
Abstract
Introduction
In this phase 4, multicentre, prospective, non-interventional PIONEER REAL Netherlands study, we assessed clinical outcomes associated with once-daily oral semaglutide use in real-world clinical practice in adults living with type 2 diabetes (T2D) naïve to injectable glucose-lowering medication.
Methods
Participants initiated on oral semaglutide were followed for 34–44 weeks. Change in glycated haemoglobin (HbA1c) from baseline (BL) to end of study (EOS) was the primary endpoint; secondary endpoints included change in body weight (BW) from BL to EOS, the proportion of participants with HbA1c < 7.0% at EOS and the composite endpoints of HbA1c reduction ≥ 1.0%-points with BW reduction ≥ 3% or ≥ 5% at EOS. Treatment satisfaction was assessed using the Diabetes Treatment Satisfaction Questionnaire (DTSQ status/change). Safety was evaluated in all participants who initiated oral semaglutide treatment.
Results
Oral semaglutide was initiated in 187 participants; 94.1% completed the study and 78.6% remained on treatment at EOS. At BL, 54.0% of participants were male, mean age was 58.8 years, mean duration of T2D was 8.7 years and mean body mass index was 35.1 kg/m2; mean HbA1c was 8.6% and mean BW was 103.1 kg. Significant improvements from BL to EOS were observed for HbA1c and BW (estimated change [95% confidence interval]: − 1.16%-points [− 1.48 to − 0.85]; p < 0.0001, and − 5.84 kg [− 6.88 to − 4.80]; p < 0.0001, respectively). At EOS, 47.5% of participants had an HbA1c level < 7.0%; 41.8% and 35.5% of participants achieved composite endpoints of HbA1c reduction ≥ 1.0%-points plus BW reduction ≥ 3% or ≥ 5%, respectively. DTSQ status and change scores improved by 2.1 (p = 0.0003) and 10.8 points (p < 0.0001), respectively. Oral semaglutide was easy or very easy to consume for 81.5% of participants. Adverse events were mostly mild/moderate, with gastrointestinal disorders being the most common.
Conclusion
In this real-world population, we reported clinically significant reductions in HbA1c and BW, improved treatment satisfaction and no new safety concerns.
A graphical abstract is available with this article.
Clinical Trial Registration
NCT04601740.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The large, phase 3 PIONEER clinical programme demonstrated clinical efficacy for oral semaglutide, reporting significant improvements in important therapeutic goals for people living with type 2 diabetes (T2D) (glycaemic control and body weight loss) versus placebo and most active comparators, with a safety profile consistent with the glucagon-like peptide-1 receptor agonist drug class. |
The PIONEER REAL programme, which included 13 non-interventional studies in different countries, was designed to investigate and provide insights into how oral semaglutide performs in routine clinical practice in a broad population of adults living with T2D, to support clinical efficacy findings and confirm tolerability of oral semaglutide in a real-world setting. |
The phase 4, multicentre, prospective, non-interventional PIONEER REAL Netherlands study is part of this programme and investigated once-daily oral semaglutide and associated clinical outcomes in routine clinical practice in adults living with T2D naïve to injectable glucose-lowering therapy in the Netherlands. |
What was learned from the study? |
Clinically significant improvements in glycaemic control, reductions in body weight and improvements in treatment satisfaction were observed in PIONEER REAL Netherlands, with no new safety signals; notably, more than one third of participants achieved a composite reduction in glycated haemoglobin ≥ 1.0%-points and body weight ≥ 5% after 38 weeks. |
Observations from PIONEER REAL Netherlands in routine clinical practice broadly reflect data reported in the PIONEER phase 3 clinical trials for oral semaglutide in terms of its efficacy and tolerability. |
Digital Features
This article is published with digital features, including a graphical abstract, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.25574685.
Introduction
It is well established that the global prevalence of diabetes continues to rise [1]. For people living with type 2 diabetes (T2D), early and effective glycaemic control after diagnosis is important to prevent or delay long-term diabetes-related complications [2]. More than 500 million people worldwide were estimated to be living with diabetes in 2021, and this is projected to reach approximately 700 million by 2045 [3]. Diabetes remains among the top 10 leading causes of death and disability globally, with T2D accounting for > 90% of cases [1]. In the Netherlands, an estimated 1.1 million people were living with diabetes in 2021 [4], the majority (> 90%) of whom were diagnosed with T2D [5]. Given its prevalence, T2D is associated with significant societal and economic burden [6], with an annual cost of €1.3 billion in 2019 reported in the Netherlands [4]. Therefore, it is imperative that we have effective therapies for the treatment and management of T2D.
The Netherlands Diabetes Federation advocates a person-centred approach to care for people living with T2D [7, 8]. Regarding pharmacotherapy, Dutch standards recommend a stepwise introduction of treatments based on glycated haemoglobin (HbA1c), starting with metformin then sequentially adding sulphonylureas, followed by the addition of insulin or glucagon-like peptide-1 receptor agonists (GLP-1RAs) or dipeptidyl peptidase-4 (DPP-4) inhibitors as required to achieve and maintain glycaemic targets [9]. Semaglutide is a GLP-1 analogue currently available as a once-weekly subcutaneous formulation (0.25, 0.5, 1.0 and 2.0 mg [it should be noted that the 2.0 mg dose is unavailable in the Netherlands]) [10] and as a once-daily oral formulation (3, 7 and 14 mg) as an adjunct to diet and exercise for improving glycaemic control in adults living with T2D [11]. Regulatory approval was based on the comprehensive phase 3 PIONEER clinical development programme, which demonstrated the efficacy of oral semaglutide versus placebo and most active comparators in achieving glycaemic control and weight loss, with a safety profile consistent with the GLP-1RA drug class [12,13,14,15,16,17]. Research has shown that, in addition to improving glycaemic control and weight loss, GLP-1RAs can impact other risk factors for cardiovascular (CV) complications, including lowering systolic blood pressure and modifying plasma lipids and markers of inflammation [18]. Both formulations of semaglutide have demonstrated non-inferiority to placebo in the PIONEER 6 and SUSTAIN 6 CV outcomes trials [19, 20]. Of note, 2023 European Society of Cardiology guidelines recommended GLP-1RAs and/or sodium-glucose cotransporter-2 (SGLT2) inhibitors for people with atherosclerotic CV disease or a high risk of CV disease, regardless of glycaemic control [21], heightening the interest in real-world evidence of early use of GLP-1RAs.
Even though GLP-1RAs are a well-established class of glucose-lowering agents for T2D, and initiation early in the disease trajectory is advocated, they are under-utilised in clinical practice [22,23,24,25]. Although randomised clinical trials (RCTs) are essential in gaining regulatory approval, data from non-interventional studies provide insights into how medication is used and performs in routine clinical practice.
The PIONEER REAL programme was undertaken to complement and contextualise the findings from the PIONEER phase 3 clinical trial programme and to assess the clinical outcomes associated with the use of once-daily oral semaglutide in routine clinical practice, how it is perceived and how it performs in the real world. PIONEER REAL includes 13 non-interventional studies, conducted in different countries across Europe, North America, the Middle East and East Asia, and is investigating the use of oral semaglutide in a real-world setting in a population of adults living with T2D naïve to injectable glucose-lowering medications.
To date, the PIONEER REAL studies in Canada, Switzerland, the Netherlands and Japan have been completed. Here, we report findings from the PIONEER REAL Netherlands study, which was designed to examine the clinical outcomes associated with the use of once-daily oral semaglutide in routine clinical practice in adults living with T2D, who had not previously been treated with injectable glucose-lowering medication, in the Netherlands (NCT04601740).
Methods
Study Design
This was a phase 4, 34- to 44-week, multicentre, prospective, non-interventional, single-arm study conducted in real-world settings at 27 sites in the Netherlands. As part of the PIONEER REAL study programme, it shares a similar study design, study procedure and study endpoints with the published PIONEER REAL Switzerland study [26]. PIONEER REAL Netherlands was initiated in November 2020 and completed in December 2022, and was impacted by the coronavirus disease 2019 (COVID-19) pandemic (Fig. S1 in the supplementary material), which meant that participants could have their end-of-study (EOS) visit beyond 44 weeks. Clinical data and patient-reported outcomes (PROs) were collected according to local clinical practice.
Study Participants
Participants aged ≥ 18 years, diagnosed with T2D, who were treatment-naïve to injectable glucose-lowering drug(s) except for short-term insulin treatment for acute illness lasting for a total of ≤ 14 days, and who had an available HbA1c value within 90 days prior to visit (V) 1 or taken at V1 in line with local clinical practice, were included. As this was a non-interventional study, the decision to initiate treatment with oral semaglutide was made by the participant/treating physician based on the local label before and independently from the decision to include the participant in this study. Exclusion criteria included previous participation in this study, treatment with an investigational drug ≤ 30 days prior to enrolment, mental incapacity, and unwillingness or language barriers preventing understanding or cooperation.
A full list of eligibility criteria is provided in Table S1 in the supplementary material.
Study Procedures and Visits
Participants received once-daily, commercially available oral semaglutide (treatment was not provided by the sponsor) according to local label, clinical practice and reimbursement status. No additional diagnostic or monitoring procedures were applied. Lifestyle interventions and prescription of other glucose-lowering treatments were at the discretion of the treating physician, who also determined the dosing schedule. Participants attended an informed consent, medical history and treatment initiation visit (V1, week 0), several intermediate visits depending on the local clinical practice (V2.X, weeks 1−33) and an EOS visit (V3, weeks 34−44) to assess efficacy and safety parameters, and PROs. The first visit within the window from weeks 34–44 was considered the EOS visit (Fig. S1 in the supplementary material). Due to the COVID-19 pandemic, EOS visits up to week 52 were allowed at the discretion of the treating physician.
Endpoints and Assessments
HbA1c and Weight Change
The primary endpoint was change in HbA1c (%-points) from baseline to EOS. Key secondary endpoints included relative change in body weight (%) and absolute change in body weight (kg) from baseline to EOS.
Other Key Secondary Endpoints
Other key secondary endpoints included the proportion of participants with an HbA1c level < 7.0% at EOS; the proportion of participants achieving both a reduction in HbA1c of ≥ 1.0%-points and body weight reduction of ≥ 5% from baseline to EOS, and the proportion of participants achieving both a reduction in HbA1c of ≥ 1.0%-points and body weight reduction of ≥ 3% from baseline to EOS.
Treatment Satisfaction
Treatment satisfaction was assessed using Diabetes Treatment Satisfaction Questionnaire (DTSQ) scores; DTSQ status (DTSQs) and DTSQ change (DTSQc) measured absolute and relative treatment satisfaction from baseline to EOS, respectively [27, 28]. DTSQs and DTSQc contained the same eight items to measure participant satisfaction with treatment. In DTSQs, participants scored each item on a Likert scale from 0 (very dissatisfied) to 6 (very satisfied), except for items 2 and 3 which were rated as 0 (never) to 6 (most of the time). All item scores, except 2 and 3, were added to give a total score (range 0–36). In DTSQc, participants rated their change in treatment satisfaction before and after treatment on a scale of − 3 (less satisfied now) to + 3 (more satisfied now), with 0 representing no change. The DTSQs was completed at both V1 and V3, whereas the DTSQc was completed at V3 only.
Exploratory Endpoints
As described previously [26], exploratory endpoints (reported at EOS) were: oral semaglutide dose, change from baseline in waist circumference, treatment with oral semaglutide, addition/removal or dose increase/reduction of a glucose-lowering medication, physician-reported clinical success (related to the reason to initiate oral semaglutide) and self-reported severe hypoglycaemia.
Statistical Analysis
As this study is part of the PIONEER REAL study programme, the statistical analysis has been previously described in detail [26]. In brief, to ensure that 145 participants had an available HbA1c measurement at EOS, 194 participants were needed to allow 90% and 99% probability of detecting changes in HbA1c of ≥ 0.46%-points and ≥ 1.0%-points from baseline, respectively. Detailed descriptions of the full analysis set (FAS), primary, secondary and sensitivity analyses have been published previously [26].
Statistical analyses were conducted using SAS v.9.4 (SAS Institute, NC, USA). Due to the COVID-19 pandemic, 25 participants were in the study for more than 44 weeks; therefore, an additional sensitivity analysis based on the FAS-EOS (all participants in the FAS who completed the EOS [V3] within the original window of 34–44 weeks) using a mixed model for repeated measurements (MMRM) was performed for the primary endpoint for the in-study observation period. Analyses were performed with crude and adjusted models.
For primary and secondary endpoints, estimated response and change in response were analysed using an adjusted MMRM with baseline HbA1c value, age, baseline body mass index (BMI), time and time-squared as covariates, and sex, oral glucose-lowering drugs at baseline, diabetes duration and site as fixed factors with random intercept and time (slope). Similar analyses were performed for the continuous secondary and exploratory endpoints, except for treatment satisfaction; frequency tables (percentages with numerator counts) were used to report categorical endpoints.
DTSQs and DTSQc endpoints were assessed at V3 and analysed using an analysis of covariance model, with change in DTSQs score from baseline to V3 and DTSQc score at V3 as dependent variables.
Descriptive summaries were used to document the extent and pattern of missing information on HbA1c. Baseline characteristics of participants with missing data were compared with participants who had no missing data. Missing data were not imputed.
Compliance with Ethics Guidelines
The study protocol was approved by appropriate health authorities according to local guidelines and by an institutional review board/independent ethics committee. The ethics committee was The Dutch Clinical Research Foundation for all study sites. The study was conducted in accordance with the Declaration of Helsinki of 1964 and its later amendments and International Council for Harmonisation Good Clinical Practice guidelines. Participants provided written informed consent prior to commencement of any study-related activity.
Results
Participant Disposition and Characteristics
Consent forms were completed by 197 participants, of whom 187 met the study eligibility criteria and were enrolled and initiated on treatment with oral semaglutide. Of the participants enrolled, 176 (94.1%) completed the study and 147 (78.6%) remained on treatment with oral semaglutide at EOS. Reasons for participants’ treatment discontinuation (n = 37 [19.8%]) included adverse events (n = 16 [8.6%]), change in treatment strategy (n = 3 [1.6%]), insufficient glycaemic control (n = 2 [1.1%]), impact of the COVID-19 pandemic (n = 1 [0.5%]) or other (n = 15 [8.0%]). A summary of the participant disposition is shown in Fig. S2 in the supplementary material.
Over half of participants were male (54.0%) and most participants were white (88.8%). Baseline characteristics are summarised in Table 1. In brief, at baseline, participants had a mean (standard deviation [SD]) age of 58.8 (11.36) years and mean duration of diabetes of 8.7 (5.92) years. Mean (SD) was 8.6 (1.28) % for HbA1c, 103.1 (19.51) kg for body weight, 118.1 (14.31) cm for waist circumference and 35.1 (5.90) kg/m2 for BMI at baseline. The proportion of participants with an HbA1c level < 7.0% at baseline was 8.0%. A total of 116 (62.0%) participants had CV-related medical history (Table 1). Almost half of enrolled participants had hypertension (n = 101 [54.0%]) and 44.4% (n = 83) had dyslipidaemia.
The mean number of anti-diabetes medications used by participants (excluding oral semaglutide) was 1.6 at baseline and 2.4 at EOS. The most frequently reported concomitant medications at baseline were metformin (81.3%) and sulphonylureas (62.6%) (Table 1). A total of 10.2% of participants were not receiving any concomitant glucose-lowering medications at baseline.
Approximately one third (34.2%) of participants were treated by diabetes specialists (endocrinologists), with 65.8% treated by non-specialists in primary care settings. The most frequent reasons physicians gave for initiating oral semaglutide were to improve glycaemic control (97.9%) and aid weight management (90.9%), followed by reasons to address CV risk factors (13.9%), overcome issues with hypoglycaemia on current treatment (8.6%) and to simplify the current treatment regimen (2.7%). Most participants were initiated on 3 mg oral semaglutide (98.4%) with just three (1.6%) participants starting on the 7 mg dose. Median (interquartile range) oral semaglutide exposure time was 37.7 (32.3–41.4) weeks.
Glycaemic Parameters and Body Weight
Of those enrolled, 168/187 (89.8%) participants had complete covariate and dependent variable information to be included in the primary analysis of the primary endpoint. There was a significant reduction in the primary endpoint of HbA1c from the observed baseline value of 8.5% to an estimated mean of 7.3% at EOS (in participants who had at least one post-baseline HbA1c value, n = 168), with an estimated change (95% confidence interval [CI]) of − 1.16%-points (− 1.48 to − 0.85; p < 0.0001; − 12.72 mmol/mol [− 16.13 to − 9.31]; p < 0.0001; Fig. 1). Secondary and sensitivity analyses of change in HbA1c were consistent with the primary analysis (Fig. S3 in the supplementary material). A post hoc analysis of change in HbA1c from baseline to EOS stratified by baseline HbA1c (≥ 7% to < 8%; ≥ 8% to < 9%; and ≥ 9%) showed significant HbA1c reductions (p < 0.0001) in each subgroup. Although higher reductions in HbA1c were seen in participants with a higher baseline HbA1c, the differences between subgroups were not significant (Tables S3 and S4 in the supplementary material).

Estimated change in HbA1c from baseline to EOS (N = 168). Data are from the in-study observation period. At week 0, observed mean HbA1c at baseline for participants with at least one post-baseline assessment is plotted. Estimated HbA1c change is analysed using an adjusted model, with baseline HbA1c, age, baseline BMI, time and time-squared as covariates, and sex, glucose-lowering agents at baseline, diabetes duration and site as fixed factors with random intercept and time (slope). The outer lines of the band represent 95% CI. BMI body mass index, CI confidence interval, EOS end of study, HbA1c glycated haemoglobin
A total of 165/187 (88.2%) participants who had a body weight measurement at baseline and post-baseline were included in the analysis of change in body weight. Body weight significantly decreased from baseline to EOS, as shown by estimated change using the adjusted MMRM (95% CI) absolute and relative changes of − 5.84 kg (− 6.88 to − 4.80; p < 0.0001) and − 5.65% (− 6.66 to − 4.64; p < 0.0001), respectively (Fig. 2). In the additional analyses performed for the FAS in-study and on-treatment observation periods for the participants who had the EOS visit within the original visit window (weeks 34–44), changes in body weight were found to be similar across all sensitivity analyses and were statistically significant (p < 0.0001).

Estimated change in body weight from baseline to EOS (N = 165). Data are from the in-study observation period. At week 0, observed mean body weight at baseline for participants with at least one post-baseline assessment is plotted. Body weight change is analysed using an adjusted model, with baseline body weight, age, baseline BMI, time and time-squared as covariates, and sex, glucose-lowering agents at baseline, diabetes duration and site as fixed factors with random intercept and time (slope). The outer lines of the band represent 95% CI. BMI body mass index, CI confidence interval, EOS end of study
Post hoc analyses were performed to investigate if there were differences in change in HbA1c and body weight according to whether participants were treated by diabetes specialists or non-diabetes specialists. The results indicated that there were no significant differences in HbA1c and body weight outcomes between treating physicians. Significant reductions in HbA1c and in body weight were noted in both physician groups (data not shown).
Approximately half (47.5%) of participants had an HbA1c level < 7.0% at EOS, whereas 41.8% and 35.5% of participants achieved the composite endpoints of HbA1c reduction ≥ 1.0%-points plus body weight reduction ≥ 3% or ≥ 5%, respectively (Fig. 3).

Proportion of participants a achieving HbA1c targets and b with composite endpoints of HbA1c and body weight reductions at EOS. a n = 160, b n = 141. EOS end of study, HbA1c glycated haemoglobin
Treatment Satisfaction
Treatment satisfaction improved significantly with oral semaglutide, as increases were observed in both DTSQs (absolute treatment satisfaction) and DTSQc (relative treatment satisfaction) scores (Fig. 4). Of 157 participants who filled the ‘dosing conditions’ questionnaire, oral semaglutide was considered easy or very easy to consume by 41 participants (26.1%) and 87 participants (55.4%), respectively.

Participant-reported satisfaction: a absolute treatment satisfaction measured with DTSQs and b relative treatment satisfaction measured with DTSQc. Data are for the in-study observation period. For DTSQs, 0 = very dissatisfied and 36 = very satisfied. For DTSQc, − 18 = much less satisfied and + 18 = much more satisfied. CI confidence interval, DTSQc Diabetes Treatment Satisfaction Questionnaire change, DTSQs Diabetes Treatment Satisfaction Questionnaire status, EOS end of study, SD standard deviation. aObserved change was 10.78 (SD 6.15)
Exploratory Endpoints
In total, 147 (78.6%) participants were being treated with oral semaglutide at EOS; of these, 79 (53.7%) were receiving 14 mg, 62 (42.2%) were receiving 7 mg and 6 (4.1%) were receiving 3 mg. The mean (SD) dose at EOS was 10.6 (3.76) mg. Some participants required changes to their baseline concomitant glucose-lowering medication during the study period; 46 (24.6%) had additional or increased dose of glucose-lowering medications whereas 58 (31.0%) had either ceased their medication or decreased the dose. Concomitant medication use at EOS is summarised in Table S2 in the supplementary material. Treating physicians considered treatment with oral semaglutide a clinical success for 139 (81.3%) participants. There were no self-reported issues with hypoglycaemia in 96.2% of participants. Estimated mean change from baseline in waist circumference was − 4.78 cm, 95% CI − 7.35 to − 2.21; p < 0.001.
Safety
Overall, 246 adverse events were reported in 109 (58.3%) participants; most were mild or moderate in severity (Table 2). Ten serious adverse events were reported in nine (4.8%) participants. Two of the serious adverse events (one incidence of cholecystitis and one anal abscess) were considered likely related to oral semaglutide by the treating physician; however, both adverse events were resolved. Consistent with the safety profile of the GLP-1RA class, gastrointestinal disorders were the most frequently reported adverse events, observed in 84 (44.9%) participants. Adverse events resulted in oral semaglutide being withdrawn in 19 (10.2%) participants, most commonly due to nausea, diarrhoea and vomiting. Oral semaglutide was interrupted in 19 (10.2%) participants; the dose was reduced in 6 (3.2%) participants due to adverse events. Seven (3.8%) participants reported severe hypoglycaemic episodes between baseline and EOS, all of whom were also taking sulphonylureas, a common side-effect of which is hypoglycaemia; however, no data relating to potential causality of the severe hypoglycaemic episodes were captured. One death occurred during the study (a 72-year-old male participant died of cardiac disorder); however, the death was deemed unlikely to be related to oral semaglutide.
Discussion
This prospective, observational, real-world study conducted in the Netherlands forms part of the wider PIONEER REAL programme undertaken to provide insights on how oral semaglutide is prescribed, used and perceived in everyday clinical practice in an adult population of participants living with T2D. The results of PIONEER REAL Netherlands further support the use of oral semaglutide for glycaemic control and body weight reduction in a real-world population, with no new safety signals, and revealed an improvement in treatment satisfaction. The oral formulation provides a useful treatment option for patients with a preference for oral therapy [22], which, when coupled with the improvement in treatment satisfaction, may encourage a wider uptake of GLP-1RAs.
Over the course of the study, clinically significant improvements in the key therapeutic goals of glycaemic control and body weight loss were achieved, with more than one third of participants achieving a reduction in both HbA1c of ≥ 1.0%-points and body weight of ≥ 5%. In addition, study participants reported a greater degree of both absolute and relative treatment satisfaction at EOS than at baseline, as measured by responses to the DTSQs and DTSQc.
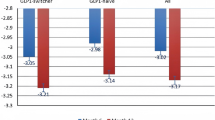
Participants enrolled in this real-world study were from both primary and secondary/tertiary care settings and their characteristics can be considered representative of the general population living with T2D. Observations for glycaemic control were consistent with those reported in RCTs for oral semaglutide. The decrease in HbA1c of 1.2%-points observed in this study (with a mean dose of 10.6 mg), reflects the reductions of between 1.2 and 1.3%-points with oral semaglutide 14 mg after 52 weeks in the PIONEER phase 3 programme (PIONEER 2, 3 [78 weeks], 4, 7 [flexibly dosed, including 3, 7 and 14 mg] and 8; based on the treatment policy estimand) [14,15,16,17, 29]. The HbA1c reduction observed in PIONEER REAL Netherlands is furthermore consistent with the reduction reported in the PIONEER REAL Switzerland study, which reported an HbA1c change of − 0.91%-points at EOS [26]. Differences in characteristics at baseline such as HbA1c levels (8.6% in this study, and 7.7% in PIONEER REAL Switzerland) and the mean number of concomitant glucose-lowering medications (1.6 in this study and 0.8 in PIONEER REAL Switzerland) may contribute to the slightly greater reduction in HbA1c observed in PIONEER REAL Netherlands than in PIONEER REAL Switzerland. Despite some differences in the populations, improvements in glycaemic control also closely reflect those reported with once-weekly subcutaneous semaglutide in the prospective 30-week real-world SURE Netherlands study, in which participants also had an HbA1c reduction of − 1.2%-points (95% CI − 1.3 to − 1.0; p < 0.0001) [30]. Reductions in HbA1c were marginally higher in this study than reported in the real-world US-based IGNITE study in which there was an HbA1c reduction of 0.9%-points (95% CI − 1.1 to − 0.6) after a mean treatment duration of 5.7 months in people prescribed oral semaglutide [31]. This may be due to differences in study population and design, such as the IGNITE study using retrospective data from electronic health records [31].
Only 8.0% of study participants had an HbA1c level < 7.0% at baseline; however, this increased to approximately half (47.5%) of participants at EOS. Although this observation is slightly lower than the range seen at 26 weeks in PIONEER trials (56–72%) [13,14,15], it is greater than reported in the SURE Netherlands study in which 36.9% of participants had an HbA1c level < 7.0% at EOS [30].
The reported body weight reduction was 5.84 kg (5.65%). This is a slightly higher weight reduction than has been previously reported in other PIONEER studies; however, it is still similar to the upper end of the range observed after 52 weeks in PIONEER 2, 4, 7 and 8 (approximately 2.9–5.0 kg for oral semaglutide 14 mg) [14,15,16,17]. The weight-loss effects of subcutaneous semaglutide at a dose of 2.4 mg have also been reported in studies in both people with overweight or obesity with or without T2D [32, 33]. There is also evidence of a reduction in major adverse CV events (MACE) with GLP-1RAs in people living with obesity and T2D; weekly GLP-1RAs (exenatide, dulaglutide and semaglutide) were reported to reduce the risk of MACE versus placebo [34]. The reduction in body weight, independent of HbA1c levels, may contribute to the CV benefits that have been associated with GLP-1RA treatment in people living with T2D [2, 35], alongside the impact of GLP-1RAs on other risk factors for CV disease such as anti-inflammatory effects, reduction in blood pressure and increased microvascular and coronary blood flow [18].
Physicians considered treatment to be a clinical success in > 80% of participants; however, clinical success is a subjective assessment and dependent on individual treatment targets for each participant. Observations from DTSQs and DTSQc assessments do, however, support physician sentiment, with satisfaction among participants significantly improved with oral semaglutide treatment.
Oral semaglutide in this real-world study was used in accordance with the label and local clinical practice in the Netherlands; most participants (98.4%) were initiated on the 3 mg dose, and three participants (1.6%) received the 7 mg dose at baseline. At EOS, only 53.7% of participants were receiving the maximum approved oral semaglutide dose of 14 mg, whereas 42.2% were treated with 7.0 mg and 4.1% with 3 mg.
Most participants reported concomitant glucose-lowering medication at baseline in this study, with only 19 (10.2%) participants reporting no medication at V1; given that duration of diabetes was reported as ≥ 1 year in over 90% of participants, high baseline treatment use was to be expected. Metformin and sulphonylureas were the most prescribed agents, with SGLT2 and DPP-4 inhibitors used in < 5% of participants. In a recent study of people with T2D in specialist care in Italy initiating oral semaglutide treatment, a similar proportion of people were naïve to glucose-lowering medication (9.6%) and 43.5% were taking two or more oral glucose-lowering treatments at the point of oral semaglutide initiation [36]. In the Italian cohort, at baseline, the most common glucose-lowering treatment was metformin (79.9%) [36], which was similar to PIONEER REAL Netherlands (81.3%). In contrast to PIONEER REAL Netherlands, the next two most common categories used by people in Italian clinical practice were DPP-4 inhibitors and SGLT2 inhibitors (24.9% and 20.9%, respectively) with sulphonylureas being used by 14.6% of people [36]. This may reflect differences in the prescription of glucose-lowering medication in a specialist versus non-specialist setting, with only 34.2% of participants in PIONEER REAL Netherlands treated by diabetes specialists. It could also be attributed to differences between countries with respect to treatment guidelines for people with T2D. In the Netherlands, stepwise treatment for T2D begins with biguanides such as metformin, followed by the addition of a sulphonylurea when biguanides do not adequately control hyperglycaemia, followed by the addition of basal insulin and eventually intensive pharmacotherapy with insulin [9]. Reimbursement policies may contribute to the pattern of therapies observed at baseline, with restrictions based on BMI and previous/ongoing therapies such as metformin, sulphonylureas and/or basal insulin [37]; at the time of the study, in the Netherlands, GLP-1RAs were restricted to reimbursement in people with a BMI ≥ 30 kg/m2 with inadequate glycaemic control on metformin and a sulphonylurea, and in people with a BMI ≥ 30 kg/m2 with inadequate glycaemic control on basal insulin for more than 3 months [37]. This pattern could also be influenced by limited familiarity of primary care physicians with GLP-1RAs, clinical inertia or physician preferences for established, generically available agents [22,23,24,25]. The results of this study may help overcome patient-held barriers to treatment and reimbursement restrictions in the Netherlands.
In PIONEER REAL Netherlands, adverse events with oral semaglutide were consistent with those observed in the PIONEER phase 3 RCTs. Reflective of the GLP-1RA class, gastrointestinal effects, including nausea, diarrhoea and vomiting, were the most common adverse events [12, 14,15,16,17, 29, 38]. The discontinuation rate (19.8%) was higher than reported for the PIONEER trials (7–13% with oral semaglutide 14 mg) [12, 14,15,16,17, 29, 38] and for the SURE Netherlands study for subcutaneous semaglutide (approximately 5%) [30]. It should be noted that this study focussed on participants naïve to treatment with injectable glucose-lowering therapy, with no individuals reporting prior GLP-1RA experience at baseline; the most common reason for discontinuation was a safety concern related to oral semaglutide. Notably, no new safety concerns were observed and the benefit–risk balance for oral semaglutide remains positive.
Participant enrolment in PIONEER REAL Netherlands was at the discretion of the treating physician. As such, the broad population included in the study may more closely reflect real-world practice, making it more generalisable than data from RCTs. It should be noted that there were slightly more male participants enrolled in the study, participants were predominantly white (reflective of the demographics of the study location) and a high proportion of participants had CV-related medical history. This study has limitations: it was an observational study that did not include a comparator arm, hence, alternative explanations for changes from baseline in HbA1c and other evaluated endpoints cannot be ruled out. In addition, data were collected as part of routine clinical practice, meaning results are less robust than those based on clinical trial data and potential confounding factors cannot be excluded. The observed changes in HbA1c may have been influenced by the clinical reason to initiate semaglutide treatment.
Observations from PIONEER REAL Netherlands add to those previously reported in the SURE Netherlands study and highlight that both formulations of semaglutide (oral and subcutaneous) can provide clinical improvements in the key management goals (glycaemic control and weight loss) for individuals living with T2D [30]. Availability of an oral formulation of semaglutide offers people living with T2D an alternative to injectable GLP-1RA therapy and could potentially improve acceptance and adherence in addition to facilitating earlier use of GLP-1RAs, even in a primary care setting [39].
Conclusion
The results of the PIONEER REAL Netherlands study show significant reductions in HbA1c and body weight after initiating oral semaglutide, as well as improvements in treatment satisfaction. These results complement data from the PIONEER trials and offer insights into how oral semaglutide performs in routine clinical practice in a broad, real-world population of adults living with T2D.
Data Availability
The data sets generated and/or analysed during the current study are available from Novo Nordisk on reasonable request. Data will be shared with bona fide researchers submitting a research proposal approved by the independent review board. Access request proposals can be found at http://novonordisk-trials.com. Data will be made available after research completion and approval of the product and product use in the European Union and the United States. Individual participant data will be shared in data sets in a de-identified/anonymised format.
References
GBD 2021 Diabetes Collaborators. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet. 2023;402:203–34.
ElSayed NA, Aleppo G, Aroda VR, et al. 6. Glycemic targets: standards of care in diabetes-2023. Diabetes Care. 2023;46(Suppl 1):S97–110.
Sun H, Saeedi P, Karuranga S, et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119.
VZInfo. Diabetes mellitus. https://www.vzinfo.nl/diabetes-mellitus. Accessed 4 Dec 2023.
VZinfo. Diabetes mellitus - age and gender [Leeftijd en geslacht]. https://www.vzinfo.nl/diabetes-mellitus/leeftijd-en-geslacht. Accessed 4 Dec 2023.
Peters ML, Huisman EL, Schoonen M, Wolffenbuttel BHR. The current total economic burden of diabetes mellitus in the Netherlands. Neth J Med. 2017;75:281–97.
Netherlands Diabetes Federation. NDF Toolkit - person-centred diabetes care and prevention [NDF Toolkit Persoonsgerichte diabeteszorg en preventie]. https://diabetesfederatie.nl/ndf-kwaliteitsagenda/ndf-persoonsgerichte-diabeteszorg. Accessed 4 Dec 2023.
Rutten GEHM, van Vugt HA, de Weerdt I, de Koning E. Implementation of a structured diabetes consultation model to facilitate a person-centered approach: results from a nationwide Dutch study. Diabetes Care. 2018;41:688–95.
van den Heuvel JM, Farzan N, van Hoek M, Maitland-van der Zee AH, Ahmadizar F. Mining treatment patterns of glucose-lowering medications for type 2 diabetes in the Netherlands. BMJ Open Diabetes Res Care. 2020;8:e000767.
European Medicines Agency. Summary of product characteristics: Ozempic. https://www.ema.europa.eu/en/documents/product-information/ozempic-epar-product-information_en.pdf. Accessed 4 Dec 2023.
European Medicines Agency. Summary of product characteristics: Rybelsus. https://www.ema.europa.eu/en/documents/product-information/rybelsus-epar-product-information_en.pdf. Accessed 4 Dec 2023.
Aroda VR, Rosenstock J, Terauchi Y, et al. PIONEER 1: randomized clinical trial of the efficacy and safety of oral semaglutide monotherapy in comparison with placebo in patients with type 2 diabetes. Diabetes Care. 2019;42:1724–32.
Mosenzon O, Blicher TM, Rosenlund S, et al. Efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment (PIONEER 5): a placebo-controlled, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7:515–27.
Pieber TR, Bode B, Mertens A, et al. Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7:528–39.
Pratley R, Amod A, Hoff ST, et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394:39–50.
Rodbard HW, Rosenstock J, Canani LH, et al. Oral semaglutide versus empagliflozin in patients with type 2 diabetes uncontrolled on metformin: the PIONEER 2 trial. Diabetes Care. 2019;42:2272–81.
Zinman B, Aroda VR, Buse JB, et al. Efficacy, safety, and tolerability of oral semaglutide versus placebo added to insulin with or without metformin in patients with type 2 diabetes: the PIONEER 8 trial. Diabetes Care. 2019;42:2262–71.
Ussher JR, Drucker DJ. Glucagon-like peptide 1 receptor agonists: cardiovascular benefits and mechanisms of action. Nat Rev Cardiol. 2023;20:463–74.
Husain M, Birkenfeld AL, Donsmark M, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2019;381:841–51.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–44.
Marx N, Federici M, Schütt K, et al. 2023 ESC guidelines for the management of cardiovascular disease in patients with diabetes. Eur Heart J. 2023;44:4043–140.
Gallwitz B, Giorgino F. Clinical perspectives on the use of subcutaneous and oral formulations of semaglutide. Front Endocrinol (Lausanne). 2021;12:645507.
Heintjes EM, Houben E, Beekman-Hendriks WL, et al. Trends in mortality, cardiovascular complications, and risk factors in type 2 diabetes. Neth J Med. 2019;77:317–29.
Honigberg MC, Chang LS, McGuire DK, Plutzky J, Aroda VR, Vaduganathan M. Use of glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes and cardiovascular disease: a review. JAMA Cardiol. 2020;5:1182–90.
Nelson AJ, O’Brien EC, Kaltenbach LA, et al. Use of lipid-, blood pressure-, and glucose-lowering pharmacotherapy in patients with type 2 diabetes and atherosclerotic cardiovascular disease. JAMA Netw Open. 2022;5:e2148030.
Kick A, M'Rabet-Bensalah K, Acquistapace F, et al. Real-world use of oral semaglutide in adults with type 2 diabetes: the PIONEER REAL Switzerland multicentre, prospective, observational study. Diabetes Ther. 2024;15:623–37.
Bradley C. Handbook of psychology and diabetes: a guide to psychological measurement in diabetes research and practice. Harwood Academic Publishers; 1994.
Bradley C. Diabetes treatment satisfaction questionnaire. Change version for use alongside status version provides appropriate solution where ceiling effects occur. Diabetes Care. 1999;22:530–2.
Rosenstock J, Allison D, Birkenfeld AL, et al. Effect of additional oral semaglutide vs sitagliptin on glycated hemoglobin in adults with type 2 diabetes uncontrolled with metformin alone or with sulfonylurea: the PIONEER 3 randomized clinical trial. JAMA. 2019;321:1466–80.
Wolffenbuttel BHR, Brugts MP, Catarig AM, et al. Once-weekly semaglutide use in type 2 diabetes: real-world data from the SURE Netherlands observational study. Adv Ther. 2023;40:920–33.
Aroda VR, Faurby M, Lophaven S, Noone J, Wolden ML, Lingvay I. Insights into the early use of oral semaglutide in routine clinical practice: the IGNITE study. Diabetes Obes Metab. 2021;23:2177–82.
Davies M, Færch L, Jeppesen OK, et al. Semaglutide 2·4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet. 2021;397:971–84.
Wilding JPH, Batterham RL, Calanna S, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384:989–1002.
Uneda K, Kawai Y, Yamada T, et al. Systematic review and meta-analysis for prevention of cardiovascular complications using GLP-1 receptor agonists and SGLT-2 inhibitors in obese diabetic patients. Sci Rep. 2021;11:10166.
Piccini S, Favacchio G, Panico C, et al. Time-dependent effect of GLP-1 receptor agonists on cardiovascular benefits: a real-world study. Cardiovasc Diabetol. 2023;22:69.
Morieri ML, Candido R, Frontoni S, et al. Clinical features, cardiovascular risk profile, and therapeutic trajectories of patients with type 2 diabetes candidate for oral semaglutide therapy in the Italian specialist care. Diabetes Ther. 2023;14:2159–72.
Malkin SJP, Hunt B, Huisman EL, Grand TS, Chubb B. The long-term cost-effectiveness of oral semaglutide in the Netherlands based on the PIONEER 2, 3 and 4 randomized controlled trials. Diabetes Res Clin Pract. 2021;175:108759.
Aroda VR, Erhan U, Jelnes P, et al. Safety and tolerability of semaglutide across the SUSTAIN and PIONEER phase IIIa clinical trial programmes. Diabetes Obes Metab. 2023;25:1385–97.
Evans M, Morgan AR, Bain SC, et al. Meeting the challenge of virtual diabetes care: a consensus viewpoint on the positioning and value of oral semaglutide in routine clinical practice. Diabetes Ther. 2022;13:225–40.
Acknowledgements
The authors thank the study participants, the investigators and study site staff who conducted the study. The authors also wish to thank Kabirdev Mandavya, Medical Advisor, Novo Nordisk Pharma AG for medical review of the manuscript.
Medical Writing/Editorial Assistance
Medical writing support was provided by Sophie Bruce and Sabah Farooq, of Apollo, OPEN Health Communications, and funded by Novo Nordisk, in accordance with Good Publication Practice (GPP) guidelines (www.ismpp.org/gpp-2022).
Funding
This study and the journal’s Rapid Service Fee was sponsored by Novo Nordisk A/S and is registered with ClinicalTrials.gov (NCT04601740).
Author information
Authors and Affiliations
Contributions
William van Houtum was responsible for data collection and was joint national lead for this PIONEER REAL Netherlands study. As such, he provided insights and information regarding the article, checked national data, and checked and corrected the article. Patrick Schrömbges was responsible for data collection and was joint national lead for this PIONEER REAL Netherlands study. As such, he provided insights and information regarding the article, checked national data, and checked and corrected the article. Hanan Amadid and Uffe C. Braae were responsible for study design and conduct. Arianne C. van Bon was a study site investigator and provided insights and information regarding the article, checked national data and checked and corrected the article. Charlotte Hoogstraten was responsible for study conduct. Hans Herrings was a study site investigator and provided insights and information regarding the article. Data were analysed by the sponsor. All authors had full access to all the data in the study, participated in interpretation of the data, actively contributed to and revised all drafts of the manuscript and made the decision to submit the manuscript for publication.
Corresponding author
Ethics declarations
Conflict of Interest
William van Houtum and Patrick Schrömbges are National Leaders of PIONEER REAL Netherlands. Hanan Amadid, Uffe C. Braae and Charlotte Hoogstraten are employees of, and shareholders in, Novo Nordisk A/S. Arianne C. van Bon has been a speaker at various training courses and conferences (sponsored and not sponsored) and a speaker for medical companies including Novo Nordisk. Hans Herrings was sponsored by Novo Nordisk to organise, collect data for and advise on, the PIONEER REAL Netherlands study.
Ethical Approval
The study protocol was approved by appropriate health authorities according to local guidelines and by an institutional review board/independent ethics committee. The ethics committee was The Dutch Clinical Research Foundation for all study sites. The study was conducted in accordance with the Declaration of Helsinki of 1964 and its later amendments and International Council for Harmonisation (ICH) Good Clinical Practice guidelines. Participants provided written informed consent prior to commencement of any study-related activity.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
van Houtum, W., Schrömbges, P., Amadid, H. et al. Real-World Use of Oral Semaglutide in Adults with Type 2 Diabetes in the PIONEER REAL Netherlands Multicentre, Prospective, Observational Study. Diabetes Ther (2024). https://doi.org/10.1007/s13300-024-01588-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13300-024-01588-5