
Abstract
The classic focus on the mechanisms of action of aldosterone was directed primarily on its role in modulating renal excretory function and maintaining volume homeostasis. In contrast, many recent studies have demonstrated a much wider and expanded role for aldosterone and for the mineralocorticoid receptor (MR). Activation of the MR promotes inflammation, collagen formation, fibrosis, and necrosis with consequent renal injury. Increasing evidence has accrued that implicates the pathophysiological overactivation of the MR as a major determinant of progression of both diabetic and nondiabetic chronic kidney disease (CKD). By promoting cascades of injury encompassing inflammation and fibrosis, MR overactivation constitutes a pivotal determinant of CKD progression and consequently its associated morbidity and mortality. Based on this mechanism of action, blockade of the MR with the nonsteroidal MR antagonist finerenone is currently being investigated as a novel treatment regimen to slow the progression of CKD. The recently reported FIDELIO-DKD (FInerenone in reducing kiDnEy faiLure and dIsease prOgression in Diabetic Kidney Disease) study demonstrated that patients with CKD and type 2 diabetes (T2D) who were treated with finerenone manifested a lower risk of a composite primary outcome event compared with patients in the placebo arm (defined as kidney failure or a sustained decrease of ≥ 40% in the estimated glomerular filtration rate from baseline, or death from renal causes). In addition, patients in the finerenone group also manifested a lower risk of a key secondary outcome event (defined as death from cardiovascular causes, nonfatal myocardial infarction, nonfatal stroke, or hospitalization for heart failure). Based on the success of these major clinical trials, finerenone was approved by the FDA on 9 July 2021 as a novel treatment for retarding CKD progression in patients with T2D (https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-drug-reduce-risk-serious-kidney-and-heart-complications-adults-chronic-kidney-disease).
Podcast Video (MP4 258973 KB)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features
This article is published with digital features, including a podcast video and audio file, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.19182245.
Podcast Transcript
Hello and welcome to a podcast with Diabetes Therapy, entitled, ‘Aldosterone and Mineralocorticoid Receptor Antagonists in 2021—a Paradigm Shift’. Today, we are delighted to be joined by Dr Murray Epstein, Professor of Medicine Emeritus in the division of Nephrology and Hypertension at the University of Miami, Miller School of Medicine.
As an introduction to proceedings, I would just like to ask for your thoughts Dr Epstein on the progress in the treatment of diabetic kidney disease over recent years.
What I’m going to do today is to share with you some major advances that have impacted not only nephrology, but the entire cardiorenal space. Really, an extraordinary forward trajectory encompassing our ability to treat patients with diabetic kidney disease (DKD) and their associated cardiovascular and renal complications.
In medicine, we have metrics, and the first slide is merely meant to convey the relevance and the immediacy of what I will be talking about. For many years now the National Kidney Foundation (NKF) and the CKD Prognosis Consortium have published the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines. These guidelines build on relatively new innovations in our ability to treat patients, and this first slide provides a timeline of recent KDIGO promulgations. One thing that stands out loudly and clearly is that, going from the KDIGO guidelines released in 2012, there was an 8-year gap before the next set of guidelines were promulgated and published, which is the norm. However, look at what happens after 2020: there have been so many innovative changes with respect to a number of new drug classes that the new guidelines will be coming out, less than 2 years later, in the first quarter of 2022.
The reason for that is, essentially, the emergence of two major drug classes: the sodium-glucose cotransporter-2 (SGLT-2) inhibitors and novel, non-steroidal mineralocorticoid receptor antagonists (MRA). In the latter class of agents, finerenone was approved by the Food and Drug Administration just a few months ago. So, the bottom line is that the 2-year interval between the KDIGO guidelines published in 2020 and those to be released in the first quarter of next year (2022) speak loudly to the rapidity of innovation in the treatment of DKD.
Moving on to today’s main topic, could we ask you to explain the role of aldosterone in renal physiology?
There has been a major paradigm shift in our concept of what aldosterone does in terms of both maintaining everyday volume homeostasis and as a modulator or a mediator of primary aldosteronism or Conn’s syndrome.
What can be seen, looking at the left-hand side of this slide, is that angiotensinogen (renin substrate) is produced by the liver. Renin, released by the kidney, then cleaves angiotensinogen and that in turn forms angiotensin 1. Angiotensin-converting enzyme (ACE) released from the lungs acts on angiotensin 1 to cleave it to form the vasoactive peptide, angiotensin 2. And angiotensin 2 acts directly on the blood vessels, promoting vasoconstriction.
The old paradigm, the one that I learned in medical school, indicated a role for aldosterone and the renin-angiotensin system in two settings: maintaining normal everyday volume homeostasis and, in addition to that, if it goes awry, to actually produce hypertension. The concept states that angiotensin II is a stimulant, a secretagogue, acting on the adrenal cortex to promote release of aldosterone and that in turn acts at the level of the kidney to promote sodium retention and, concomitantly, potassium excretion in the distal convoluted tubule. This very neat theory has changed dramatically in the last 2 years, and it is not merely of academic interest but rather, speaks loudly to redefine to the everyday clinical practice of primary care physicians and healthcare providers as well as specialists. And that is one of the messages that I would like to share today.
How has the understanding of the role of aldosterone changed over the past 30 years?
The next slide contrasts the role of aldosterone over the last 30 years: what most of us learnt in medical school in 1990–1995 and our current updated understanding of the pathophysiology today in 2021.
The right side of the slide focusses on a number of new innovations and an enhanced understanding for the role of aldosterone. That angiotensin is merely one of several drivers of aldosterone secretion. That, 30 years ago, we thought that aldosterone was the only secretagogue, but now it is recognised as one of several physiological ligands for the mineralocorticoid receptor (MR). That, while we were taught years ago that aldosterone primarily elevates blood pressure by sodium-retaining effects, thereby leading to volume expansion with consequent blood pressure elevation, we now know that such a formulation is a simplification. Indeed, we now realize that aldosterone raises blood pressure by multiple mechanisms, but primarily by its actions on the vasculature and, indeed, on the central nervous system.
We also were taught that MRAs, and the one that most of us learnt back in medical school, the steroidal MRA spironolactone, acts by blocking the binding of aldosterone to the MR. We now know that overactivation of the MR is a major determinant of progression of chronic kidney disease (CKD) via an array of multiple mechanisms, thereby promoting injury, specifically, inflammation and fibrosis.
Importantly, and this relates to how we manage patients, there has been a paradigm shift in our understanding in the role of aldosterone and the importance of MR activation. And we now realize that this is where major benefits can be achieved in terms of a wide array of target organ injury.
Could I just ask you to talk a little about primary aldosteronism and its prevalence?
The classic view, dating back 50 years to the seminal work of Jerome Conn at that time, at the University of Michigan, is that primary aldosteronism was manifested by hypertension. This new entity called primary aldosteronism (later named Conn’s syndrome), was caused by an adrenal tumor secreting excessive amounts of the adrenal hormone aldosterone, As originally defined, primary aldosteronism was associated with hypokalaemia, present in approximately 40% of patients, and usually there was a lack of oedema in this setting. The reason for that was the mechanism of aldosterone escape, which I will return to later, leading to an increase in natriuresis and diuresis. And, finally, it was believed that the excess of aldosterone resulted in metabolic alkalosis, hypomagnesaemia, and mild hypernatraemia. Again, if we were sitting in an office with our patient in 1990, the tell-tale signs we would look for would be hypokalaemia and substantial hypertension and metabolic alkalosis. And we were also taught that primary aldosteronism is a rare and categorical disease.
Recently, there has been a huge paradigm shift, and our current understanding of primary aldosteronism has been completely revised. This was best summarised in a landmark publication by a group of major investigators in the field published in the Annals of Internal Medicine in 2020. Brown et al. showed that the prevalence of primary aldosteronism is high and, importantly for all of us in clinical medicine, remains largely unrecognised. Furthermore, that severe hypertension and hypokalaemia are not prerequisites for the diagnosis of primary aldosteronism; rather, it can frequently be detected not only in normokalaemic hypertensive patients, but, indeed, across a very wide spectrum of blood pressure alterations.
Essentially, these observations have served to radically redefine primary aldosteronism. From a disease that, historically, was identified as a rare, infrequent cause of hypertension to what we now know it as today: a common syndrome that plays a pivotal role in the pathogenesis of hypertension.
How are aldosterone and the MR involved in the progression of CKD?
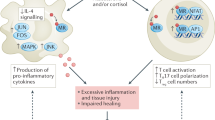
We were originally taught that overactivation of the MR was modulated by a single ligand, aldosterone, but we now know that there are an array of ligands as well as non-ligand activators of the MR. The lower left-hand corner of the slide shows that in terms of ligands it is not only aldosterone but also cortisol that activates the MR. In addition, there are a range of non-ligand activators of the MR. Rac 1 and hyperglycaemia can do it, which is highly relevant to our management of patients with DKD and sodium excess via a high salt diet can activate the MR as well.
When the MR is activated, a number of inflammatory and fibrotic pathways are implemented, and current treatment is aimed at blocking inflammation and fibrosis.
The right side of the slide shows a number of drivers of CKD and type 2 diabetes progression. Inflammation and fibrosis, importantly, but also alterations in haemodynamics, for example an increase in intraglomerular pressure, which in turn can drive downstream fibrosis and inflammation. And, finally, on the lower right-hand corner of the slide, metabolic disarray, most prominently exemplified by poor glycaemic control can contribute to progression of CKD and type 2 diabetes.
Overall, what we have is a convergence of several mechanisms: aldosterone or MR-activated or mediated inflammation and fibrosis, abnormal haemodynamics, and, finally, metabolic disarray, primarily inappropriate glycaemic control, all of which act in concert to promote progression of DKD.
We know that ACE inhibitors and angiotensin receptor blockers are widely used in the treatment of CKD, so why not just rely solely on these agents and not aldosterone blockers to prevent progression of CKD?
In theory, treatment with an ACE inhibitor or an angiotensin 2 receptor blocker (ARB) should prevent progression of CKD. However, there is a “fly in the ointment”, and it is depicted in this next slide. This study by Staessen et al., conducted over 40 years ago, involved treating patients with very large doses of captopril, the first of the ACE inhibitors. Please note that they prescribed doses as high as 600 mg a day, which would obviously be considered inappropriately high today, and treatment was associated with an initial reduction in plasma aldosterone levels. However, after a few months, despite continuing treatment with massive ACE inhibition, aldosterone levels increased. Essentially, aldosterone escaped from the inhibitory effects of ACE inhibition and reverted back to, and even exceeded, pre-study levels. The take-home message is that RAS inhibition alone does not suffice to block aldosterone.
Another study by Schjoedt et al. of the Steno Diabetes Center, in Copenhagen, Denmark, investigated what the relevance of aldosterone escape might be to subsequent progression of CKD. Patients with type 1 diabetes with diabetic nephropathy were divided into two groups: those that escaped the inhibitory effects of ACE inhibition (shown in blue in the slide) and a non-escape group (shown in green). Patients who experienced aldosterone escape with reversion back to or exceeding pre-study levels demonstrated a greater decline in GFR than those in the non-escape group. In conclusion, the take-home message is that prevalent aldosterone levels, which are not blocked, promote a more rapid decline of eGFR and are likely to be associated with greater progression in CKD.
How do the mechanisms of action of nonsteroidal MRAs differ from those of steroidal MRAs?
I previously mentioned that one of the reasons for updating and publishing the new KDIGO guidelines in the first quarter of 2022 is the emergence of a new class of agents: novel non-steroidal MRAs. This next slide describes how the mechanisms of action of nonsteroidal MRAs differ from those of first-generation steroidal MRAs—to do this I have compared the classic steroidal MRA, which has been a mainstay of clinical practice for many years, spironolactone, with the new, recently approved third-generation MRA, finerenone.
Pharmacologically, finerenone has a bulky nature that allows it to bind to the MR in a way that prevents the MR from becoming or adopting an agonist conformation. In addition, finerenone has full MR antagonist activity compared to the classic steroidal MRAs like spironolactone, and more recently eplerenone, which have partial MR agonist activity that is undesirable.
It is therefore important to understand that the new, non-steroidal MRAs are able to act or to promote blockade in a more unfettered or unchained mechanism which, obviously, is relevant for progression for CKD.
How is finerenone hypothesised to provide kidney and cardiovascular protection?
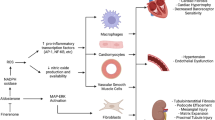
The final slide summarises the multiple mechanisms whereby the new, third-generation non-steroidal MRAs, typified by finerenone, may provide both kidney and cardiovascular protection. Essentially, finerenone acting on the MR blocks the transcription of pro-inflammatory profibrotic genes in various cell types. As an example, if we look at the right upper-hand corner, we can see the kidney and we can see the specific cell types affected: podocytes, mesangial cells, macrophages, and fibroblasts, as well as epithelial cells on the tubule. All of these are impacted in a beneficial manner to retard or to interdict injury, inflammation and fibrosis. That, in turn, acts to slow or to retard progression of kidney disease. So, what we are doing is buying an extraordinary amount of time for the patient to delay the day of reckoning in terms of progression to end-stage kidney disease (ESKD) with its requirement for initiating maintenance haemodialysis.
In summary, I propose that what we have reviewed today, the reappraisal of the role of aldosterone and the MR, and initiating treatment with the novel nonsteroidal MRAs, constitutes a major innovation in our treatment armamentarium—a significant contribution to our treatment paradigm. And, allow me to bring it up to date to the summer of 2021 when based on the ability to reach the pre-specified endpoint in two major clinical trials conducted globally in 45–50 different countries with thousands of patients (FIDELIO and FIGARO), the Food and Drug Administration in the United States approved finerenone for the management of patients with CKD in the setting of diabetes. So, again, a major step forward that will be incorporated into the new KDIGO guidelines that I alluded to in my introductory remarks. Thank you all for your interest and your attention.
I think that concludes our podcast. Thank you, Dr Epstein, for your time and insights today. We hope that this has been of interest to our listeners.
Acknowledgements
The author thanks David L. Epstein, MD, for his thoughtful discussions and critique and John Bilbruck of Envision Pharma Group for expert editorial support in the preparation of this transcript. John Bilbruck also acted as the interviewer for this podcast.
Funding
Murray Epstein received no personal funding for this submitted article. The Rapid Service Fee was funded by Bayer Corporation.
Medical Writing Assistance
Medical writing assistance was provided by John Bilbruck of Envision Pharma Group and was funded by Bayer Corporation. Envision Pharma Group’s services complied with international guidelines for Good Publication Practice (GPP3).
Author Contributions
Murray Epstein, MD, meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, takes responsibility for the integrity of the work as a whole, and has given his approval for this version to be published. Murray Epstein, MD, participated substantively in the drafting, writing, critical revision, and approval of the final version.
Disclosures
Murray Epstein, MD, reports personal fees from Vifor Pharma, Alnylam Pharmaceuticals, Inc., and Bayer Healthcare outside the submitted article.
Compliance with Ethics Guidelines
This article does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Epstein, M. A Podcast Discussing Aldosterone and Mineralocorticoid Receptor Antagonists in 2021: A Paradigm Shift. Diabetes Ther 13, 583–588 (2022). https://doi.org/10.1007/s13300-022-01236-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-022-01236-w