
Abstract
Introduction
Untreated nonalcoholic fatty liver may progress to nonalcoholic steatohepatitis (NASH) and cirrhosis and induce hepatocellular carcinoma and liver failure. Type 2 diabetes mellitus (T2DM), often complicated with nonalcoholic fatty liver disease (NAFLD), is a driver of NAFLD progression. Thus, efficacious treatment strategies for patients with coexisting NAFLD and T2DM are important for preventing NAFLD progression. Although previous studies have demonstrated that either sodium-glucose transporter 2 inhibitors (SGLT2is) or glucagon-like peptide 1 receptor agonists (GLP-1 RAs) benefit NASH patients with T2DM, the rate of NASH resolution has not sufficiently improved. Therefore, we developed a protocol for a randomized controlled trial to examine whether the addition of an SGLT2i to the treatment regimen of patients receving a GLP-1 RA (combination therapy), within the therapeutic dose range for T2DM, increases the rate of NASH resolution in patients with coexisting NASH and T2DM.
Methods
This open-label, randomized, parallel-group study commenced in June 2021, will conclude recruitment in May 2023, and will end by March 2025. Sixty patients with NASH complicated by T2DM are enrolled at the Ehime University Hospital in Toon, Japan. Participants will be randomized into: (1) an intervention group receiving combination therapy with the SGLT2i luseogliflozin 2.5 mg, once daily (Taisho Pharmaceutical, Tokyo, Japan) and the GLP-1 RA semaglutide 0.5 mg, once per week (Novonordisk, Copenhagen, Denmark); and (2) a control group receiving monotherapy with the GLP-1 analog semaglutide. The primary endpoints, which will be ascertained by liver biopsy, are: (1) NASH resolution rate from baseline without worsening of liver fibrosis after 52 weeks of intervention; (2) rate of improvement from baseline of at least 1 point in the NAFLD activity score without worsening of liver fibrosis after 52 weeks of intervention; and (3) rate of improvement from baseline of at least one fibrosis stage without worsening of NASH after 52 weeks of intervention.
Trial Registration
University Hospital Medical Information Network Clinical Trial Registry (UMIN-CTR) number: UMIN000045003. Japan Registry of Clinical Trials registration number: jRCTs061210009.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
As type 2 diabetes mellitus (T2DM) not only often complicates nonalcoholic fatty liver disease (NAFLD) but also contributes to the progression of the pathogenesis of NAFLD, developing optimal treatment strategies for patients with NAFLD complicated by T2DM is important to prevent the progression of NAFLD. |
Several treatments for NAFLD have been suggested, but the rate of nonalcoholic steatohepatitis (NASH) resolution has not sufficiently improved. |
What will be learned from the study? |
This study may identify a new treatment option for NASH complicated by T2DM. |
Introduction
An unhealthy lifestyle can lead to various metabolic diseases, such as obesity, type 2 diabetes mellitus (T2DM), and dyslipidemia [1]. Metabolic diseases are risk factors for cardiovascular disease and cancer and can affect a healthy life expectancy and life prognosis [2,3,4]. Nonalcoholic fatty liver disease (NAFLD), a metabolic disease phenotype of the liver, is a global health problem [5,6,7,8]. An untreated nonalcoholic fatty liver may progress to nonalcoholic steatohepatitis (NASH) and cirrhosis and may induce hepatocellular carcinoma and liver failure [9,10,11]. Therefore, it is necessary to identify NAFLD and provide appropriate interventions for patients at high risk of progression to a serious disease.
T2DM is often complicated with NAFLD and is a driver of its progression [12,13,14,15]. Thus, treatment strategies for patients with NAFLD with concomitant T2DM are important to prevent the progression of NAFLD. Several treatments for NAFLD have been suggested [16,17,18,19,20,21,22,23,24,25,26,27,28]. Weight loss as a result of lifestyle improvements is an effective and ideal treatment for NAFLD [17, 18]. However, as many clinicians have observed, it is very difficult to convince patients to continue losing body weight, and it is important to supplement interventions in their lifestyles with pharmacotherapy [18]. A meta-analysis has shown that the use of the peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist pioglitazone improves histological findings, such as fibrosis, steatosis, ballooning degeneration of hepatocytes, and lobular inflammation [19]. Nonetheless, in terms of long-term prognosis, pioglitazone is a worrisome treatment option because it may put patients at risk of heart failure, which is associated with T2DM and NAFLD [29,30,31]. Recently, the efficacy of sodium-glucose cotransporter-2 inhibitors (SGLT2is) or glucagon-like peptide-1 receptor agonists (GLP-1 RAs) for the treatment of NASH has received much attention [20,21,22,23,24,25,26,27,28]. A randomized controlled trial (RCT) showed that both SGLT2i monotherapy for NAFLD, including NASH, and GLP-1 RA monotherapy for NASH improved histological findings [20,21,22]. Additionally, SGLT2is or GLP-1 RAs are effective against cardiovascular diseases, which are major complications of NASH and T2DM [32, 33]. However, the resolution rate of NASH following pharmacological intervention with these drugs is approximately 40–65% [20,21,22,23,24,25,26,27]. Therefore, improved treatment is needed to increase the resolution rate of NASH.
One of the most promising treatments for NASH, as indicated by a previous study, is the administration of an SGLT2i in addition to GLP-1 RA therapy [20,21,22,23,24,25,26,27,28]. To assess the efficacy of this treatment, we developed a protocol for a RCT to examine whether the addition of an SGLT2i to the therapeutic regimen of patients already receiving a GLP-1 RA, within the therapeutic dose range for T2DM, will increase the resolution rate of NASH in patients with NASH and T2DM.
Methods
Study Design
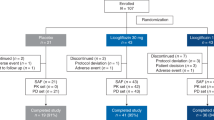
The trial, entitled “Luseogliflozin and semaglutide for patients with nonalcoholic steatohepatitis and T2DM: an open-label, randomized, parallel-group study,” commenced in June 2021 at the Ehime University Hospital and will conclude in March 2025. The schedule for this study is shown in Fig. 1. The enrollment of patients was initiated in June 2021 and will end in May 2023. The study will be explained to patients with NAFLD complicated by T2DM, and informed consent will be obtained before the screening tests are conducted. Patients who meet the eligibility criteria of the pre-screening test will be randomized into the intervention and control groups.

Flowchart of the study schedule
Compliance with Ethical Guidelines
This study and its protocols have been assessed and approved by the Certified Review Board of Ehime University (Toon, Ehime, Japan; E-mail: rinri@m.ehime-u.ac.jp), which had obtained certification from the Minister of Health, Labour and Welfare in Japan (approval ID number: jRCTs061210009; University Hospital Medical Information Network ID: UMIN000045003). The study will be conducted in accordance with the Declaration of Helsinki of 1964 and its later amendments, the Ethical Guidelines for Medical and Health Research Involving Human Patients issued by the Ministry of Health, Labour and Welfare in Japan, the Clinical Trials Act, and other current legal regulations in Japan. Informed consent will be obtained from all the study patients.
Sample Size Calculation
The efficacy of combination therapy with a SGLT2i and a GLP-1 RA for NASH is unknown. Therefore, we used the results of the therapeutic effects of SGLT2i and GLP-1 RA monotherapies for NASH and calculated the sample size. Based on the results reported in studies on SGLT2is [22,23,24,25,26,27], we estimated the efficacy of treatment with luseogliflozin 2.5 mg (SGLT2; i.e. the percentage of patients with NASH resolution at 24–52 weeks of treatment with the study drug) to be 37% and that of semaglutide 0.5 mg (GLP-1 RA) to be 30% [21]. Also, based on these results, we estimated that the efficacy of the respective therapy would be 67% in the intervention group (combination therapy) and 30% in the control group (semaglutide monotherapy). The sample size required to maintain 80% power in a two-tailed test (5% level of significance) will be 29 cases in the control group and 29 cases in the intervention group, with a total of 58 cases. The target number of patients will be set at 60 in total (30 in each group), with consideration given to the small percentage of patients who may discontinue or drop out of the study.
Eligibility Criteria
The inclusion criteria are: (1) patients aged 20–75 years at the time of obtaining consent to participate; (2) patients with T2DM who were diagnosed with NASH with stage ≥ 1 and NAFLD activity score (NAS) ≥ 4 according to the classification of the NASH Clinical Research Network (NASH CRN) by liver biopsy within 6 months (180 days) before obtaining consent or the preliminary examination [34]; (3) patients with glycated hemoglobin (HbA1c) ≥ 6.5% (HbA1c ≥ 6% for those undergoing drug treatment) and ≤ 10.5% in the preliminary examination; and (4) patients who provided written consent for their participation in this study.
The exclusion criteria are: (1) patients who received SGLT2is or GLP-1 RAs within 3 months (90 days) before starting the drug administration study or after liver biopsies; (2) patients who used other SGLT2is or GLP-1 RAs during the study period; (3) patients with histories of serious adverse reactions to SGLT2is or GLP-1 RAs; (4) patients with decompensated liver cirrhosis (Child–Pugh score: B or C); (5) patients with serious renal diseases (serum creatinine levels > 2.0 mg/dL or chronic kidney disease stage 4 or higher); (6) patients with malignancies; (7) patients with histories of severe hypoglycemia; (8) patients with histories of ketoacidosis; (9) patients with histories of cerebral infarctions with paralysis; (10) patients with urinary tract/genital infections or histories of recurring urinary tract/genital infections; (11) patients with the following complications: hepatitis due to other causes, such as viral hepatitis, autoimmune hepatitis, primary cholestatic cholangitis, psychiatric disorders, seizures, and paroxysmal diseases; (12) patients who were hospitalized for acute coronary syndromes, unstable anginas, acute myocardial infarctions, acute cerebral infarctions, or transient ischemic attacks within 3 months (90 days) of providing informed consent; (13) pregnant or lactating women, women who may become pregnant during the study period, or women who planned to become pregnant; (14) patients who had commenced or changed their doses of pioglitazone or vitamin E within 6 months (180 days) of starting the study drug or within 3 months (90 days) of liver biopsies and commencing the study drug; (15) patients who use oral steroids or injectable steroids regularly; (16) patients who are participating or intend to participate in other clinical studies using other study drugs while participating in this study; (17) decision by the principal investigator or sub-investigator that participation in the research is not in the best interest of the patient (e.g., to the detriment of the welfare of the patient), or that their inclusion is considered to interfere with, limit, or confuse the specific evaluation of the clinical research protocol; or (18) persons working under the direction of the principal investigator or the medical institution conducting the research, an employee of the principal investigator or of the medical institution directly involved in this or other clinical research, or family members of such employees or of the principal investigator.
Recruitment and Obtaining Consent
The principal investigator and sub-investigator will provide an explanatory document, which is approved by the Certified Review Board, Ehime University, to the patients and also provide sufficient written and oral explanations. Written, free, and voluntary consent will be obtained from the patients. We will perform the screening tests only for patients who provide informed consent, and we will enroll only those patients who meet the eligibility criteria at the Ehime University Hospital.
Registration of Patients and Random Allocations
After obtaining written consent from a candidate patient, the principal investigator or sub-investigator will conduct an examination and obtain information on the patient’s background, body composition, blood chemistry tests, urinalysis, FibroScan (Echosens, Paris, France), and liver biopsies to confirm that the patient meets the eligibility criteria and that none of the exclusion criteria apply.
Patients meeting the eligibility criteria will be assigned randomly in a 1:1 ratio to the control and intervention groups. A minimization method will be used for randomization, with liver fibrosis stage (1–3 vs. 4) and NAS (4–6 vs. 7–8) at screening being used as allocation factors. Allocation will be performed using the University Hospital Medical Information Network (UMIN) Internet Data and Information Center for Medical Research cloud version. The patient will be assigned an identification code that does not contain any information that can identify them.
Study Intervention
Intervention Group
Semaglutide (0.25 mg) will be injected subcutaneously once per week. After 2 weeks of treatment, luseogliflozin (2.5 mg) will additionally be administered once daily before or after breakfast. After 4 weeks of treatment, the dose of semaglutide will be increased from 0.25 to 0.5 mg (the maintenance dose for patients with T2DM) and injected subcutaneously once a week. If a scheduled semaglutide administration is missed and the time until the next scheduled administration is 2 days (48 h) or longer, semaglutide will be administered as soon as the missed dose of semaglutide is identified. Subsequently, it should be administered on a predetermined day of the week. However, if the time until the next scheduled administration of semaglutide is less than 2 days (48 h), the subsequent dose will be administered on the next predetermined day. If the patient forgets to take luseogliflozin as scheduled, the patient is instructed to report the missed dose to the investigator or sub-investigator as soon as they become aware of it. At this point, the patient will receive instructions on whether or not to take the medication. The medication will be administered for 52 weeks.
Control Group
Semaglutide (0.25 mg) will be injected subcutaneously once per week. After 4 weeks of treatment, the semaglutide dose will be increased from 0.25 to 0.5 mg and injected subcutaneously once a week. In cases where semaglutide administration is missed and the time until the next administration is 2 days (48 h) or longer, semaglutide will be administered as soon as it is observed that the dose of semaglutide has been missed. Subsequently, it should then be administered on a predetermined day of the week. However, if the time until the next administration of semaglutide is less than 2 days (48 h), the next dose should be administered on the next predetermined day. The medication will be administered for 52 weeks.
Dose, Dose Increase, and Dose Decrease Methods
If the principal investigator or sub-investigator considers that it is difficult to increase the dose of semaglutide due to the occurrence of gastrointestinal adverse events (AEs) or for other reasons at the fourth week of administration, the dose increase can be postponed. The dose increase will be attempted based on the patient’s symptoms and medical condition; however, if it is not tolerated, a dose of 0.25 mg will be continued. In the intervention group, the dose of luseogliflozin will be increased to 5 mg if the principal investigator or sub-investigator concludes that the 2.5-mg dose of luseogliflozin did not sufficiently improve the pathological condition by decreasing the levels of the liver enzymes aspartate aminotransferase (AST), alanine transaminase (ALT), and gamma-glutamyl transferase (GGT) to normal levels.
Concomitant Drugs and Therapy
The dosage of drugs for T2DM, other than luseogliflozin and semaglutide, can be increased or decreased. The dose of pioglitazone and vitamin E, however, is not changed.
Concomitant Therapy Restrictions
Concomitant use of SGLT2 inhibitors other than luseogliflozin and that of GLP-1 receptor agonists other than semaglutide is prohibited. Additionally, regular use of oral steroids or injectable steroids is prohibited.
Data Collection and Schedule
Data will be collected on the following items: patient background (sex, birthdate, age, height, body weight, body mass index [BMI], comorbidities, medical histories, current medical histories, and medication statuses); vital signs (blood pressure and pulse rates); body composition (body fat and skeletal muscle mass measured using InBody 720 [Biospace Corporation Limited, Seoul, Korea]); blood chemistry tests (white blood cell counts, red blood cell counts, hematocrits, platelet counts, prothrombin times, and fasting plasma glucose, HbA1c, total cholesterol, triglyceride, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, creatinine, blood urea nitrogen, sodium, potassium, chloride, uric acid, total bilirubin, direct bilirubin, AST, ALT, GGT, total protein, albumin, insulin, and C-peptide levels); urinalysis (urinary albumin, qualitative results [sugar, protein, occult blood, ketone, pH], sediment); liver stiffness and steatosis measured using FibroScan (Echosens); AEs from medical interviews; and histological diagnoses of liver specimens from liver biopsies. A liver biopsy will be obtained from the right liver lobe guided by laparoscopy or an abdominal ultrasound. Pathological specimens should be at least 1.5 cm in size if possible and stained with hematoxylin and eosin or silver impregnation. The pathological diagnosis will be made by two Japanese specialists (Masanori Abe and Yoshio Tokumoto) from the Society of Hepatology who are blinded to the patient background and blood test results (Table 1). During the study, examinations will be performed before the commencement of the intervention and every 13 weeks thereafter. The frequency of outpatient visits for the study is three (weeks 13, 26, and 39). Liver biopsies and FibroScans will be performed before the commencement of the intervention and at the end of the treatment period to confirm the histology (Table 1).
Endpoints
As this is an exploratory trial, we will set three primary endpoints: (1) resolution rate from the baseline of NASH (defined by the NASH CRN as no more than mild residual inflammatory cells [score of 0 or 1] and no hepatocyte ballooning [score of 0]) without the worsening of liver fibrosis, with worsening defined as an increase of one stage or more on the Kleiner fibrosis classification scale [34]) after 52 weeks of intervention; (2) improvement rates from baseline of at least 1 point in NAFLD activity score without any worsening of liver fibrosis after 52 weeks of intervention; and (3) improvement rates from baseline of at least one fibrosis stage without any worsening of NASH (with worsening defined as an increase of ≥ 1 point in either the lobular inflammation score or the hepatocyte ballooning score according to the NASH CRN criteria) after 52 weeks of intervention. The secondary endpoints are: (1) proportion of cases with change in liver fibrosis from baseline (improvement/instability/worsening) after 52 weeks of intervention; (2) change from baseline liver stiffness determined by FibroScan after 52 weeks of intervention; (3) change from the baseline steatosis determined by FibroScan after 52 weeks of intervention; (4) change from baseline body weight, BMI, and body composition after 52 weeks of intervention; (5) change from baseline HbA1c values after 52 weeks of intervention; and (6) change from baseline in liver enzymes (AST, ALT, and GGT) after 52 weeks of intervention. The safety endpoint is the frequency of AEs. The exploratory endpoints are: (1) change from baseline in the total NAFLD activity score, steatosis score, lobular inflammation score, hepatocyte ballooning score, and fibrosis stage and the rate of resolution of steatosis score, lobular inflammation score, and hepatocyte ballooning score after 52 weeks of intervention; (2) within-group comparison of the change from baseline in body weight, HbA1c, and liver enzymes (AST, ALT, and GGT) after 52 weeks of intervention; (3) within-group comparison of the change from baseline in liver stiffness according to FibroScan results after 52 weeks of intervention; (4) within-group comparison of the change from baseline in blood chemistry (white blood cell count, red blood cell count, hematocrit, platelet count, prothrombin time, fasting plasma glucose, total cholesterol, triglyceride, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, creatinine, blood urea nitrogen, sodium, potassium, chloride, uric acid, total bilirubin, direct bilirubin, total protein, albumin, insulin, and C-peptide levels) and urinalysis (urinary albumin) after 52 weeks of intervention.
Statistical Analyses
All statistical analyses will be performed by a biostatistician at an independent laboratory (Masahiro Yamada at Soiken Inc.).
Efficacy analyses of primary endpoints, secondary endpoints, and exploratory endpoints
The full analysis set (FAS) is the population of all study patients who are randomized, excluding the following: (1) patients who did not meet the eligibility criteria; (2) patients who had not received any experimental treatment after randomization; (3) patients with no measurements after randomization; (4) serious violations of the research protocol (failure to obtain consent, enrollment outside the contract period, etc.). Additionally, since we would like to assess the stability of the analysis results, efficacy analyses will also be conducted on each protocol set, which is the FAS excluding the following: (1) patients for whom the measurement of the primary endpoint was not available; (2) serious violations of the research protocol (e.g., wrong allocation, failure to meet the eligibility criteria, concomitant use of prohibited drugs, and non-compliance with medication). Regarding the primary endpoint, multiple imputation will be performed for missing data. For other endpoints, missing data will not be completed, but when possible, a sensitivity analysis, such as a mixed-effects model for repeated measures, will be conducted to evaluate the stability of the analysis results. The analysis of the safety endpoint will include patients who are enrolled in the study and receive at least one dose of the study drug.
At the primary endpoints, the rate of NASH resolution and the rate of improvement in NAFLD activity score and fibrosis of each group from baseline (pre-test) at 52 weeks of treatment will be calculated, and comparisons between groups will be performed using the Cochran–Mantel–Haenszel test, with adjustments for background factors, such as fibrosis stage (1–3, 4) and NAS (4–6, 7–8), at a 5% significance level (two-sided).
At the secondary endpoints, the percentage of cases with progression of liver fibrosis and changes from baseline in liver stiffness and steatosis, body weight, BMI, body composition, HbA1c, and liver enzymes after 52 weeks of intervention will be calculated for each group, and comparisons between groups will be performed using the Cochran–Mantel–Haenszel test for discrete values and analysis of covariance for the continuous value with adjustment of background factors, such as fibrosis stage (1–3, 4) and NAS (4–6, 7–8), at a 5% significance level (two-sided).
Regarding the characteristics of patients at baseline, the mean, standard deviation, and minimum, median, and maximum values will be calculated using the measured value data by group, and frequency tabulation will be performed for the total value data by group. For quantitative data, the unpaired t-test or Wilcoxon test will be used. The χ2 test will be used for discrete value analysis. A two-sided p value < 0.05 will be considered to be statistically significant.
At the safety endpoint, the number and incidence of AEs and diseases will be calculated during the treatment period, and the incidence rates will be compared.
At the exploratory endpoints, the change from baseline in the total NAFLD activity score, steatosis score, lobular inflammation score, hepatocyte ballooning score, fibrosis stage, and rate of improvement of steatosis score, lobular inflammation score, and hepatocyte ballooning score in each group from baseline (pre-test) at 52 weeks of treatment will be calculated, and comparisons between the groups will be performed using the analysis of covariance for continuous values and the Cochran–Mantel–Haenszel test for discrete values with adjustments of background factors, such as fibrosis stage (1–3, 4) and NAS (4–6, 7–8), at a 5% significance level (two-sided). For within-group comparisons, the changes from baseline with respect to body weight, liver stiffness and steatosis, blood chemistry, and urinalysis after 52 weeks of intervention in each group will be calculated, and the paired t-test will be used. A two-sided p value < 0.05 (two-sided) will be considered to be statistically significant.
Report of AEs
Any unfavorable medical event that occurs in a patient during this study, including exacerbation of an existing disease, will be treated as an AE during the study period. If the efficacy index worsens within the standard range, it will not be treated as an AE. When an AE occurs, the details of the AEs will be recorded in the case report form. If any AE is observed, it will be followed up until normalization or recovery to a level that cannot be regarded as an AE in principle or until the symptoms are stabilized or fixed in the case of irreversible AEs caused by organic disorders.
Stopping Guidelines
Criteria for the Discontinuation of the Study for Patients
The principal investigator or sub-investigator will stop administering the study drug to a patient and discontinue the patient’s participation in the research when they consider that it is impossible for the patient to continue in the study due to any of the following reasons: (1) the patient withdraws consent to participate in the research; (2) the patient does not meet the eligibility criteria after enrollment; (3) the administration of the study drug is undesirable for the patient due to worsening of the primary disease; (4) it is difficult for the patient to continue participating in the study due to exacerbation of complications; (4) it is difficult for the patient to continue participating in the study due to an AE; (5) the patient is pregnant; (6) adherence to the study protocol is poor (e.g., when it is considered that the patient is using < 75% of the total number of study drugs); (7) the entire study is discontinued; or (8) the physician considers that it is not appropriate for the patient to continue participating in the study for other reasons.
Criteria for Discontinuation of the Entire Study
When any of the following events occur, the principal investigator or sub-investigator will consider whether or not to continue the research: (1) significant information regarding the quality, safety, or efficacy of the study drug is obtained; (2) it is considered to be difficult to recruit the planned number of patients within the study period; (3) the Review Committee instructs changes to the research plan that are difficult to accept; (4) facts or information that undermine the ethical validity or scientific rationale of the research are obtained; (5) facts or information that undermine the appropriateness of conducting the research or the reliability of the research results are found; or (6) the review committee recommends or directs that the research is stopped.
Strengths and Limitations
Our study has several strengths. First, it is the first RCT to evaluate the efficacy of combination therapy with an SGLT-2i and a GLP-1 RA. Second, the patients enrolled in the study will be patients with T2DM. A previous study has shown that T2DM is frequently associated with NAFLD. NASH and advanced NASH are more common in patients with NAFLD complicated by T2DM than in those without T2DM [12,13,14,15]. Therefore, it is important to clarify the effects of treatment in patients with NASH complicated by T2DM. Finally, our study is the first trial to compare the effects of SGLT-2i and GLP-1 RA combination therapy with those of GLP-1 RA monotherapy for NASH within the therapeutic dose range for T2DM. T2DM promotes progression from NAFLD to NASH and worsens fibrosis [12,13,14,15]. In patients with undiagnosed NASH, who often visit T2DM outpatient clinics, resolving NASH along with achieving glycemic control may be important to reduce the number of future patients with advanced NASH. Therefore, it is important to demonstrate the efficacy of the drugs against NASH along with the treatment of T2DM. However, our study has a number of limitations. First, since our study is an exploratory study, we calculated group sample sizes according to the effects of SGLT-2i and GLP-1 RA monotherapy on NASH rather than the effect of combination therapy. Second, our study will be conducted at a single institution in Japan. Hence, the target population may be limited to a certain group. Therefore, in order to confirm the additional effect of luseogliflozin on semaglutide in patients with nonalcoholic steatohepatitis, it is necessary to conduct a multiethnic study involving more centers.
References
Alberti KG, Zimmet P, Shaw J, IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—a new worldwide definition. Lancet. 2005;366:1059–62. https://doi.org/10.1016/S0140-6736(05)67402-8.
Hess PL, Al-Khalidi HR, Friedman DJ, et al. The metabolic syndrome and risk of sudden cardiac death: The atherosclerosis risk in communities study. J Am Heart Assoc. 2017;6:e006103. https://doi.org/10.1161/JAHA.117.006103.
Steele CB, Thomas CC, Henley SJ, et al. Vital signs: Trends in incidence of cancers associated with overweight and obesity—United States, 2005–2014. MMWR Morb Mortal Wkly Rep. 2017;66:1052–8. https://doi.org/10.15585/mmwr.mm6639e1.
Jayedi A, Soltani S, Zargar MS, Khan TA, Shab-Bidar S. Central fatness and risk of all cause mortality: Systematic review and dose–response meta-analysis of 72 prospective cohort studies. BMJ. 2020;370:m3324. https://doi.org/10.1136/bmj.m3324.
Younossi ZM, Tampi RP, Racila A, et al. Economic and clinical burden of nonalcoholic steatohepatitis in patients with type 2 diabetes in the U.S. Diabetes Care. 2020;43:283–9. https://doi.org/10.2337/dc19-1113.
Marchesini G, Brizi M, Bianchi G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–50. https://doi.org/10.2337/diabetes.50.8.1844.
Simon TG, Roelstraete B, Hartjes K, et al. Non-alcoholic fatty liver disease in children and young adults is associated with increased long-term mortality. J Hepatol. 2021;S0168–8278:01883–93.
Terai S, Buchanan-Hughes A, Ng A, Lee IH, Hasegawa K. Comorbidities and healthcare costs and resource use of patients with nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) in the Japan medical data vision database. J Gastroenterol. 2021;56:274–84. https://doi.org/10.1007/s00535-021-01759-2.
Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–23. https://doi.org/10.1053/jhep.2003.50161.
Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384–91. https://doi.org/10.1016/j.jhep.2011.10.027.
Hashimoto E, Tokushige K. Prevalence, gender, ethnic variations, and prognosis of NASH. J Gastroenterol. 2011;46(Suppl 1):63–9. https://doi.org/10.1007/s00535-010-0311-8.
Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol. 2019;71:793–801. https://doi.org/10.1016/j.jhep.2019.06.021.
McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J Hepatol. 2015;62:1148–55. https://doi.org/10.1016/j.jhep.2014.11.034.
Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149:389-97.e10. https://doi.org/10.1053/j.gastro.2015.04.043.
Tilg H, Moschen AR, Roden MM. Nafld and diabetes mellitus. Nat Rev Gastroenterol Hepatol. 2017;14:32–42. https://doi.org/10.1038/nrgastro.2016.147.
Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. https://doi.org/10.1056/NEJMoa0907929.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149(367):e5. https://doi.org/10.1053/j.gastro.2015.04.005 (quiz e14).
Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–9. https://doi.org/10.1002/hep.23276.
Boettcher E, Csako G, Pucino F, Wesley R, Loomba R. Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2012;35:66–75. https://doi.org/10.1111/j.1365-2036.2011.04912.x.
Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679–90. https://doi.org/10.1016/S0140-6736(15)00803-X.
Newsome PN, Buchholtz K, Cusi K, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113–24. https://doi.org/10.1056/NEJMoa2028395.
Takahashi H, Kessoku T, Kawanaka M, et al. Ipragliflozin improves the hepatic outcomes of patients with diabetes with NAFLD. Hepatol Commun. 2022;6:120–32. https://doi.org/10.1002/hep4.1696.
Lai LL, Vethakkan SR, Nik Mustapha NR, Mahadeva S, Chan WK. Empagliflozin for the treatment of nonalcoholic steatohepatitis in patients with type 2 diabetes mellitus. Dig Dis Sci. 2020;65:623–31. https://doi.org/10.1007/s10620-019-5477-1.
Akuta N, Watanabe C, Kawamura Y, et al. Effects of a sodium-glucose cotransporter 2 inhibitor in nonalcoholic fatty liver disease complicated by diabetes mellitus: Preliminary prospective study based on serial liver biopsies. Hepatol Commun. 2017;1:46–52. https://doi.org/10.1002/hep4.1019.
Akuta N, Kawamura Y, Watanabe C, et al. Impact of sodium glucose cotransporter 2 inhibitor on histological features and glucose metabolism of non-alcoholic fatty liver disease complicated by diabetes mellitus. Hepatol Res. 2019;49:531–9. https://doi.org/10.1111/hepr.13304.
Akuta N, Kawamura Y, Fujiyama S, et al. SGLT2 inhibitor treatment outcome in nonalcoholic fatty liver disease complicated with diabetes mellitus: the long-term effects on clinical features and liver histopathology. Intern Med. 2020;59:1931–7. https://doi.org/10.2169/internalmedicine.4398-19.
Fujimori N, Tanaka N, Kimura T, et al. Long-term luseogliflozin therapy improves histological activity of non-alcoholic steatohepatitis accompanied by type 2 diabetes mellitus. Clin J Gastroenterol. 2020;13:83–9. https://doi.org/10.1007/s12328-019-01018-1.
Sato T. Liver stiffness in 10 patients with non-alcoholic fatty liver disease before and after luseogliflozin administration. Kanzo. 2018;59:554–62. https://doi.org/10.2957/kanzo.59.554 (in Japanese).
Yen FS, Wei JC, Chiu LT, Hsu CC, Hou MC, Hwu CM. Thiazolidinediones were associated with higher risk of cardiovascular events in patients with type 2 diabetes and cirrhosis. Liver Int. 2021;41:110–22. https://doi.org/10.1111/liv.14714.
Sinha B, Ghosal S. Assessing the need for pioglitazone in the treatment of patients with type 2 diabetes: A meta-analysis of its risks and benefits from prospective trials. Sci Rep. 2020;10:15781. https://doi.org/10.1038/s41598-020-72967-8.
Mantovani A, Byrne CD, Benfari G, Bonapace S, Simon TG, Targher G. Risk of heart failure in patients with nonalcoholic fatty liver disease: JACC review topic of the week. J Am Coll Cardiol. 2022;79:180–91. https://doi.org/10.1016/j.jacc.2021.11.007.
Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in Type 2 diabetes. N Engl J Med. 2015;373:2117–28. https://doi.org/10.1056/NEJMoa1504720.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–44. https://doi.org/10.1056/NEJMoa1607141.
Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. https://doi.org/10.1002/hep.20701.
Acknowledgements
We wish to thank all participants in this study and all the medical staff who cooperated with us in this study.
Funding
This study is supported by Taisho Pharmaceutical Co., Ltd. Taisho Pharmaceutical Co., Ltd. also paid the journal’s Rapid Service Fees, including the fees for the organization and submission of the results to the journal. Taisho Pharmaceutical Co., Ltd. is not involved in the planning, implementation, data management, statistical analysis, evaluation, or writing related to this study.
Medical Writing and Editorial Assistance
We would like to thank Masahiro Yamada at Soiken Inc. who helped us with the statistical analysis as well as Editage (www.editage.com) for English language editing.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Conceptualization: Teruki Miyake and Osamu Yoshida. Methodology: Teruki Miyake and Osamu Yoshida. Software, SAS Institute, Inc. (Cary, NC, USA). Validation: Teruki Miyake and Yoichi Hiasa. Formal analysis: Shinya Furukawa. Investigation: Teruki Miyake, Osamu Yoshida, Yohei Koizumi, Takao Watanabe, Eiji Takeshita, Kotaro Sunago, Atsushi Yukimoto, Kyoko Watanabe, Masumi Miyazaki, Sayaka Kanzaki, Hironobu Nakaguchi, Mitsuhito Koizumu, Yasunori Yamamoto, Teru Kumagi, and Yoichi Hiasa. Histopathological evaluation: Masanori Abe and Yoshio Tokumoto. Data curation: Teruki Miyake, Osamu Yoshida, and Masashi Hirooka. Writing—original draft preparation: Teruki Miyake and Osamu Yoshida. Writing—review and editing: Teruki Miyake, Osamu Yoshida, Bunzo Matsuura, and Yoichi Hiasa. Supervision: Yoichi Hiasa. Project administration: Teruki Miyake, Osamu Yoshida, and Yoichi Hiasa. Funding acquisition: Teruki Miyake, Osamu Yoshida, and Yoichi Hiasa.
Disclosures
Shinya Furukawa has received lecture fees from Novo Nordisk Pharma Ltd., Boehringer Ingelheim Pharmaceuticals, Inc., Sumitomo Dainippon Pharma Co., Ltd., Ono Pharmaceutical Co., LTD., Eli Lilly and Company, and Sanofi. None of the funding entities are involved in the planning, implementation, data management, statistical analysis, evaluation, or writing related to this study. Teruki Miyake, Osamu Yoshida, Bunzo Matsuura, Masashi Hirooka, Masanori Abe, Yoshio Tokumoto, Yohei Koizumi, Takao Watanabe, Eiji Takeshita, Kotaro Sunago, Atsushi Yukimoto, Kyoko Watanabe, Masumi Miyazaki, Sayaka Kanzaki, Hironobu Nakaguchi, Mitsuhito Koizumu, Yasunori Yamamoto, Teru Kumagi, and Yoichi Hiasa have nothing to disclose.
Compliance with Ethics Guidelines
This study and its protocols have been assessed and approved by the Certified Review Board of Ehime University (Toon, Ehime, Japan; E-mail: rinri@m.ehime-u.ac.jp), which had obtained certification from the Minister of Health, Labour and Welfare in Japan (approval ID number: jRCTs061210009; University Hospital Medical Information Network ID: UMIN000045003). The study will be conducted in accordance with the Declaration of Helsinki of 1964 and its later amendments, the Ethical Guidelines for Medical and Health Research Involving Human Patients issued by the Ministry of Health, Labour and Welfare in Japan, the Clinical Trials Act, and other current legal regulations in Japan. Informed consent will be obtained from all the study patients.
Data Availability
Regarding the data obtained in this study, there is no description of data sharing to a third party without the research funder, Taisho Pharmaceutical Co., Ltd., after the end of the study in the research plan. Approval for data sharing by the Certified Review Board, Ehime University, has not been obtained. Therefore, it is not open to the public. However, the research funder has been approved to use the research data upon their request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Miyake, T., Yoshida, O., Matsuura, B. et al. Additional Effect of Luseogliflozin on Semaglutide in Nonalcoholic Steatohepatitis Complicated by Type 2 Diabetes Mellitus: An Open-Label, Randomized, Parallel-Group Study. Diabetes Ther 13, 1083–1096 (2022). https://doi.org/10.1007/s13300-022-01239-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-022-01239-7