
Abstract
The comprehensive molecular characterization of gastric and gastroesophageal junction adenocarcinomas has led to the improvement of targeted and more effective treatments. As a result, several biomarkers have been introduced into clinical practice and the implementation of innovative diagnostic tools is under study. Such assessments are mainly based on the evaluation of limited biopsy material in clinical practice. In this setting, the pathologist represents a key player in the selection of patients facilitating precision medicine approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) represents a major health issue worldwide. With over one million new cases in 2020 and 769,000 related deaths, GC ranks globally fifth for incidence and fourth for mortality among malignancies [1]. Incidence and mortality vary across different regions and ethnicities. GC is the most commonly diagnosed malignant neoplasm and the first cause of cancer-related death in several South-Central Asian countries. Moreover, Eastern Asia and Eastern Europe have the highest GC incidence rates, while Northern America and Northern Europe have the lowest [1]. It is also noteworthy that a decrease in distal GC incidence has been observed during the last 50 years in Western countries. This trend may be explained by the decreased prevalence of Helicobacter Pylori infections and the improvement of health and hygiene standards [2]. On the other hand, a higher incidence of proximal GC (i.e. of gastric cardia and gastroesophageal junction) has been reported [3], probably related to the increasing incidence of gastroesophageal reflux disease and obesity [4].
Histological and molecular classification of gastric cancer
GC is a heterogeneous neoplasm from both a phenotypical and a molecular viewpoint. In the last 60 years, many histopathological and, more recently, molecular classifications have tried to dissect the biological complexity of GC. Molecular characterizations of GC also allowed to set the stage for the development of molecular targeted therapies and the discovery of predictive biomarkers.
Gastric/gastroesophageal junction adenocarcinoma (GAC) accounts for more than 90% of GCs, representing the most common histotype among gastric malignancies. Other types of GC are squamous cell carcinoma, adenosquamous carcinoma, undifferentiated carcinoma, gastroblastoma and neuroendocrine neoplasms [5].
Among GAC’s morphological classifications, the WHO and Laurén’s are two of the most widely used.
2019 WHO classification recognizes five main subtypes of GAC: (i) tubular adenocarcinoma, (ii) papillary adenocarcinoma, (iii) mucinous adenocarcinoma, (iv) poorly cohesive carcinoma (PCC) and (v) mixed adenocarcinoma. Other rarer variants are GAC with lymphoid stroma, hepatoid carcinoma, micropapillary carcinoma and GAC of fundic-gland type [5].
Laurén’s classification subdivides GAC into intestinal (53%), diffuse (33%) and indeterminate (14%) types [6]. Despite having a lesser degree of sophistication than the WHO classification, Lauren’s classification effectively emphasizes crucial aspects of GAC and is commonly used by clinicians and surgeons. In fact, the intestinal and diffuse subtypes present not only different morphologic appearances but also different molecular background: intestinal-type GAC is usually associated with Helicobacter Pylori and Correa’s cascade, while the diffuse type is characterized by loss of E-cadherin expression [7].
However, in the era of personalized oncology, the morphology-based evaluation must be integrated with additional molecular biomarkers assessment. In this context, two NGS-based molecular classifications have been introduced: The Cancer Genome Atlas (TCGA; 2014) and the Asian Cancer Research Group (ACRG; 2015).
According to TCGA classification, GAC may be subdivided into: (i) tumors positive for Epstein-Barr virus (EBV) (9%), (ii) microsatellite instability-high (MSI) tumors (21%), (iii) genomically stable (GS) tumors (20%) and (iv) tumors with chromosomal instability (CIN) (50%) [8].
EBV-positive GACs commonly arise in the gastric body or fundus and usually present high levels of DNA promoter hypermethylation and of CpC island methylation (CIMP). Specific molecular alterations of these tumors are mutations of PIK3CA (80%), ARID1A (55%), and BCOR (23%), together with CDKN2A promoter hypermethylation (100%) [8]. Moreover, overexpression of Programmed Death Ligands 1 and 2 (PD-L1 and PD-L2) and significant lymphoid infiltration are commonly found in EBV-positive GACs [9].
MSI GACs are defined by loss of mismatch repair (MMR) functions, usually related to hypermethylation of MLH1 promoter region [10]. These neoplasms typically occur in the gastric antrum of older patients, but they may arise in the context of Lynch syndrome and non-polyposis colorectal cancer syndrome [11]. The majority of MSI GACs show intestinal-type morphology and have been associated with a lower rate of nodal metastasis and a more favorable prognosis [12].
GS GACs usually develop in the distal stomach of younger patients (median age 59). Usually associated with Laurén’s diffuse histotype, GS tumors have the worst overall survival and prognosis among TCGA subtypes [8]. This group is characterized by low copy number alterations and a low mutation rate, and the most common mutated genes are ARID1, RHOA, and CDH1. In about 15% of GS GACs a fusion between CLDN18 and ARHGAP26 does occur, which is mutually exclusive with RHOA mutations [8].
CIN GACs usually arise at the gastroesophageal junction and commonly present an intestinal histotype [13]. Characteristic of these neoplasms are DNA aneuploidy, highly variable chromosomal copy numbers, and mutations of TP53, with consequent chromosomal instability. Moreover, genomic amplifications of EGFR, ERBB2, ERBB3, MET, FGFR2, and VEGF-A, are frequent [8].
ACRG classification proposes four other molecular subtypes of GACs, i.e.: (i) microsatellite unstable (MSI) tumors (23%); (ii) microsatellite stable with epithelial-mesenchymal transition features (MSS/EMT) tumors (15%); (iii) microsatellite stable with TP53 active (MSS/TP53+) tumors (26%); and iv) microsatellite stable with TP53 inactive (MSS/ TP53−) tumors (36%) [14].
MSI GACs usually have an antral location and intestinal histotype. Often diagnosed at an early stage, MSI GAC is characterized by the best prognosis among ACRG subtypes. Hypermutation, loss of MLH1 expression and elevated DNA methylation signature are characteristic of this subtype. Commonly mutated genes in MSI GACs are ARID1A (44.2%), PI3K-PTEN-mTOR pathway-related genes (42%), KRAS (23.3%), and ALK (16.3%) [14].
MSS/EMT GACs are associated with an earlier onset and with diffuse histotype. MSS/EMT GC is the ACRG subtype with the worst prognosis and is associated with advanced stage at diagnosis, a high percentage of recurrence and a high frequency of peritoneal metastases. This subgroup is characterized by loss of E-cadherin expression and by a low tumoral mutational burden [14].
MSS/TP53+ GACs are associated with intestinal histotype, EBV infection and a high percentage of liver-limited metastasis. Mutations in ARID1A, PIK3CA, SMAD4, KRAS, and APC are commonly reported [14].
MSS/TP53− tumors usually present an intestinal histotype and are more common in males. Among ACRG subtypes, MSS/TP53− GACs have the highest prevalence of TP53 mutations (60%). Moreover, recurrent amplification of ERBB2, EGFR, CCNE1, CCND1, MDM2, ROBO2, and GATA6 have been described [14].
Despite every proposed classification that shed light on different aspects of GC, a flawless and unifying classification is still lacking [15]. More efforts are required to dissect GC heterogeneity to develop more efficient predictive biomarkers and targeted therapies.
Through the analysis of large molecular data from different platforms, the landmark study by TCGA in 2017 demonstrated that GEJ cancers and GCs have notable molecular similarities, with a progressive increase of chromosomal instability phenotype proximally [16]. Although GEJ cancer has been considered separate from GC according to a model whereby EAC originates from Barrett’s esophagus and thus is not of gastric origin, this growing body of evidence indicates that they should be considered as a single entity for clinical trials of neoadjuvant, adjuvant or systemic therapies.
Targeted therapies in clinical practice
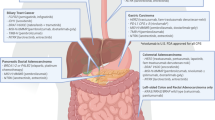
The scientific and medical community is making great efforts toward the development of new molecular targeted therapies (Table 1). At present, the use of anti-Her2 targeted therapies and immune check-point inhibitors (ICIs) is well-established in the clinical setting (Fig. 1). Other promising/appealing molecular targeted therapies are currently being evaluated in clinical trials. In high-incidence countries, such as Japan and Korea, multiple biomarkers, including Her2, MMR proteins, EBV, PD-L1, EGFR, FGFR2, and CLDN18.2, are routinely evaluated by means of immunohistochemistry or in situ hybridization in all GC patients [17]. In lower-incidence Western countries, Her2 and PD-L1 assessment on biopsy samples should be mandatory before initiation of the first-line treatment in all patients with GC. The immunohistochemical assessment of the MMR proteins and EBV status is recommended, according to tissue availability.

Immunohistochemical evaluation of predictive biomarkers in gastric/gastroesophageal adenocarcinoma. A Heterogeneous Her2 expression in a gastroesophageal junction adenocarcinoma showing the concomitant presence of a 3+ positive and a negative biopsy. B Loss of the mismatch repair complex (MMR) protein PMS2. C PD-L1 Combined Positive Score (CPS) > 10. D Membranous expression of CLDN18. (E) EBER (Epstein–Barr virus-encoded small RNAs) in situ hybridization in an EBV-associated adenocarcinoma
Anti-Her2 targeted therapy
The member of the epidermal growth factor receptor (EGFR) family, Her2 or ERBB2 (erb-b2 receptor tyrosine kinase 2) is involved in transmembrane signaling, and its overexpression is responsible for increased cell proliferation, growth, and cell survival [18]. Her2 overexpression has been associated with intestinal histotype and proximal GACs, being reported up to 30% of gastroesophageal junction’s cancers [19, 20]. Assessment of Her2 status is based on immunohistochemistry (score 0, 1+, 2+, and 3+); for tumors with Her2 score 2+, fluorescent in situ hybridization (FISH) should be performed [21]. Her2 -positive tumors are defined by a Her2 score 3+ or by a Her2 score 2+ with FISH-detected HER2 amplification [20].
Following the results of the ToGa trial, tumor biopsies from all patients with GAC at an advanced stage should be assessed for Her2 status [22]. In fact, in patients with advanced Her2-positive GAC, a combination of trastuzumab and fluoropyrimidine plus cisplatin chemotherapy significantly improves overall survival (OS: 13.8 vs 11.1 months) [22]. Care must be taken in the assessment of Her2 status, since preinvasive lesions may also harbor HER2 amplifications and overexpression [23]. Moreover, Her2 expression may be extremely heterogenous in GAC, even within individual neoplasms, with a great impact on trastuzumab efficacy. A heterogeneous expression of Her2 in primary GAC has been associated with shorter progression-free survival (PFS) after trastuzumab-containing first-line chemotherapies [24,25,26]. Therefore, it is highly recommended to acquire tumoral material representative of the entire lesion, performing at least 5 biopsies [27]. Moreover, 1–14% of GACs also present a discordant Her2 expression between primary and metastatic tumors [28,29,30,31,32,33,34]. According to the GASTHER1 study [35], patients with Her2 positive disease identified in repeated biopsies of recurrent and/or metastatic lesions in the context of initially Her2 negative primary GAC, have PFS comparable to those with Her2 positive disease on initial assessment, after trastuzumab-containing chemotherapy. Hence, it is recommended to repeat the Her2 status assessment in primary and metastatic lesions in patients with advanced GAC and initially Her2 negative primary neoplasm.
Primary resistance to trastuzumab is reported in about 50% of GAC patients treated with combined therapy [22]. It has been suggested this phenomenon may be linked to alterations in RTK-RAS-PI3K/AKT pathway and related genes (e.g., EGFR, FGFR2, MET, and KRAS) [8, 36, 37].
The effectiveness of trastuzumab in responder patients may also be compromised by the development of acquired resistance, the causes of which have not been completely elucidated yet [9]. Heterogeneity of Her2 expression can partially explain this phenomenon: trastuzumab-containing chemotherapies, while eradicating Her2 positive neoplastic clones, also confer a proliferative advantage to Her2 negative malignant cells [38, 39]. Moreover, loss of ERBB2 amplification, focal ERBB2 exon 16 deletion and alterations in the RTK/RAS/PI3K pathway have been reported to occur in GAC patients after trastuzumab-containing chemotherapy [37]. In a study by Kim and colleagues [40], CCNE1 amplifications have been linked to a lower response rate to lapatinib combined with capecitabine and oxaliplatin as first-line neoadjuvant therapy in Her2 positive GAC patients. In the same study, analysis of circulating tumor DNA (ctDNA) revealed the progressive onset of MYC, EGFR, FGFR2, and MET amplifications in the progression phase of the disease.
To overcome the issue of primary and acquired resistance to trastuzumab, new Her2-targeted molecules and combinations have been developed.
The antibody–drug conjugate trastuzumab-deruxtecan (T-Dx) is composed of a molecule of trastuzumab attached to a cytotoxic topoisomerase I inhibitor by a cleavable tetrapeptide-based linker. This drug is able to affect not only Her2 positive cells but also the surrounding Her2 negative neoplastic clones [41]. According to Shitara and colleagues [42], T-Dx significantly improves OS (12.5 vs 8.4 months) in patients with Her2 positive advanced-stage GACs previously treated with at least two lines of therapy, including trastuzumab. Of note, a significant antitumoral activity of T-Dx has also been reported in patients with Her2-low GACs (i.e. GACs with Her2 score 2+ and FISH negative) [43]. Ongoing clinical trials (NCT03821233, NCT03602079 and NCT03255070) are trying to evaluate the efficacy of other antibody-drug conjugates against Her2-positive GACs [20].
A synergistic antineoplastic activity between anti-Her2 and anti-PD-1 agents has been reported [44]. This effect could be explained because anti-Her2 drugs may induce PD-1/PD-L1 expression, increase tumor-infiltrating immune cells and promote cross-presentation by dendritic cells [45, 46]. These observations have represented the rationale for several clinical trials. Results from a randomized phase III trial (NCT03615326) support the efficacy of pembrolizumab plus trastuzumab-including chemotherapy in Her2 positive GACs, with a significant reduction in tumor size and improvement in objective response rate (ORR); complete responses in some participants have also been reported [47]. A combination of the anti-Her2 antibody margetuximab and pembrolizumab is also being under evaluation in patients with previously treated, Her2 positive GACs [48]. Moreover, the phase II/III MAHOGANY trial [49] is currently evaluating margetuximab plus retifanlimab with or without chemotherapy and margetuximab plus tebotelimab with chemotherapy in first-line unresectable metastatic or locally advanced GACs. A combination of T-Dx plus the anti-PD-1 antibody is currently under evaluation in phase I/II trial NCT04379596 [50].
Other new anti-HER2 molecules include tucatinib and ZW25, and their efficacy in the treatment of Her2-positive GC is currently under investigation [20, 51].
It has to be noted that Her2 immunohistochemical evaluation still remains a superior predictive biomarker than more sophisticated next-generation technologies. This further underlines the role of the pathologist’s expertise in the therapeutic stratification of GAC patients.
Immunotherapy
ICIs act by reducing neoplastic immune tolerance. ICIs include antibodies against PD1 (pembrolizumab, nivolumab), PD-L1 (atezolizumab, avelumab, durvalumab) and CTLA-4 (ipilimumab) [52].
Potential biomarkers for response to ICIs include PD-L1 expression, microsatellite instability, positive EBV status and high tumor mutational burden (TMB) [53]. PD-L1 expression in GC is determined immunohistochemically by using Combined Positive Score (CPS), which is defined as the number of PD-L1 positive tumor cells, macrophages and lymphocytes, divided by the total number of neoplastic cells, multiplied by 100. GAC is considered PD-L1 positive if CPS ≥ 1.
It must be noted that only invasive neoplasia must be assessed for PD-L1 expression: dysplasia (formerly known as intraepithelial neoplasia) must not be considered when assessing immunohistochemical biomarkers. In fact, PD-L1 has also been described in gastric dysplastic lesions [7, 54]. Of note, our group [55] first described a case of an EBV-associated foveolar gastric dysplasia, which may represent another rare but possible pitfall in the assessment of patients’ eligibility for immunotherapy.
ICIs have been approved in the treatment of selected patients with advanced GAC, but with limited benefit. Pembrolizumab has been approved in the USA as third-line (or greater) therapy in patients with GACs with CPS score ≥ 1 [56]. Nivolumab is used in Japan as third-line option to treat patients with advanced GACs disregarding PD-L1 status [57]. In the phase III CheckMate-649 trial, first-line treatment with nivolumab plus chemotherapy has shown significantly improved survival benefit in previously untreated patients with PD- L1 positive advanced GAC, when compared to chemotherapy alone (OS: 14.4 vs 11.1 months) [58].
On the other hand, some randomized trials have failed to demonstrate the superiority of ICIs above chemotherapy in terms of clinical advantages. In the KEYNOTE-061 study, pembrolizumab has not significantly improved OS compared with paclitaxel as second-line therapy for advanced GACs with PD-L1 CPS score ≥ 1 [59]. According to the KEYNOTE-062 trial’s results, pembrolizumab alone or pembrolizumab plus chemotherapy were not superior to chemotherapy in terms of OS and PFS [60].
Loss of MMR proteins’ function is mostly caused by mutational inactivation or epigenetic silencing of DNA MMR genes, resulting in microsatellite instability. In clinical practice, microsatellite status is evaluated by molecular assays or, as a surrogate, by immunohistochemical analysis of the expression of MMR proteins (MLH1, PMS2, MSH2 and MSH6) [10]. FDA approved the use of pembrolizumab as second-line therapy against MSI-GC [61], but it is worth remembering the majority of MSI-GC are localized and only 3–4% of metastatic GACs are MSI [56, 59].
Amplification of genes encoding for PD-L1 (CD274) and PD-L2 (PDCD1LG2) have been described in EBV-positive GCs [8], suggesting the use of ICIs in this molecular subtype. In a study by Kim and colleagues [62], a significant response to pembrolizumab as a second or greater line of therapy in patients with EBV-positive GCs has been observed (ORR of 100%). In the same study, an ORR of 85.7% in MSI-GCs has also been reported.
TMB may represent a novel predictive biomarker for ICIs, but its efficacy has not been established yet, and further studies are needed. Kim and colleagues have investigated the value of TMB as a potential biomarker of response to pembrolizumab in advanced GAC [62]. However, results were unsatisfactory: high TMB (i.e., > 10–15 mutations per megabase according to different adopted sequencing panels) was associated with MSI subtype, but not all patients with high TMB attained an objective response. Conversely, according to the results of the phase II KEYNOTE-158 study [63], high TMB may identify a subgroup of GAC patients who could significantly respond to pembrolizumab monotherapy. Finally, Greally and colleagues [64] reported an association between TMB and improved survival only in MSI GAC patients.
Other possible ICIs-based therapies are currently under investigation. Results from early phase trials have suggested that combinations of anti-PD-1 antibody plus tyrosine kinase inhibitors (TKIs) may be effective against GAC. Reported ORRs have, respectively, been 44% for regorafenib plus nivolumab [65], and 69% of lenvatinib plus pembrolizumab [66].
In phase I/II NivoRam trials [67], a combination of nivolumab plus the anti-VEGFR-2 ramucirumab in second-line treatment for advanced GC has been evaluated, with promising results. Finally, a combination of nivolumab and ipilimumab is currently being studied in patients with GAC [53].
Future directions
Anti-Claudin 18.2 therapies
Claudin 18.2 (CLDN18.2) is a component of intercellular junctions specific for gastric epithelial cells and is expressed in a significant percentage of primitive and metastatic GACs [68, 69]. As discussed above, up to 15% of GS GACs harbor CLDN18–ARHGAP26/6 fusions [8], and the presence of the fusion is related to CLDN18.2 expression in almost all cases [70]. Expression of CLDN18.2 is extremely heterogenous in GC, and 6 biopsies should be considered as a standard of sampling for an adequate immunohistochemical profiling [71]. Since CLDN18.2 is a trans-membranous protein, it represents an ideal target for monoclonal antibodies [68]. Zolbetuximab is a recently developed antibody that binds CLDN18.2 with consequent antibody-dependent and complement-dependent cell death [72]. Phase IIa MONO trial reports an ORR of 9% and a disease control rate of 23% using zolbetuximab as a single agent in patients with recurrent or refractory advanced GACs [73]. In phase II study FAST, the combination of zolbetuximab and EOX (epirubicin, oxaliplatin and capecitabine) as first-line chemotherapy significantly improves PFS (7.5 vs 5.3 months) and OS (13.0 vs 8.4 months) in patients with CLDN18.2 positive GACs [74]. Moreover, phase III studies NCT03504397 and NCT03653507 are currently evaluating the addition of zolbetuximab to first-line chemotherapy in GACs with high levels of CLDN18.2 expression [20].
Finally, the use of CLDN18.2-targeted chimeric antigen receptor (CAR) T against CLDN18.2 positive gastric PADX models has been investigated, with preliminary but encouraging results [20, 75].
PARP inhibitors
About 10% of GACs harbor mutations in genes involved in homologous DNA repair (e.g. BRCA1/BRCA2 and ATM) [8, 14, 76]. Poly (ADP-ribose) polymerase (PARP) inhibitors target a crucial component of the base excision repair pathway appear to be effective against tumors with homologous recombination repair deficiencies. For example, a maintenance regimen with PARP inhibitor olaparib improves PFS in patients with germline BRCA1/2-mutated pancreas carcinoma [77]. According to Bang and colleagues [78], a combination of olaparib and paclitaxel improved OS (13.1 vs 8.3 months) in patients with advanced GAC. OS benefit was greater in patients with tumor lacking ATM mutations, but the difference was not statistically significant. These results were not confirmed by the phase III GOLD trial in selected Asian patients with advanced GAC [79], but the study may have been limited by the very small number of ATM-low participants [80]. Despite the contrasting results, the use of PARP inhibitors in GAC is currently under study. For example, the NCT03427814 trial is now evaluating the efficacy of pamiparib-including maintenance therapy in patients with advanced-stage GAC, while trials NCT03008278, NCT04209686 and NCT02678182 are investigating the combination of PARP inhibitors with angiogenesis inhibitors and ICIs [20].
EGFR-targeted therapies
EGFR amplification and EGFR overexpression are reported in approximately 5–10% of GACs and are considered markers of poor prognosis [81, 82]. Anti-EGFR therapies have already been investigated in GAC, but with disheartening results. Phase III trials EXPAND [83] and REAL-3 [84] have failed to demonstrate significant survival benefits when adding an anti-EGFR antibody to first-line chemotherapy in unselected patients with metastatic GAC. On the other hand, it has been suggested that anti-EGFR therapies have significant activity against tumors with high levels of EGFR expression, supporting the need for a better characterization of the EGFR status for future clinical trials [20, 84, 85]. However, Smyth and colleagues [86] report a particularly poor prognosis for EGFR-amplified GACs treated with EGFR inhibitors in combination with chemotherapy, probably due to an antagonistic effect between anti-EGFR agents and the chemotherapeutic drug epirubicin. Other mechanisms of resistance to anti-EGFR therapies are co-amplification of HER2, NRAS, KRAS, MYC, or CCNE1, and mutation of KRAS or GNAS [84, 86].
FGFR-targeted therapies
FGFR2 amplifications occur in 5–10% of GACs and have been associated with poor prognosis and with CIN and GS molecular subtypes [20, 87, 88]. AZD4547 represents one of the first selective pan-FGFR TKIs developed. Despite encouraging preclinical results [89], phase II trial (SHINE) failed to demonstrate improved PFS with AZD4547 compared with paclitaxel in the second-line treatment of patients with metastatic GACs harboring FGFR2 polysomy or FISH-detected amplifications [90].
However, intratumor heterogeneity of FGFR2 amplification and poor concordance between FGFR2 amplification/polysomy and expression may hamper the case selection of clinical trials [90].
Others FGFR-targeted therapies are currently under investigation. The FGFR1-4 irreversible inhibitor futibatinib is being tested in a phase II trial involving patients with advanced-stage solid tumors harboring FGFR alterations (NCT02052778) [91]. Bemarituzumab is a monoclonal antibody against the FGFR2b splice variant that is frequently overexpressed in FGFR2-amplified GACs, and its use has been investigated with promising results [20]. Results from phase II FIGHT trials suggest that the addition of bemarituzumab to mFOLFOX6 led to clinically meaningful and statistically significant improvements in PFS (9.5 vs 7.4 months), OS (not reached vs 12.9 months) and ORR (47% vs 33%) in patients with previously untreated, FGFR2b-overexpressing advanced-stage GACs [92]. Phase III FIGHT trial is currently ongoing [93].
MET-targeted therapies
Alteration of the MET/ hepatocyte growth factor (HGF) pathway has been associated with more aggressive disease and poorer prognosis in different gastrointestinal neoplasms [94].
A combination of mFOLFOX6 plus HGF-signaling inhibitor onartuzumab has been investigated in a METGastric trial [95], without any significant improvement in OS, PFS or ORR. Moreover, the RILOMET-1 trial [96] failed to prove the significant clinical benefits of c-MET-signaling inhibitor rilotumumab in patients with MET-positive GACs. Despite these results, some studies suggest that MET TKIs have significant antitumor activity in patients with a high level of MET amplification [97, 98]. The potential efficacy of anti-MET therapies is probably undermined by the development of acquired resistance. According to Frigault and colleagues [99], MET D1228 V/N/H and Y1230C mutations or high copy number MET gene amplifications are responsible for the acquisition of resistance to savolitinib in patients with MET-amplified GAC.
Other possible therapeutic scenarios
Anti-angiogenic therapies have been investigated in GAC, with contrasting results.
The addition of anti-VEGFA antibody bevacizumab to chemotherapy as first-line treatment has not been associated with an improvement in OS [100, 101]. On the other hand, studies evaluating the effectiveness of ramucirumab in the second-line setting have provided some interesting results. In the phase III RAINBOW trial [102], a combination of ramucirumab with paclitaxel has been reported to increase OS (9.6 vs 7.4 months), when compared with placebo plus paclitaxel. Moreover, according to Fuchs and colleagues [103], ramucirumab monotherapy significantly improves OS (5.2 vs 3.8 months) in GAC patients. The VEGFR-2 TKI apatinib has been associated with increased overall and PFS compared to placebo in patients with advanced GAC refractory to at least two lines of chemotherapy [104]. However, these results have not been confirmed in the phase III ANGEL trial [105].
Regorafenib is a multi-kinase inhibitor that also targets angiogenic receptor tyrosine kinases and is currently used in colorectal cancer and GIST patients [106]. In the phase II INTEGRATE trial [107], regorafenib has been associated with longer PFS (2.6 vs 0.9 months) in previously treated GC compared to placebo. Phase III INTEGRATE trial (NCT02773524) is currently ongoing [20].
Overexpression of Matrix Metallo Proteinase-9 (MMP-9) in GAC has been associated with poor prognosis [108]. The addition of MMP-9 inhibitor andecaliximab to FOLFOX presented encouraging antitumoral activity against GAC in the phase I/Ib trial [109], but those results have not been confirmed in subsequent studies (GAMMA-1) [110].
Finally, a combination of pembrolizumab and a modulator of the Wnt and PI3KeAKT signaling pathways Dickkopf-1 (DKK1) has been tested in GC patients, with promising results [111].
Liquid biopsy
Intratumoral and intertumoral heterogeneity may fuel therapeutic resistance and treatment failure in cancer patients. Longitudinal profiling of serial blood samples may help to identify and characterize acquired drug resistance and track cancer evolution over time. In this context, the application of liquid biopsy in the clinic may be a powerful tool to guide therapeutic-decision progress in case of genomically discordant tissue samples or whether biopsy sampling cannot be performed. In a study by Pectasides and colleagues, genomic tissue profiling identified discordance of common alterations (HER2, MYC, CCND1, EGFR) between primary tumor and metastasis in 32% of patients. However, in most discordant cases, the analysis of circulating-free DNA (cfDNA) was concordant with the metastatic tissue, suggesting the potential for cfDNA profiling to more effectively guide the therapeutic choice [112]. In a cohort of 42 gastrointestinal cancer patients with acquired resistance to targeted therapy, post-progression cfDNA profiling was found to identify more accurately resistance alterations when compared to tissue analysis [113]. A recent study by Nakamura and colleagues [114] compared trial enrollment using circulating tumor DNA (ctDNA) sequencing to enrollment using tumor tissue sequencing in 1,687 patients with advanced gastrointestinal cancer. According to the results, ctDNA profiling significantly shortened the screening duration and improved the trial enrollment rate compared to tissue genotyping, thus representing a possible source of advance and innovation in the delivery of precision oncology.
In the metastatic setting, the levels of cfDNA and ctDNA positively correlate with tumor burden. For this reason, liquid biopsy has emerged to predict recurrence or relapse in gastric cancer following surgical resection [115].
Conclusions
GAC still remains an aggressive and deadly neoplasm. The great efforts made to develop novel effective molecular targeted therapies have not been followed by completely satisfying results. A lesson that can be learned from unsuccessful clinical trials is that adequate patients’ selection should always be a priority in both clinical studies and daily practice. Cornerstone of this process is a wise use of predictive biomarkers, which are necessary tools to identify those patients that will benefit from targeted therapy.
On the other hand, the use of biomarkers in clinics and in clinical research settings has several limitations. In fact, a single biomarker is not representative of the biologic complexity of the neoplasm and of the high level of genomic and molecular interpatient heterogeneity.
In the era of biomarker-driven oncology, the pathologist plays a central role in the process of therapeutic choice, which means that they must correctly perform the evaluation and avoid possible pitfalls. For this reason, in the context of gastroesophageal malignancies, biomarker assessment should always be performed by an expert and dedicated gastrointestinal pathologist.
The effectiveness of targeted therapies may be negatively affected by the high levels of spatial and temporal intratumor heterogeneity found in GAC. It is, therefore, crucial to perform adequate sampling that is representative of the entire neoplasm. However, heterogeneity is not a static concept since new molecular alterations may appear with time. Moreover, therapies can induce a selective pressure that gives proliferative advantages to resistant subclones, changing the proportion of different cellular populations that made up the primitive tumor. This implies that the initial biomarkers’ status may not be considered definitive, and re-assessment is highly recommended in recurrent and metastatic lesions. In this setting, a liquid biopsy may represent a powerful tool to guide the therapeutical decision process.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249
Anderson WF, Camargo MC, Fraumeni JF, Correa P, Rosenberg PS, Rabkin CS (2010) Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA 303(17):1723–1728
Gullo I, Grillo F, Mastracci L, Vanoli A, Carneiro F, Saragoni L et al (2020) Precancerous lesions of the stomach, gastric cancer and hereditary gastric cancer syndromes. Pathologica 112(3):166–185
Rubenstein JH, Taylor JB (2010) Meta-analysis: the association of oesophageal adenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment Pharmacol Ther 32(10):1222–1227
WHO Classification of Tumours Editorial Board (2019) Digestive system tumours. International Agency for Research on Cancer, Lyon
Lauren P (1965) The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. an attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 64:31–49
Businello G, Angerilli V, Parente P, Realdon S, Savarino E, Farinati F et al (2021) Molecular landscapes of gastric pre-neoplastic and pre-invasive lesions. Int J Mol Sci 22(18):9950
Network CGAR (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513(7517):202–209
Pellino A, Riello E, Nappo F, Brignola S, Murgioni S, Djaballah SA et al (2019) Targeted therapies in metastatic gastric cancer: current knowledge and future perspectives. World J Gastroenterol 25(38):5773–5788
Fassan M, Scarpa A, Remo A, De Maglio G, Troncone G, Marchetti A et al (2020) Current prognostic and predictive biomarkers for gastrointestinal tumors in clinical practice. Pathologica 112(3):248–259
Gylling A, Abdel-Rahman WM, Juhola M, Nuorva K, Hautala E, Järvinen HJ et al (2007) Is gastric cancer part of the tumour spectrum of hereditary non-polyposis colorectal cancer? A molecular genetic study. Gut 56(7):926–933
Velho S, Fernandes MS, Leite M, Figueiredo C, Seruca R (2014) Causes and consequences of microsatellite instability in gastric carcinogenesis. World J Gastroenterol 20(44):16433–16442
Ahn S, Lee SJ, Kim Y, Kim A, Shin N, Choi KU et al (2017) High-throughput protein and mRNA expression-based classification of gastric cancers can identify clinically distinct subtypes, concordant with recent molecular classifications. Am J Surg Pathol 41(1):106–115
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS et al (2015) Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 21(5):449–456
Businello G, Galuppini F, Fassan M (2021) The impact of recent next generation sequencing and the need for a new classification in gastric cancer. Best Pract Res Clin Gastroenterol 50–51:101730
Network CGAR, University AWGA, Agency BC, Hospital BaWs, Institute B, University B et al (2017) Integrated genomic characterization of oesophageal carcinoma. Nature 541(7636):169–175
Nakamura Y, Fujisawa T, Taniguchi H, Bando H, Okamoto W, Tsuchihara K et al (2021) SCRUM-Japan GI-SCREEN and MONSTAR-SCREEN: path to the realization of biomarker-guided precision oncology in advanced solid tumors. Cancer Sci 112(11):4425–4432
Ménard S, Pupa SM, Campiglio M, Tagliabue E (2003) Biologic and therapeutic role of HER2 in cancer. Oncogene 22(42):6570–6578
Van Cutsem E, Bang YJ, Feng-Yi F, Xu JM, Lee KW, Jiao SC et al (2015) HER2 screening data from ToGA: targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer 18(3):476–484
Nakamura Y, Kawazoe A, Lordick F, Janjigian YY, Shitara K (2021) Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: an emerging paradigm. Nat Rev Clin Oncol 18(8):473–487
Bartley AN, Washington MK, Colasacco C, Ventura CB, Ismaila N, Benson AB et al (2017) HER2 testing and clinical decision making in gastroesophageal adenocarcinoma: guideline from the college of American pathologists, American society for clinical pathology, and the american society of clinical oncology. J Clin Oncol 35(4):446–464
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A et al (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376(9742):687–697
Fassan M, Mastracci L, Grillo F, Zagonel V, Bruno S, Battaglia G et al (2012) Early HER2 dysregulation in gastric and oesophageal carcinogenesis. Histopathology 61(5):769–776
Yagi S, Wakatsuki T, Yamamoto N, Chin K, Takahari D, Ogura M et al (2019) Clinical significance of intratumoral HER2 heterogeneity on trastuzumab efficacy using endoscopic biopsy specimens in patients with advanced HER2 positive gastric cancer. Gastric Cancer 22(3):518–525
Wakatsuki T, Yamamoto N, Sano T, Chin K, Kawachi H, Takahari D et al (2018) Clinical impact of intratumoral HER2 heterogeneity on trastuzumab efficacy in patients with HER2-positive gastric cancer. J Gastroenterol 53(11):1186–1195
Kaito A, Kuwata T, Tokunaga M, Shitara K, Sato R, Akimoto T et al (2019) HER2 heterogeneity is a poor prognosticator for HER2-positive gastric cancer. World J Clin Cases 7(15):1964–1977
Gullo I, Grillo F, Molinaro L, Fassan M, De Silvestri A, Tinelli C et al (2015) Minimum biopsy set for HER2 evaluation in gastric and gastro-esophageal junction cancer. Endosc Int Open 3(2):E165–E170
Marx AH, Tharun L, Muth J, Dancau AM, Simon R, Yekebas E et al (2009) HER-2 amplification is highly homogenous in gastric cancer. Hum Pathol 40(6):769–777
Kim MA, Lee HJ, Yang HK, Bang YJ, Kim WH (2011) Heterogeneous amplification of ERBB2 in primary lesions is responsible for the discordant ERBB2 status of primary and metastatic lesions in gastric carcinoma. Histopathology 59(5):822–831
Bozzetti C, Negri FV, Lagrasta CA, Crafa P, Bassano C, Tamagnini I et al (2011) Comparison of HER2 status in primary and paired metastatic sites of gastric carcinoma. Br J Cancer 104(9):1372–1376
Fassan M, Ludwig K, Pizzi M, Castoro C, Guzzardo V, Balistreri M et al (2012) Human epithelial growth factor receptor 2 (HER2) status in primary and metastatic esophagogastric junction adenocarcinomas. Hum Pathol 43(8):1206–1212
Fusco N, Rocco EG, Del Conte C, Pellegrini C, Bulfamante G, Di Nuovo F et al (2013) HER2 in gastric cancer: a digital image analysis in pre-neoplastic, primary and metastatic lesions. Mod Pathol 26(6):816–824
Cho EY, Park K, Do I, Cho J, Kim J, Lee J et al (2013) Heterogeneity of ERBB2 in gastric carcinomas: a study of tissue microarray and matched primary and metastatic carcinomas. Mod Pathol 26(5):677–684
Cappellesso R, Fassan M, Hanspeter E, Bornschein J, d’Amore ES, Cuorvo LV et al (2015) HER2 status in gastroesophageal cancer: a tissue microarray study of 1040 cases. Hum Pathol 46(5):665–672
Park SR, Park YS, Ryu MH, Ryoo BY, Woo CG, Jung HY et al (2016) Extra-gain of HER2-positive cases through HER2 reassessment in primary and metastatic sites in advanced gastric cancer with initially HER2-negative primary tumours: results of GASTric cancer HER2 reassessment study 1 (GASTHER1). Eur J Cancer 53:42–50
Pietrantonio F, Fucà G, Morano F, Gloghini A, Corso S, Aprile G et al (2018) Biomarkers of primary resistance to trastuzumab in HER2-positive metastatic gastric cancer patients: the AMNESIA case-control study. Clin Cancer Res 24(5):1082–1089
Janjigian YY, Sanchez-Vega F, Jonsson P, Chatila WK, Hechtman JF, Ku GY et al (2018) Genetic predictors of response to systemic therapy in esophagogastric cancer. Cancer Discov 8(1):49–58
Pietrantonio F, Caporale M, Morano F, Scartozzi M, Gloghini A, De Vita F et al (2016) HER2 loss in HER2-positive gastric or gastroesophageal cancer after trastuzumab therapy: implication for further clinical research. Int J Cancer 139(12):2859–2864
Seo S, Ryu MH, Park YS, Ahn JY, Park Y, Park SR et al (2019) Loss of HER2 positivity after anti-HER2 chemotherapy in HER2-positive gastric cancer patients: results of the GASTric cancer HER2 reassessment study 3 (GASTHER3). Gastric Cancer 22(3):527–535
Kim ST, Banks KC, Pectasides E, Kim SY, Kim K, Lanman RB et al (2018) Impact of genomic alterations on lapatinib treatment outcome and cell-free genomic landscape during HER2 therapy in HER2+ gastric cancer patients. Ann Oncol 29(4):1037–1048
Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K et al (2017) Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol 18(11):1512–1522
Shitara K, Bang YJ, Iwasa S, Sugimoto N, Ryu MH, Sakai D et al (2020) Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N Engl J Med 382(25):2419–2430
Yamaguchi K, Bang Y, Iwasa S, Sugimoto N, Ryu M, Sakai D et al (2020) 1422MO-Trastuzumab deruxtecan (T-DXd; DS-8201) in patients with HER2-low, advanced gastric or gastroesophageal junction (GEJ) adenocarcinoma: results of the exploratory cohorts in the phase II, multicenter, open-label DESTINY-Gastric01 study. Ann Oncol 31:S899–S900
Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H et al (2011) Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci USA 108(17):7142–7147
Gall VA, Philips AV, Qiao N, Clise-Dwyer K, Perakis AA, Zhang M et al (2017) Trastuzumab increases HER2 uptake and cross-presentation by dendritic cells. Cancer Res 77(19):5374–5383
Iwata TN, Sugihara K, Wada T, Agatsuma T (2019) [Fam-] trastuzumab deruxtecan (DS-8201a)-induced antitumor immunity is facilitated by the anti-CTLA-4 antibody in a mouse model. PLoS ONE 14(10):e0222280
Janjigian YY, Kawazoe A, Yañez P, Li N, Lonardi S, Kolesnik O et al (2021) The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 600(7890):727–730
Catenacci DVT, Kang YK, Park H, Uronis HE, Lee KW, Ng MCH et al (2020) Margetuximab plus pembrolizumab in patients with previously treated, HER2-positive gastro-oesophageal adenocarcinoma (CP-MGAH22-05): a single-arm, phase 1b–2 trial. Lancet Oncol 21(8):1066–1076
Catenacci DV, Rosales M, Chung HC, Yoon H H, Shen L, Moehler M et al (2021) MAHOGANY: margetuximab combination in HER2+ unresectable/metastatic gastric/gastroesophageal junction adenocarcinoma. Future Oncol 17(10):1155–1164
Kotani D, Shitara K (2021) Trastuzumab deruxtecan for the treatment of patients with HER2-positive gastric cancer. Ther Adv Med Oncol 13:1758835920986518
Kulukian A, Lee P, Taylor J, Rosler R, de Vries P, Watson D et al (2020) Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Mol Cancer Ther 19(4):976–987
Smyth EC, Wotherspoon A, Peckitt C, Gonzalez D, Hulkki-Wilson S, Eltahir Z et al (2017) Mismatch repair deficiency, microsatellite instability, and survival: an exploratory analysis of the medical research council adjuvant gastric infusional chemotherapy (MAGIC) trial. JAMA Oncol 3(9):1197–1203
Attia H, Smyth E (2021) Evolving therapies in advanced oesophago-gastric cancers and the increasing role of immunotherapy. Expert Rev Anticancer Ther 21(5):535–546
Fassan M, Brignola S, Pennelli G, Alberti G, Angerilli V, Bressan A et al (2020) PD-L1 expression in gastroesophageal dysplastic lesions. Virchows Arch 477(1):151–156
Angerilli V, Galuppini F, Pennelli G, Fanelli GN, d’Amore ESG, Michelotto M et al (2021) Epstein-Barr virus associated gastric dysplasia: a new rare entity? Virchows Arch. https://doi.org/10.1007/s00428-021-03206-2
Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M et al (2018) Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol 4(5):e180013
Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K et al (2017) Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390(10111):2461–2471
Pietrantonio F, Randon G, Di Bartolomeo M, Luciani A, Chao J, Smyth EC et al (2021) Predictive role of microsatellite instability for of PD-1 blockade in patients with advanced gastric cancer: a meta-analysis of randomized clinical trials. ESMO Open 6(1):100036
Shitara K, Özgüroğlu M, Bang YJ, Di Bartolomeo M, Mandalà M, Ryu MH et al (2018) Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 392(10142):123–133
Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW et al (2020) Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: the KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol 6(10):1571–1580
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD et al (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372(26):2509–2520
Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K et al (2018) Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 24(9):1449–1458
Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K et al (2020) Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 21(10):1353–1365
Greally M, Chou JF, Chatila WK, Margolis M, Capanu M, Hechtman JF et al (2019) Clinical and molecular predictors of response to immune checkpoint inhibitors in patients with advanced esophagogastric cancer. Clin Cancer Res 25(20):6160–6169
Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M et al (2020) Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J Clin Oncol 38(18):2053–2061
Kawazoe A, Fukuoka S, Nakamura Y, Kuboki Y, Wakabayashi M, Nomura S et al (2020) Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): an open-label, single-arm, phase 2 trial. Lancet Oncol 21(8):1057–1065
Hara H, Shoji H, Takahari D, Esaki T, Machida N, Nagashima K et al (2019) Phase I/II study of ramucirumab plus nivolumab in patients in second-line treatment for advanced gastric adenocarcinoma (NivoRam study). J Clin Oncol 37(4_suppl):129
Sahin U, Schuler M, Richly H, Bauer S, Krilova A, Dechow T et al (2018) A phase I dose-escalation study of IMAB362 (Zolbetuximab) in patients with advanced gastric and gastro-oesophageal junction cancer. Eur J Cancer 100:17–26
Coati I, Lotz G, Fanelli GN, Brignola S, Lanza C, Cappellesso R et al (2019) Claudin-18 expression in oesophagogastric adenocarcinomas: a tissue microarray study of 523 molecularly profiled cases. Br J Cancer 121(3):257–263
Nakayama I, Shinozaki E, Sakata S, Yamamoto N, Fujisaki J, Muramatsu Y et al (2019) Enrichment of CLDN18-ARHGAP fusion gene in gastric cancers in young adults. Cancer Sci 110(4):1352–1363
Pellino A, Brignola S, Riello E, Niero M, Murgioni S, Guido M et al (2021) Association of CLDN18 protein expression with clinicopathological features and prognosis in advanced gastric and gastroesophageal junction adenocarcinomas. J Pers Med. 11(11):1095
Singh P, Toom S, Huang Y (2017) Anti-claudin 18.2 antibody as new targeted therapy for advanced gastric cancer. J Hematol Oncol 10(1):105
Türeci O, Sahin U, Schulze-Bergkamen H, Zvirbule Z, Lordick F, Koeberle D et al (2019) A multicentre, phase IIa study of zolbetuximab as a single agent in patients with recurrent or refractory advanced adenocarcinoma of the stomach or lower oesophagus: the MONO study. Ann Oncol 30(9):1487–1495
Sahin U, Türeci Ö, Manikhas G, Lordick F, Rusyn A, Vynnychenko I et al (2021) FAST: a randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann Oncol 32(5):609–619
Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G et al (2019) Claudin18.2-specific chimeric antigen receptor engineered T cells for the treatment of gastric cancer. J Natl Cancer Inst 111(4):409–418
Alexandrov LB, Nik-Zainal S, Siu HC, Leung SY, Stratton MR (2015) A mutational signature in gastric cancer suggests therapeutic strategies. Nat Commun 6:8683
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ et al (2019) Maintenance olaparib for germline. N Engl J Med 381(4):317–327
Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH et al (2015) Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol 33(33):3858–3865
Bang YJ, Xu RH, Chin K, Lee KW, Park SH, Rha SY et al (2017) Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 18(12):1637–1651
Ku GY (2020) Next generation sequencing in gastric or gastroesophageal adenocarcinoma. Transl Gastroenterol Hepatol 5:56
Nagatsuma AK, Aizawa M, Kuwata T, Doi T, Ohtsu A, Fujii H et al (2015) Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer 18(2):227–238
Liao JB, Lee HP, Fu HT, Lee HS (2018) Assessment of EGFR and ERBB2 (HER2) in gastric and gastroesophageal carcinomas: EGFR amplification is associated with a worse prognosis in early stage and well to moderately differentiated carcinoma. Appl Immunohistochem Mol Morphol 26(6):374–382
Lordick F, Kang YK, Chung HC, Salman P, Oh SC, Bodoky G et al (2013) Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol 14(6):490–499
Maron SB, Alpert L, Kwak HA, Lomnicki S, Chase L, Xu D et al (2018) Targeted therapies for targeted populations: anti-EGFR treatment for EGFR-amplified gastroesophageal adenocarcinoma anti-EGFR therapy for EGFR-amplified gastroesophageal cancer. Cancer Discov 8(6):696–713
Petty RD, Dahle-Smith A, Stevenson DAJ, Osborne A, Massie D, Clark C et al (2017) Gefitinib and EGFR gene copy number aberrations in esophageal cancer. J Clin Oncol 35(20):2279–2287
Smyth EC, Vlachogiannis G, Hedayat S, Harbery A, Hulkki-Wilson S, Salati M et al (2020) EGFR amplification and outcome in a randomised phase III trial of chemotherapy alone or chemotherapy plus panitumumab for advanced gastro-oesophageal cancers. Gut. https://doi.org/10.1136/gutjnl-2020-322658
Su X, Zhan P, Gavine PR, Morgan S, Womack C, Ni X et al (2014) FGFR2 amplification has prognostic significance in gastric cancer: results from a large international multicentre study. Br J Cancer 110(4):967–975
Matsumoto K, Arao T, Hamaguchi T, Shimada Y, Kato K, Oda I et al (2012) FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer 106(4):727–732
Xie L, Su X, Zhang L, Yin X, Tang L, Zhang X et al (2013) FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin Cancer Res 19(9):2572–2583
Van Cutsem E, Bang YJ, Mansoor W, Petty RD, Chao Y, Cunningham D et al (2017) A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol 28(6):1316–1324
Hollebecque A, Doi T, Saavedra O, Takahashi O, He H, Benhadji KA et al (2020) A phase II study of futibatinib (TAS-120) in patients (pts) with advanced (adv) solid tumors harboring fibroblast growth factor receptor (FGFR) genomic aberrations. J Clin Oncol 38(4_suppl):TPS470
Wainberg ZA, Enzinger PC, Kang YK, Yamaguchi K, Qin S, Lee KW et al (2021) Randomized double-blind placebo-controlled phase 2 study of bemarituzumab combined with modified FOLFOX6 (mFOLFOX6) in first-line (1L) treatment of advanced gastric/gastroesophageal junction adenocarcinoma (FIGHT). J Clin Oncol 39(3):160
Catenacci DV, Tesfaye A, Tejani M, Cheung E, Eisenberg P, Scott AJ et al (2019) Bemarituzumab with modified FOLFOX6 for advanced FGFR2-positive gastroesophageal cancer: FIGHT Phase III study design. Future Oncol 15(18):2073–2082
Oga T, Yamashita Y, Soda M, Kojima S, Ueno T, Kawazu M et al (2019) Genomic profiles of colorectal carcinoma with liver metastases and newly identified fusion genes. Cancer Sci 110(9):2973–2981
Shah MA, Bang YJ, Lordick F, Alsina M, Chen M, Hack SP et al (2017) Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: the METGastric randomized clinical trial. JAMA Oncol 3(5):620–627
Catenacci DVT, Tebbutt NC, Davidenko I, Murad AM, Al-Batran SE, Ilson DH et al (2017) Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 18(11):1467–1482
Shitara K, Kim TM, Yokota T, Goto M, Satoh T, Ahn JH et al (2017) Phase I dose-escalation study of the c-Met tyrosine kinase inhibitor SAR125844 in Asian patients with advanced solid tumors, including patients with. Oncotarget 8(45):79546–79555
Kawazoe A, Shitara K (2019) Next-generation sequencing and biomarkers for gastric cancer: what is the future? Ther Adv Med Oncol 11:1758835919848189
Frigault MM, Markovets A, Nuttall B, Kim KM, Park SH, Gangolli EA et al (2020) Mechanisms of acquired resistance to savolitinib, a selective MET inhibitor in MET-amplified gastric cancer. JCO Precis Oncol. https://doi.org/10.1200/PO.19.00386
Ohtsu A, Shah MA, Van Cutsem E, Rha SY, Sawaki A, Park SR et al (2011) Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 29(30):3968–3976
Shen L, Li J, Xu J, Pan H, Dai G, Qin S et al (2015) Bevacizumab plus capecitabine and cisplatin in Chinese patients with inoperable locally advanced or metastatic gastric or gastroesophageal junction cancer: randomized, double-blind, phase III study (AVATAR study). Gastric Cancer 18(1):168–176
Wilke H, Muro K, Van Cutsem E, Oh SC, Bodoky G, Shimada Y et al (2014) Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 15(11):1224–1235
Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C et al (2014) Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 383(9911):31–39
Li J, Qin S, Xu J, Xiong J, Wu C, Bai Y et al (2016) Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol 34(13):1448–1454
Kang YK, Kang WK, Di Bartolomeo M, Chau I, Yoon HH, Cascinu S et al (2019) LBA43 - Randomized phase III ANGEL study of rivoceranib (apatinib) + best supportive care (BSC) vs placebo + BSC in patients with advanced/metastatic gastric cancer who failed ≥2 prior chemotherapy regimens. Ann Oncol 30:v877–v878
Thangaraju P, Singh H, Chakrabarti A (2015) Regorafenib: a novel tyrosine kinase inhibitor: a brief review of its therapeutic potential in the treatment of metastatic colorectal carcinoma and advanced gastrointestinal stromal tumors. Indian J Cancer 52(3):257–260
Pavlakis N, Sjoquist KM, Martin AJ, Tsobanis E, Yip S, Kang YK et al (2016) Regorafenib for the treatment of advanced gastric cancer (INTEGRATE): a multinational placebo-controlled phase II trial. J Clin Oncol 34(23):2728–2735
Chen J, Chen LJ, Zhou HC, Yang RB, Lu Y, Xia YL et al (2014) Prognostic value of matrix metalloproteinase-9 in gastric cancer: a meta-analysis. Hepatogastroenterology 61(130):518–524
Shah MA, Starodub A, Sharma S, Berlin J, Patel M, Wainberg ZA et al (2018) Andecaliximab/GS-5745 alone and combined with mFOLFOX6 in advanced gastric and gastroesophageal junction adenocarcinoma: results from a phase I study. Clin Cancer Res 24(16):3829–3837
Shah MA, Yanez Ruiz EP, Bodoky G, Starodub A, Cunningham D, Yip D et al (2019) A phase III, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of andecaliximab combined with mFOLFOX6 as first-line treatment in patients with advanced gastric or gastroesophageal junction adenocarcinoma (GAMMA-1). J Clin Oncol 37(4_suppl):4
Klempner S, Bendell J, Villaflor VM, Tenner L, Stein S, Sirard C et al (2018) Safety and efficacy of a DKK1 inhibitor (DKN-01) in combination with pembrolizumab (P) in patients (Pts) with advanced gastroesophageal (GE) malignancies. Ann Oncol 29(8):vii222
Pectasides E, Stachler MD, Derks S, Liu Y, Maron S, Islam M et al (2018) Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov 8(1):37–48
Parikh AR, Leshchiner I, Elagina L, Goyal L, Levovitz C, Siravegna G et al (2019) Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 25(9):1415–1421
Nakamura Y, Taniguchi H, Ikeda M, Bando H, Kato K, Morizane C et al (2020) Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat Med 26(12):1859–1864
Pu WY, Zhang R, Xiao L, Wu YY, Gong W, Lv XD et al (2016) Prediction of cancer progression in a group of 73 gastric cancer patients by circulating cell-free DNA. BMC Cancer 16(1):943
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement. MF is supported by a grant from the Italian Health Ministry/Veneto region research program NET-2016–02363853 and AIRC 5 per mille 2019 (ID. 22759 program).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
SL reports personal fees (as speaker bureau or advisor) from Amgen, Merck Serono, Lilly, Astra Zeneca, Incyte, Daiichi-Sankyo, Bristol-Myers Squibb, Servier, MSD, Roche, Pierre-Fabre, GSK, and received research grants from Amgen, Merck Serono, Bayer, Roche, Lilly, Astra Zeneca, Bristol-Myers Squibb, unrelated to the current work. MF reports personal fees (as speaker bureau or advisor) from Roche, MSD, GSK, Astellas Pharma, Diaceutics and received research grants from Astellas Pharma, QED therapeutics, and macrophage pharma, unrelated to the current work. ES has served as a speaker for Abbvie, AGPharma, Alfasigma, EG Stada Group, Fresenius Kabi, Grifols, Janssen, Innovamedica, Malesci, Pfizer, Reckitt Benckiser, Sandoz, SILA, Sofar, Takeda, Unifarco, served as a consultant for Alfasigma, Amgen, Biogen, Bristol-Myers Squibb, Celltrion, Diadema Farmaceutici, Falk, Fresenius Kabi, Janssen, Merck & Co, Reckitt Benckiser, Regeneron, Sanofi, Shire, SILA, Sofar, Synformulas GmbH, Takeda, Unifarco and he received research support from Reckitt Benckiser, SILA, Sofar, Unifarco, unrelated to the current work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Businello, G., Angerilli, V., Lonardi, S. et al. Current molecular biomarkers evaluation in gastric/gastroesophageal junction adenocarcinoma: pathologist does matter. Updates Surg 75, 291–303 (2023). https://doi.org/10.1007/s13304-022-01330-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13304-022-01330-5