge of personalized targeted therapy in glioblastoma is further complicated by the relative rarity of the disease in contrast to other cancers; progress has been further slowed by poor accrual to clinical trials. The ability to develop personalized therapy in glioblastoma will require ongoing molecular profiling efforts, and success will be contingent upon targeted therapy being administered to a properly selected group of patients harboring the correct molecular target or pathway of interest.
The complex disease heterogeneity of glioblastoma must be met with creative clinical trial designs that incorporate both clinical and molecular factors that are efficient and hypothesis based. The integration of these factors into clinical trial designs may help accelerate our progress towards the development of targeted therapy in glioblastoma. There will need to be a collaborative movement on a multi-institutional level to make prospective collection of blood for genomic testing and tumor tissue from patients on clinical trials a standard of care. Lessons and results from other trials should be used to inform and enhance the development of brain tumor clinical trials. We must also further our understanding of why our current clinical trials fail in order to educate us on what to do better the next time. Although challenges are clearly evident, they are not insurmountable if there is a collaborative effort rooted deep in the common goal of improving outcomes for our patients.
References
Druker, B.J., et al., Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med, 2001. 344(14): p. 1038-1042.
Stupp, R., et al., Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med, 2005. 352(10): p. 987-996.
Gilbert, M.R., et al., Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol, 2013. 31(32): p. 4085-4091.
Wong, E.T., et al., Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol, 1999. 17(8): p. 2572.
Lamborn, K.R., et al., Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol, 2008. 10(2): p. 162-170.
Norden, A., et al., Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology, 2008. 70(10): p. 779-787.
Yung, W.K., et al., A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer, 2000. 83(5): p. 588-593.
Gilbert, M.R., et al., A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med, 2014. 370(8): p. 699-708.
Chinot, O.L., et al., Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med, 2014. 370(8): p. 709-722.
Stupp, R., et al., Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol, 2014. 15(10): p. 1100-1108.
Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 2008. 455(7216): p. 1061-1068.
Kim, J., et al., Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell, 2015. 28(3): p. 318-328.
Bonavia, R., et al., Heterogeneity maintenance in glioblastoma: a social network. Cancer Res, 2011. 71(12): p. 4055-4060.
Sottoriva, A., et al., Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A, 2013. 110(10): p. 4009-4014.
Verhaak, R.G., et al., Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 2010. 17(1): p. 98-110.
Furnari, F.B., et al., Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev, 2007. 21(21): p. 2683-2710.
Phillips, H.S., et al., Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell, 2006. 9(3): p. 157-173.
Brennan, C.W., et al., The somatic genomic landscape of glioblastoma. Cell, 2013. 155(2): p. 462-477.
Goodenberger, M.L. and R.B. Jenkins, Genetics of adult glioma. Cancer Genet, 2012. 205(12): p. 613-621.
Bedard, P.L., et al., Tumour heterogeneity in the clinic. Nature, 2013. 501(7467): p. 355-364.
Patel, A.P., et al., Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 2014. 344(6190): p. 1396-1401.
Zhou, B.B., et al., Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov, 2009. 8(10): p. 806-823.
Liu, J., et al., Glioblastoma tumor initiating cells: therapeutic strategies targeting apoptosis and microRNA pathways. Curr Mol Med, 2013. 13(3): p. 352-357.
Kim, H., et al., Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res, 2015. 25(3): p. 316-327.
Slamon, D., et al., Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med, 2011. 365(14): p. 1273-1283.
Iyer, L., et al., Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest, 1998. 101(4): p. 847-854.
Lu, C.Y., et al., Clinical implication of UGT1A1 promoter polymorphism for irinotecan dose escalation in metastatic colorectal cancer patients treated with bevacizumab combined with FOLFIRI in the first-line setting. Transl Oncol, 2015. 8(6): p. 474-479.
Armstrong, T.S., et al., Risk analysis of severe myelotoxicity with temozolomide: the effects of clinical and genetic factors. Neuro Oncol, 2009. 11(6): p. 825-832.
Hegi, M.E., et al., MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med, 2005. 352(10): p. 997-1003.
Rapkins, R.W., et al., The MGMT promoter SNP rs16906252 is a risk factor for MGMT methylation in glioblastoma and is predictive of response to temozolomide. Neuro Oncol, 2015. 17(12): p. 1589-1598.
Nathanson, D.A., et al., Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science, 2014. 343(6166): p. 72-76.
Stommel, J.M., et al., Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science, 2007. 318(5848): p. 287-290.
Pao, W. and J. Chmielecki, Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer, 2010. 10(11): p. 760-774.
Brandes, A.A., et al., Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res, 2008. 14(4): p. 957-960.
Inda, M.M., et al., Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev, 2010. 24(16): p. 1731-1745.
Vivanco, I., et al., Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov, 2012. 2(5): p. 458-471.
Wood, E.R., et al., A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res, 2004. 64(18): p. 6652-6659.
Karaman, M.W., et al., A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol, 2008. 26(1): p. 127-132.
Reardon, D.A., et al., A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res, 2013. 19(4): p. 900-908.
DiMasi, J.A. and H.G. Grabowski, Economics of new oncology drug development. J Clin Oncol, 2007. 25(2): p. 209-216.
Murgo, A.J., et al., Designing phase 0 cancer clinical trials. Clin Cancer Res, 2008. 14(12): p. 3675-3682.
Herbst, R.S., et al., Lung Master Protocol (Lung-MAP)-a biomarker-driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin Cancer Res, 2015. 21(7): p. 1514-1524.
Kim, E.S., et al., The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov, 2011. 1(1): p. 44-53.
Barker, A.D., et al., I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther, 2009. 86(1): p. 97-100.
Kaplan, R., et al., Evaluating many treatments and biomarkers in oncology: a new design. J Clin Oncol, 2013. 31(36): p. 4562-4568.
Conley, B.A. and J.H. Doroshow, Molecular analysis for therapy choice: NCI MATCH. Semin Oncol, 2014. 41(3): p. 297-299.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Weathers, SP.S., Gilbert, M.R. Toward Personalized Targeted Therapeutics: An Overview. Neurotherapeutics 14, 256–264 (2017). https://doi.org/10.1007/s13311-016-0496-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0496-5