
Abstract
Introduction
Type 2 diabetes (T2D) management has reached a point where not only optimal glycaemic control is necessary, but also additional interventions with proven cardiovascular risk reduction benefit. Subcutaneous semaglutide has been shown to provide cardiovascular protection, but its use may be limited by its injection formulation. To overcome this limitation, an oral semaglutide tablet has been developed, which could potentially be of the same value as its injection counterpart, but in a much wider group of patients with T2D, thereby allowing for broader cardiovascular risk reduction in this vulnerable patient population.
Methods
A total of 100 consecutive patients with T2D and a disease duration of up to 10 years, without manifest cardiovascular disease, who are treated with metformin (± sulphonylurea) and optimal cardioprotective therapy, will be recruited in a single-blinded, randomized trial named “Semaglutide Anti-atherosclerotic Mechanisms of Action Study (SAMAS).” After 1:1 randomization, patients will receive either oral semaglutide 14 mg daily or placebo for 1 year. The primary outcome comprises changes in atherosclerosis-related structural and functional characteristics of the arterial wall, namely: reduction of the carotid intima-media thickness, improvement of endothelial function and decrease in arterial stiffness. Secondary outcomes are changes in atherogenic small dense low-density lipoproteins, glucose control (HbA1c) and inflammatory markers (hsCRP). Possible correlations between primary endpoints and changes in lipids, HbA1c and high-sensitivity C-reactive protein will be sought.
Discussion
This is the first study to investigate the direct and indirect anti-atherosclerotic mechanisms of oral semaglutide. The results are expected to confirm the position of oral semaglutide in the multifactorial management of T2D with an emphasis on cardiovascular disease prevention.
Trial Registration
ClinicalTrials.gov Identifier: NCT05147896.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Current type 2 diabetes (T2D) management requires additional interventions to achieve proper cardiometabolic benefit. |
Oral semaglutide is the first glucagon-like peptide-1 receptor agonist available on the market, potentially allowing for a significant cardiometabolic risk reduction in a broader population of patients with T2D due to its innovative oral formulation. |
We designed an innovative clinical trial to explore the possible anti-atherosclerotic effects of oral semaglutide, and its effects associated with arterial structure and function parameters in patients with T2D in primary cardiovascular prevention. |
This is the first study to investigate the direct and indirect anti-atherosclerotic mechanisms of oral semaglutide, thus solidating its position in the multifactorial T2D management strategy, with emphasis on cardiovascular disease prevention. |
Introduction
Cardiovascular risk assessment and cardiovascular disease prevention are urgently needed to decrease morbidity and mortality, improve quality of life and decrease global expenditure on diabetes and its chronic complications in the vulnerable population of people with diabetes [1]. Based on the results of cardiovascular outcome trials (CVOT) and other trials on new antidiabetic drugs, a new perspective on diabetes management has emerged. Drugs with proven cardiovascular risk reduction characteristics are now referred to as diabetes/disease-modifying drugs, since they possess several beneficial effects beyond glycaemic control, such as cardiovascular system protection, renal protection, weight reduction, blood pressure lowering and protection against hypoglycaemia [2]. Representatives of these drugs include sodium glucose co-transporter 2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists, whose protective benefits have been established in CVOTs, and international scientific guidelines have clearly recommended their prescribing to most patients with type 2 diabetes (T2D), regardless of their risk factors or glycaemic control targets.
Therefore, the European Society of Cardiology (ESC) guidelines suggest using either SGLT-2 inhibitors or GLP-1 receptor agonists with proven cardiovascular risk reduction benefits as the first-line antidiabetic therapy in naïve patients with T2D with overt atherosclerotic cardiovascular disease or with high or very high cardiovascular risk. If patients with T2D have been previously treated with metformin, the guidelines suggest adding either of the two drugs to the metformin therapy. In both cases, introduction of these drugs should be irrespective of the initial glycated haemoglobin (HbA1c) values [3]. The American College of Cardiology (ACC) 2020 expert consensus on novel therapies for cardiovascular risk reduction in T2D similarly suggests using either SGLT-2 inhibitors or GLP-1 receptor agonists with proven cardiovascular benefit in patients with T2D who are older than 18 years and who are at high risk for atherosclerotic cardiovascular disease or have one of the following: atherosclerotic cardiovascular disease, heart failure or diabetic kidney disease. These suggestions are also irrespective of the initial HbA1c values and metformin is not recommended as an obligatory primary therapy [4]. Both recommendations allow for other antidiabetic drugs to be added to the therapeutic regimen as necessary to achieve adequate glycaemic control [3, 4]. Nevertheless, as presented at the last American Diabetes Association meeting in 2021, from the diabetologists’ point of view, metformin still remains the first-line therapy in T2D [5]. This new, simple approach in patients with T2D and either established atherosclerotic cardiovascular disease or risk factors for atherosclerotic cardiovascular disease initially suggests combined therapy with the combination of metformin and GLP-1 receptor agonist [2].
GLP-1 receptor agonists have an excellent impact on glycaemic control, and they also promote body weight loss, slight decreases in blood pressure and modest reductions in total and low-density lipoprotein (LDL) cholesterol as well as triglycerides, in addition to a number of other beneficial effects [6,7,8]. GLP-1 receptors are present on cardiomyocytes and endothelial cells and in the autonomic nervous system [9]. Nevertheless, the cardiovascular protective mechanisms of GLP-1 receptor agonists are still poorly understood, even though some have been proposed, such as anti-inflammatory properties, improvement of endothelial dysfunction and reduction of myocardial ischaemia injury; however, not all GLP-1 receptor agonists have proven to have cardio-renal protective actions [10]. Those with a proven ability to reduce major adverse cardiovascular events (MACE) are liraglutide, dulaglutide, albiglutide and subcutaneous semaglutide; in contrast, oral semaglutide was found to show only a trend towards MACE reduction, but this effect was not statistically significant as the cardiovascular noninferiority study lasted < 2 years [11,12,13]. Further studies are ongoing that will show whether treatment with oral semaglutide could reach the same beneficial effect on MACE as its injectable counterpart [11, 14]. However, as both drug formulations contain the same active agent, one could deduce that the impact on MACE should and will prove to be similar.
The established beneficial cardiovascular outcomes of GLP-1 receptor agonists seem to be mediated through their actions on endothelial function, blood lipids, blood pressure decrease and anti-inflammatory activity [11, 14, 15]. Some GLP-1 receptor agonists with proven cardiovascular benefit have a beneficial action on the very first step of the atherosclerotic cascade, reducing atherogenic small dense LDL [16, 17]. This benefit is in line with the proposed direct anti-atherosclerotic action of liraglutide, which showed cardiovascular benefit in the LEADER trial [18], since evidence from preclinical and clinical studies suggest that this GLP-1 receptor agonist can reduce atherosclerosis formation and progression [19]. Similar effects with semaglutide are to be expected, based on the first preclinical observations with this innovative agent [20]. A clinical trial investigating the effect of injectable semaglutide on coronary atherosclerosis progression is also ongoing [21]. One of the frequent barriers to the introduction of GLP-1 receptor agonists in clinical practice is the injection treatment; therefore, the development of an oral semaglutide formulation (tablet) allows this obstacle to be bypassed. Oral semaglutide is the first GLP-1 receptor agonist to be produced in the form of a once-daily oral formulation, and is the only oral GLP-1 receptor agonist currently available on the market. In the present study, we therefore focussed on oral semaglutide. Its pharmaceutical innovation includes the coformulation of semaglutide with sodium N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC) [22]. The latter attenuates enzyme digestion, increases local gastric pH and is the absorption enhancer that enables efficient bioavailability of the oral formulation [22].
The beneficial effects of GLP-1 receptor agonists on the process of atherosclerosis have not yet explored for the more novel agents and the mechanisms involved remain to be fully elucidated. To date, studies have been performed for injectable GLP-1 receptor agonists, but not for the only oral agent available in this class, semaglutide. Therefore, the aim of the study described herein is to explore the possible anti-atherosclerotic effects of oral semaglutide, and its beneficial effects associated with arterial structure and functional parameters in patients with T2D without overt cardiovascular disease, with a specific focus on its possible action on primary cardiovascular disease prevention.
Objectives
The objectives of the study, all related to patients with T2D, are:
-
Objective 1: to explore whether treatment with oral semaglutide can reduce carotid intima-media thickness (cIMT);
-
Objective 2: to study whether treatment with oral semaglutide can improve endothelial function and/or decrease arterial stiffness;
-
Objective 3: to investigate whether treatment with oral semaglutide can lead to improvement of atherogenic dyslipidaemia;
-
Objective 4: to investigate whether treatment with oral semaglutide can influence the levels of high sensitivity C-reactive protein (hsCRP);
-
Objective 5: to explore whether objectives 1–3 are correlated with each other and if they correlate with changes in lipids, HbA1c and hsCRP, thereby, at least partially, explaining the anti-atherogenic potential and underlying mechanisms of the oral semaglutide formulation (tablet).
Methods
Study Design
The SAMAS study is designed as a single, blinded, randomized trial of 12 months duration, with the aim to investigate the anti-atherosclerotic mechanisms of oral semaglutide versus placebo in patients with T2D. The participants will be randomized in a 1:1 ratio to the oral semaglutide or placebo treatment groups. They will be recruited from the diabetes outpatient clinic of the University Medical Centre Ljubljana, Ljubljana, Slovenia. The study was approved on 23 August 2021 by the Slovenian National Medical Ethics Committee (Approval No. 0120–245/2021/3), and it will be conducted in accordance with the Declaration of Helsinki and Good Clinical practice guidelines. The study is supported by a grant of the University Medical Centre Ljubljana, Ljubljana, Slovenia (Grant No. 20210043). Participants will have to sign informed consent to participate.
Sample Selection
The aim is to recruit 100 consecutive patients with T2D of both genders, aged between 30 and 60 years. They will have to fulfil all of the inclusion criteria while not meeting any of the exclusion criteria (listed in Table 1). As basic antidiabetic therapy, participants will have to be treated with at least the optimal dose of metformin (i.e. 2000 mg daily) with or without sulphonylurea. The required glycaemic control at the entry to the study, based on HbA1c, will be ≤ 8.5%. The rationale underlying this choice is based on these patients being naïve for antidiabetic therapy with additional protective effects on the cardiovascular system (e.g. for treatment with SGLT-2 inhibitors). The presence of additional cardiovascular risk factors (smoking, arterial hypertension, dyslipidaemia and chronic kidney disease stage 1–3), but not overt cardiovascular disease, allow the patients to be eligible for acceptance into the trial. In this regard, other cardioprotective therapy, i.e., angiotensin-converting enzyme inhibitors/sartans, statins or beta blockers, will be maintained during the study, with the aim to strict and optimal regulation of cardiovascular risk factors.
Recruitment Strategy
Patients will be recruited from the diabetes outpatient clinic, where we already have a large pool of patients who are registered as having T2D. Additionally, patients are continuously referred to our outpatient clinic by general practitioners, and these patients also represent additional pool of possible recruitment candidates. Therefore, in accordance with the inclusion and exclusion criteria, we expect the pool of patients to be sufficient. The patients will receive written and oral information about the study at the outpatient clinic, they will have sufficient time to consider enrolling up to the first visit, when they will sign written informed consent. At the first visit, all the inclusion and exclusion criteria (Table 1) will be rechecked. Patients will be randomized to the study, after signing a written informed consent form, in 1:1 fashion to receive either oral semaglutide or placebo. They will be followed for 12 months, i.e. the treatment period. The inclusion frequency is expected to be stable and continuous and will be regularly evaluated.
Withdrawal From the Study
All participants will be free to withdraw from the study at any time. They would not be requested to provide any reason for their withdrawal, and their withdrawal from the study will not influence their further treatment at the diabetes outpatient clinic of the University Medical Centre Ljubljana. Anticipated reasons for withdrawal may include: consent withdrawal, adverse events, pregnancy discovered during the trial or failure to comply with the trial medication or protocol. Missing ≤ 5 drug doses will not be the reason for exclusion, as once the steady state is achieved, the therapeutic concentration of the drug in plasma remains stable for a certain amount of time and missing some pills does not influence it. Persons with newly diagnosed carotid plaques will be withdrawn from the study and, if necessary, referred for further diagnostics. Also, all the participants with major pathologic findings on clinical examination will be withdrawn from the trial at the discretion of the investigator. In the case of findings that merit further attention, the participants will be referred to the appropriate specialist(s). If additional cardioprotective therapy needs to be introduced in the treatment regimen, the participants will also be withdrawn from the study.
Sample Size
The sample size calculation was performed for the primary endpoint of our study, i.e. to explore whether treatment with oral semaglutide can reduce the cIMT; the cIMT was calculated based on data from a previous study [22], and the standard deviation was estimated to be 0.29 mm. Setting the targeted statistical power of analysis at 90% with two-sided test alpha (α) value of 0.05 and aiming to show a decrease of 15% after treatment with oral semaglutide, the required number of patients was calculated to be 92. Based on our previous experience, the drop-out rate is estimated to be around 8–10%; therefore, our aim is to randomize a total of 100 participants to either the oral semaglutide or placebo arm.
Randomization
The participants would be randomized into two groups by personnel performing the randomization process; these persons will not be blinded. In contrast, the investigators, those performing the ultrasonography and other metrics, those assessing outcomes, those performing the data analysis and patients will remain blinded through the whole course of the study. Unblinding of the participants will be permissible in case of medical emergencies that require knowledge of treatment allocation and in the cases of serious adverse events or suspected unexpected serious adverse reaction(s).
Intervention
Patients will be assigned to two groups, with one receiving the active agent and the other receiving placebo. The active substance will be the oral GLP-1 receptor agonist semaglutide (Rybelsus 3 mg, 7 mg, 14 mg tablets; Novo Nordisk A/S Bagsvaerd, Denmark). The drug will have to be taken on an empty stomach in the morning after at least an 8 h fasting period. After the oral semaglutide tablet is ingested, the subjects will have to wait at least 30 min before eating, drinking or taking other oral medications. The oral semaglutide tablet must be taken with no more than a half glass of water (equivalent to 120 ml), must be swallowed whole and can not be split, crushed or chewed. The starting dose of oral semaglutide will be 3 mg; after 30 days, a dose escalation to 7 mg is anticipated; and after another 30 days, dose escalation to 14 mg, i.e., the maintenance dose, is expected. A dose escalation regimen is needed to avoid the development of the potential side effects (listed in Table 2). Dose escalation will be encouraged by notifying the patient by phone, with the call made 1 day prior to the day on which the new dose starts. If treatment with oral semaglutide is associated with unacceptable gastrointestinal side effects (moderate to severe), dose reduction or extension of dose escalation intervals will be allowed as judged by the unblinded nurse involved in the randomization process.
The placebo and semaglutide tablets will be provided in identical cases, will be identically shaped and will be packed in identically shaped bottles that will be labelled with numbers. The patients will be advised to contact the study team in case of any problems or adverse events during the study.
Treatment Allocation
After the screening, the participants will be randomized to one of the two study groups in 1:1 ratio. A non-blinded study nurse (otherwise not associated with the trial) will allocate study participants into the groups evenly. Patients will be given a sufficient number of tablets (placebo or semaglutide) in bottles labelled by numbers, for the first 6 months of therapy. An additional drug package will be provided to each patient at the 6-month interval.
Trial Visits and Examinations
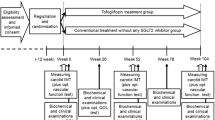
The trial will consist of three visits (V1, V2, V3) and three phone calls (P1, P2, P3) (Fig. 1). After initial screening for suitable candidates at the diabetes outpatient clinic, those eligible will be invited for the V1 (day 0) at which time a complete clinical examination (including blood pressure measurement and anthropometric measurements) will be performed, followed by ultrasound and endothelial function measurements as well as determination of arterial stiffness. A package of drugs (comprising of 6 bottles labelled 1–6) for the first 6 months will be handed over. Detailed instructions will be provided on the use of the oral semaglutide tablets (as explained above) and on the sequence at which the numbered bottles should be opened and the tablets taken. After the imaging and functional studies, blood samples will be collected. Similar procedures will be performed at V2 (6 months after the V1) and at the V3 (12 months after V1, i.e. the end of the study). At V2, glycaemic control will be re-evaluated, and in the case of insufficient control, rescue therapy with basal insulin will be initiated. Patients will also be educated on the use of insulin and dose titration.

Study protocol. cIMT Carotid intima-media thickness, EndoPAT device allowing non-invasive measurement of vasoreactivity, V1, V2, V3 visits 1 (day 0), 2, 3, respectively, P1, P2, P3 phone call 1, 2, 3, respectively, SphygmoCor technology assessing arterial stiffness
The glycaemic control at V2 is defined as insufficient if the HbA1c value decreases by < 15% compared to the value at V1, or if the HbA1c value persists at or rises > 8.0%. At P1 (30 days after V1), an assessment of patient adherence and potential side effects will be made, and encouragement for dose titration (opening of the next numbered bottle) from day 31 will be given. The same steps will be taken on day 60, with encouragement given for the higher dose (opening of the next numbered bottle) to be initiated on day 61. At 30 days after the full oral dose of semaglutide is reached, the patient will receive P3, with inquiries about adherence and potential side effects. Also, from V1 onwards, in case of hypoglycaemia events or significant blood glucose reduction, patients will be encouraged to reduce the sulphonylurea dose or discontinue using it completely, while still adhering to treatment with oral semagultide, as this agent alone or in combination with metformin does not cause hypoglycaemia.
Measurements
All measurements will be performed in a standardized, comfortable, warm environment, under the same conditions at every visit. All patients will be asked to overnight fast for at least 8 h prior to each examination. The measurements will be performed in the early morning before the patient’s first meal. Patients will also be asked to refrain from smoking and drinking alcoholic beverages or coffee for the same amount of time.
History and Clinical Examination
Detailed history and standard clinical examination of all organ systems will be performed. In the case that certain major pathologic features associated with the exclusion criteria are found, the participant will be withdrawn from the study and referred to the appropriate specialist(s).
Anthropometric Measurements
Anthropometric measurements will include height, weight, waist circumference. Height will be measured using the standard wall-mounted stadiometer and expressed in centimetres. Weight will be measured using the standard body scale and expressed in kilograms. Body mass index will then be calculated and expressed in kilograms per square meter. Waist circumference will be measured using non-rigid measurement tape and expressed in centimetres.
Blood Pressure Measurement
An automated sphygmomanometer will be used for blood pressure measurements and expressed in millimetres of mercury (mmHg).
Ultrasound Measurements of the Carotid Intima Media Thickness
Ultrasound cIMT measurements will be performed by the single investigator who is blinded to the patient’s therapy; the investigator will also not have access to the previous ultrasound scans when follow-up measurements are made. The ultrasound examination will be performed using the General Electric Logiq F6 (GE Healthcare, Chicago, IL, USA) ultrasound machine. The L6-12-RS linear probe with the frequency of 3.7–13.0 MHz will be used for vascular examination, as per manufacturer's instructions. The cIMT measurements will be performed in the standard manner, i.e. supine position of the participant and standard angles of insonation of carotid arteries.
Complete wall segments of the carotid artery branches will be carefully scanned bilaterally, starting with the common carotid artery just above the clavicle, sliding upwards towards the bifurcation and along the internal carotid artery. Near and far planes of each of the arterial segments will be scanned, while measurements will be made starting 1.0 cm proximal to the bifurcation, followed by the bifurcation itself and then 1.0 cm of the proximal internal carotid artery segment [23]. The auto-IMT modality of the ultrasound machine software will be used, which will automatically—and independently of the observer—detect the cIMT, considering that the distance between the echogenic line represents the intimal blood interface and the outer echogenic line represents the adventitial junction. The maximum cIMT value, expressed in millimetres, will be used for analysis, and determined as the mean of all six measurements performed bilaterally [23, 24].
Measurement of Endothelial Function
The endothelial function measurement will be performed on the peripheral arterial tone using the EndoPAT 2000 device (Itamar Medical Ltd., Caesarea, Israel); this device has been approved by the US Food and Drug Administration (FDA) for determination of endothelial dysfunction. It will produce standardized measurements that are mostly independent of the investigator. Patients will be lying in a semi-supine position, with their forearms extended on the foam arm rest at a 40º angle as per manufacturer’s instructions. Pneumatic probes will be placed on the index fingers bilaterally and inflated to 70 mmHg to squeeze the veins and prevent the veno-arteriolar vasoconstrictor reflex. At this point of time, a baseline signal will be recorded. After 5 min of baseline recording, a blood pressure cuff on one forearm will be inflated to at least 60 mmHg above the patient's systolic blood pressure for another 5 min. The blood pressure cuff will then be rapidly deflated, causing reactive hyperaemia, and the signal will then be followed for the last 5 min. The reactive hyperaemia index (RHI) will be calculated using the manufacturer’s algorithm based on the ratios between pulse wave amplitudes during the reactive hyperaemia and baseline phases [25,26,27].
Measurement of Arterial Stiffness
Arterial stiffness will be measured using the SphygmoCor device with CvMS software (version 9) (AtCor Medical Pty Ltd., West Ryde, Australia). For this measurement, the investigator will again be blinded regarding the therapy that a patient is taking as well as the results of the previous measurements. The measurements will be performed according to the Consensus document on arterial stiffness for measurement of carotid-femoral pulse wave velocity (cfPWV) [28]. The pulse waveforms will be collected at two sides on the right-hand side of the body, with the patient lying in the supine position. Pulse waveforms will first be obtained at the common carotid artery, then on the femoral artery. The distances from the sternal notch to the common carotid artery and from the sternal notch to the femoral artery will be measured. The CvMS software will then calculate the cfPWV automatically as the ratio of the pulse wave distance travelled and the transit time needed [28, 29].
Laboratory Analyses
Venous blood samples will be taken after an overnight fast of at least 8 h. If required, selected blood samples will be centrifuged within 30 min after collection. Aliquots of both serum and plasma will be collected, and some will be used for immediate analyses and will be immediately frozen at − 80 °C.
HbA1c, fasting blood glucose, creatinine, electrolytes, hepatic enzymes and lipids (total cholesterol, triglycerides and high-density lipoproteins) will be measured by standard laboratory methods using the VITRO 5.1FS Chemistry System (Ortho Clinical Diagnostics, Raritan, NJ, USA). LDL will be calculated using the Friedewald formula. HsCRP will be measured in serum using the VITROS Chemistry Products hsCRP Reagent (Ortho- Clinical Diagnostics, Inc., Rochester, NY, USA). Uric acid will be determined by enzymatic reaction with uricase and Trinder-like endpoint product detection (ADVIA Chemistry Systems, Siemens Healthcare Diagnostics Inc., Tarrytown, NY, USA). The CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) formula will be used to calculate estimated glomerular filtration rate. Parameters of inflammation will also be analysed, and additional parameters of coagulation will be determined.
The insulin resistance index (HOMA-IR) will be calculated from fasting insulin (FI) values in venous blood samples and fasting blood glucose (FBG) according to the following equation: FBG × FI/22.5.
For analyses of the lipoprotein subclasses, the samples will be stored at − 80 °C immediately after collection and then analysed at a later time point to determine at least 11 lipoprotein subclasses, including very-low-density lipoproteins, intermediate-density lipoprotein subclasses (IDL-A, IDL-B and IDL-C) and seven LDL subclasses (LDL1–7); the US FDA-approved non-denaturing, linear polyacrylamide gel electrophoresis system (Lipoprint; Quantimetrix Corp., Redondo Beach, CA, USA) will be used for determiniation of the LDL subclasses [17]. Electrophoresis will be performed for 60 min at 3 mA in a gel tube, with each chamber having two quality controls; after electrophoresis, the gel tubes will be scanned using the ArtixScan F1 densitometric flatbed scanner (Microtek Int. Corp., Hsinchu, Taiwan) combined with a Mac personal computer (Apple Inc, Cupertino, CA, USA) with the ScanWizard Pro software installed (Microtek Int. Corp.) [17]. The relative area of each lipoprotein band will be determined and multiplied by the total cholesterol concentration of the sample, and LDL subclasses will be defined as seven bands (LDL1-7, respectively) [30, 31].
Planned Outcomes
The main objectives of the present study, as explained above, are to examine the mechanisms responsible for anti-atherosclerotic and cardioprotective actions of the oral semaglutide formulation in patients with T2D without manifested cardiovascular disease.
Primary Outcome
The anticipated primary outcome is the reduction in cIMT, improvement of endothelial function and reduction of arterial stiffness, after 12 months of treatment with oral semaglutide.
Secondary Outcomes
The anticipated secondary outcomes comprise reduction in atherogenic small dense LDL, improvement of HbA1c and reduction of hsCRP, after 12 months treatment with oral semaglutide. Additionally, possible correlations between the primary and secondary endpoints will be sought. Also, the revaluation visit at 6 months will enable the pace of the oral semaglutide’s anti-atherosclerotic effect to be evaluated.
Data Collection
Participants will be identified by a personal study identification (ID). All venous samples will also be labelled by a study ID. Study data (history and clinical examination data and anthropometric measurements) will be collected as paper case report forms that will be stored securely. The data will also be entered into the electronic database on an on-going basis. The results of measurements of cIMT, endothelial function examinations and arterial stiffness examinations will be supported by data from the measurement devices. Laboratory data will be printed out from the laboratory analyses and stored in paper form; the data will also be transferred electronically to the electronic database system of the study. The electronic database will be stored in secured hard drives with backups. The trial adheres to the Data Protection Act that requires the data anonymisation as soon as this is possible in the study.
Data Management
All data and information relating to the study subjects will be saved and recorded on password-protected computers and will only be accessible to the research team. A database will also enable the research team to track each participant’s progress throughout the trial, including the scheduling of visits and appointments.
Data Analysis and Statistical Analysis
Statistical analysis of the stored data, using the two-way analysis of variance test, will be performed at the end of the study. Where significant differences will be found, subsequent tests will be performed. Spearman’s test will be used to assess the associations between different parameters. P values < 0.05 will be considered to be statistically significant. Microsoft Office Excel (Microsoft Corp., Redmond, WA, USA), GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA) and SPSS (SPSS, IBM Corp., Armonk, NY, USA) software will be used for the statistical analysis assessment.
Discussion
The SAMAS study is designed to examine the potential and expected beneficial effects of the first oral GLP-1 receptor agonist, semaglutide, beyond its glycaemic control capabilities, in patients with T2D without manifested cardiovascular disease. To the best of our knowledge, this is the first study of this kind. More specifically, the primary goal of the SAMAS study is to evaluate the possible effect of oral semaglutide on cIMT reduction, endothelial function improvement and arterial stiffness reduction. Therefore, this is an exploratory study that evaluates these markers. We believe that GLP-1 receptor agonists with proven cardiovascular benefit exert their beneficial action by reducing atherosclerosis formation and progression, as previously discussed [19]. This is the reason why we included in our study not only the crude measurement of cIMT but also the evaluation of arterial wall parameters, such as endothelial function and arterial stiffness. This beneficial action is expected to be supported with additional beneficial effects on improvements in lipid profiles, mainly through reduction of the atherogenic small dense LDL subclasses, which have a specific physico-chemical composition and metabolic properties, including a reduced affinity for the LDL receptor, lower antioxidant capabilities and higher susceptibility to oxidation [32]. Small dense LDLs have greater arterial entry and retention abilities, and their oxidation in the intima is considered to represent the very first step of the atherosclerotic cascade [33]. Additionally, HbA1c improvement as well as reduction in hsCRP will be studied. Correlations between these findings are also to be sought.
Patients with diabetes are at significantly increased risk for cardiovascular complications compared to the general population. As this risk is only partially associated with glycaemic control, additional risk reduction strategies remain of outmost importance in this particularly vulnerable patient population; consequently, management approaches targeting optimal glycaemic control (as defined by individualized HbA1c) as well as multifactorial cardiovascular risk factors optimization have become the basis of the modern diabetes management [34]. The therapeutic possibilities in this field have become even more exciting with the availability of antidiabetic agents with proven beneficial effects on cardiovascular risk reduction. Namely, it has become evident that some GLP-1 receptor agonists and some SGLT-2 inhibitors provide significant cardiovascular risk reduction in patients with diabetes, regardless of the glycaemic control achieved [35]. These surprising and favourable findings have generated a complete change in the paradigm of T2D treatment perception. The term diabetes disease-modifying drugs has emerged, as it is perceived that these drugs could potentially change the progression of diabetes itself, allowing for good glycaemic control, which in turn leads to a reduction in microvascular complications and, importantly, a great reduction in cardiovascular risk, ultimately resulting in more chronic complication-free years and better quality of life [34]. This approach consists of metformin, as an everlasting pillar of antidiabetic therapy, in conjunction with either a GLP-1 receptor agonist, an SGLT-2 inhibitor or both. This simple approach is based on the relative safety of all the drugs used, not only in terms of adverse effects and kidney function, but also due to low or no hypoglycaemia risk. Based on all of the described benefits, there only remain local hurdles of social security institutions to be overcome for the present approach to come to life [2, 35].
Nevertheless, an obstacle for the proper use of GLP-1 receptor agonists in clinical practice may be the injectable form of these drugs, despite the clear international recommendations issued by both diabetologists and cardiologists [36]. It is known that the fear level of patients with diabetes increases with the use of injections, resulting in resistance to therapy intensification. We believe that this would significantly change with the emergence of the first oral GLP-1 receptor agonist, namely oral semaglutide. Applicability of the GLP-1 receptor agonist as a pill will allow for a simple initiation, maintenance and/or intensification of therapy, easing the fear of patients for injection therapy. Therefore, better adherence and less therapeutic inertia is expected with the use of oral semaglutide [35, 37]. Based on the authors' clinical practice experiences, we have observed that individuals generally do not fully understand the difference between non-insulin and insulin injections. Indeed, many patients perceive any injectable therapy as similar to insulin therapy, thus rejecting any injectable therapy. This has been proposed by many authors as an explanation for the slow adoption in clinical practice of the most recent recommendations from international scientific guidelines on diabetes, which have emphasized for several years the importance of early treatment with GLP-1 receptor agonists [36, 38]. We are aware that most patients usually prefer oral pills and not injections, with the majority afraid of needles, even if the injection is once weekly.
GLP-1 receptor agonists, particularly those that are analogous to the human GLP-1, have been proven to have cardio-renal protective actions (liraglutide, dulaglutide and semaglutide) [35]. Interestingly, oral semaglutide has been proven to provide cardiovascular safety without the superior cardiovascular risk reduction benefit that was shown for its injectable counterpart. Nevertheless, as both formulations have the same active substance, we suggest that this effect has not been proven due to the design of the very short and event-driven CVOT, PIONEER 6, which was primarily performed to prove its cardiovascular safety [39, 40]. Oral semaglutide showed a clear trend towards reducing the risk of MACE, by 21%, which was statistically nonsignificant compared to 26% for injectable semaglutide, both versus placebo [39]. Nevertheless, oral semaglutide provided 49% relative risk reduction in all-cause mortality versus placebo [39]. These numbers speak for themselves and merit evaluation in other studies, one of which, the SOUL study (NCT03914326), is well under way, to prove oral semaglutide’s superiority in cardiovascular risk reduction.
Based on all of the evidence to date, we believe there is sufficient ground to perform the SAMAS study with the oral formulation of semaglutide. Mechanisms of its cardiovascular protective action have not yet been explained. We believe that it might act through the improvement of the preclinical pathophysiologic changes associated with atherosclerosis, i.e. improvement of endothelial function and reduction of arterial stiffness, both of which represent the first functional atherosclerosis-associated changes in this complex process. Additionally, we believe that reduction of very atherogenic small dense LDL might also reduce the cIMT, which could be the first morphological atherosclerotic change [41]. Similarly, cIMT reduction has been proven for liraglutide [17]. However, conflicting results on the atherosclerosis process (cIMT reduction, plaque volume and composition) were published for exenatide, as expected [11,12,13].
If the results of the SAMAS study are positive, we believe that it will add an additional tile to the mosaic of modern T2D treatment approaches. We believe that treating diabetes with modern, high-quality drugs with proven benefits on glycemia control and cardiovascular risk reduction will provide long-term improvement in patients’ lives, delaying and preventing micro- as well as macrovascular complications and improving quality of life per se and, finally, allow for long-term reduction of healthcare expenditure (which is high in most countries).
The main limitation of this study is its single blinded design. Another potential limitation may be its relative short duration of 12 months, which may be too short for certain cardiovascular risk parameters to change significantly since the study population will not include subjects with overt cardiovascular diseases. Still, the proposed SAMAS study has several strengths:
-
This is the first study to investigate the direct anti-atherosclerotic effects of the oral semaglutide formulation (tablet) by an independent investigator study, without any role of the industry.
-
The study will investigate the direct and indirect mechanisms underlying the beneficial effects of oral semaglutide on arterial function.
-
The study will provide insight into the primary cardiovascular prevention capabilities of oral semaglutide since we will recruit patients without known cardiovascular diseases and naïve for treatment with antidiabetic drugs with proven cardiovascular benefit.
References
Einarson TR, Acs A, Ludwig C, Panton UH. Economic burden of cardiovascular disease in type 2 diabetes: a systematic review. Value Health. 2018;21(7):881–90. https://doi.org/10.1016/j.jval.2017.12.019.
Mosenzon O, Del Prato S, Schechter M, et al. From glucose lowering agents to disease/diabetes modifying drugs: a “SIMPLE” approach for the treatment of type 2 diabetes. Cardiovasc Diabetol. 2021;20(1):92. https://doi.org/10.1186/s12933-021-01281-y.
Cosentino F, Grant PJ, Aboyans V, et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J. 2020;41(2):255–323. https://doi.org/10.1093/eurheartj/ehz486.
Das SR, Everett BM, Birtcher KK, et al. 2020 Expert consensus decision pathway on novel therapies for cardiovascular risk reduction in patients with type 2 diabetes. J Am Coll Cardiol. 2020;76(9):1117–45. https://doi.org/10.1016/j.jacc.2020.05.037.
American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes—2021. Diabetes Care. 2021;44(Suppl 1):S111-S24. https://doi.org/10.2337/dc21-S009
Rizzo M, Nauck MA, Mantzoros CS. Incretin-based therapies in 2021—current status and perspectives for the future. Metabolism. 2021;122:154843. https://doi.org/10.1016/j.metabol.2021.154843.
Hjerpsted JB, Flint A, Brooks A, Axelsen MB, Kvist T, Blundell J. Semaglutide improves postprandial glucose and lipid metabolism, and delays first-hour gastric emptying in subjects with obesity. Diabetes Obes Metab. 2018;20(3):610–9. https://doi.org/10.1111/dom.13120.
Sun F, Wu S, Wang J, et al. Effect of glucagon-like peptide-1 receptor agonists on lipid profiles among type 2 diabetes: a systematic review and network meta-analysis. Clin Ther 2015;37(1):225–41.e8. https://doi.org/10.1016/j.clinthera.2014.11.008
Zhao X, Wang M, Wen Z, et al. GLP-1 receptor agonists: beyond their pancreatic effects. Front Endocrinol (Lausanne). 2021;12: 721135. https://doi.org/10.3389/fendo.2021.721135.
Sachinidis A, Nikolic D, Stoian AP, et al. Cardiovascular outcomes trials with incretin-based medications: a critical review of data available on GLP-1 receptor agonists and DPP-4 inhibitors. Metabolism. 2020;111: 154343. https://doi.org/10.1016/j.metabol.2020.154343.
Husain M, Bain SC, Jeppesen OK, et al. Semaglutide (SUSTAIN and PIONEER) reduces cardiovascular events in type 2 diabetes across varying cardiovascular risk. Diabetes Obes Metab. 2020;22(3):442–51. https://doi.org/10.1111/dom.13955.
Koska J, Migrino RQ, Chan KC, Cooper-Cox K, Reaven PD. The effect of exenatide once weekly on carotid atherosclerosis in individuals with type 2 diabetes: an 18-month randomized placebo-controlled study. Diabetes Care. 2021;44(6):1385–92. https://doi.org/10.2337/dc20-2014.
Patti AM, Nikolic D, Magan-Fernandez A, et al. Exenatide once-weekly improves metabolic parameters, endothelial dysfunction and carotid intima-media thickness in patients with type-2 diabetes: an 8-month prospective study. Diabetes Res Clin Pract. 2019;149:163–9. https://doi.org/10.1016/j.diabres.2019.02.006.
Nauck MA, Quast DR. Cardiovascular safety and benefits of semaglutide in patients with type 2 diabetes: findings from SUSTAIN 6 and PIONEER 6. Front Endocrinol (Lausanne). 2021;12:645566. https://doi.org/10.3389/fendo.2021.645566.
Marquis-Gravel G, Tardif JC. Glucagon-like peptide 1 receptor agonists, carotid atherosclerosis, and cardiovascular outcomes. Diabetes Care. 2021;44(6):1252–3. https://doi.org/10.2337/dci20-0076.
Anholm C, Kumarathurai P, Pedersen LR, et al. Liraglutide in combination with metformin may improve the atherogenic lipid profile and decrease C-reactive protein level in statin treated obese patients with coronary artery disease and newly diagnosed type 2 diabetes: a randomized trial. Atherosclerosis. 2019;288:60–6. https://doi.org/10.1016/j.atherosclerosis.2019.07.007.
Nikolic D, Giglio RV, Rizvi AA, et al. Liraglutide reduces carotid intima-media thickness by reducing small dense low-density lipoproteins in a real-world setting of patients with type 2 diabetes: a novel anti-atherogenic effect. Diabetes Ther. 2021;12(1):261–74. https://doi.org/10.1007/s13300-020-00962-3.
Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–22. https://doi.org/10.1056/NEJMoa1603827.
Rizzo M, Nikolic D, Patti AM, et al. GLP-1 receptor agonists and reduction of cardiometabolic risk: potential underlying mechanisms. Biochim Biophys Acta Mol Basis Dis. 2018;1864(9 Pt B):2814–21. https://doi.org/10.1016/j.bbadis.2018.05.012
Rakipovski G, Rolin B, Nohr J, et al. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE(-/-) and LDLr(-/-) mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci. 2018;3(6):844–57. https://doi.org/10.1016/j.jacbts.2018.09.004.
Hamal S, Cherukuri L, Shaikh K, et al. Effect of semaglutide on coronary atherosclerosis progression in patients with type II diabetes: rationale and design of the semaglutide treatment on coronary progression trial. Coron Artery Dis. 2020;31(3):306–14. https://doi.org/10.1097/MCA.0000000000000830.
Eliaschewitz FG, Canani LH. Advances in GLP-1 treatment: focus on oral semaglutide. Diabetol Metab Syndr. 2021;13(1):99. https://doi.org/10.1186/s13098-021-00713-9.
Rizzo M, Rizvi AA, Patti AM, et al. Liraglutide improves metabolic parameters and carotid intima-media thickness in diabetic patients with the metabolic syndrome: an 18-month prospective study. Cardiovasc Diabetol. 2016;15(1):162. https://doi.org/10.1186/s12933-016-0480-8.
General Electric Company Versana Club Europe (2021) Auto IMT: Assess atherosclerosis risk. https://www.versanaclub.net/emea/auto-imt (2021). Accessed 27 Nov 2021.
Kuvin JT, Patel AR, Sliney KA, et al. Assessment of peripheral vascular endothelial function with finger arterial pulse wave amplitude. Am Heart J. 2003;146(1):168–74. https://doi.org/10.1016/S0002-8703(03)00094-2.
Moerland M, Kales AJ, Schrier L, van Dongen MG, Bradnock D, Burggraaf J. Evaluation of the EndoPAT as a tool to assess endothelial function. Int J Vasc Med. 2012;2012:904141. https://doi.org/10.1155/2012/904141.
Bruyndonckx L, Radtke T, Eser P, et al. Methodological considerations and practical recommendations for the application of peripheral arterial tonometry in children and adolescents. Int J Cardiol. 2013;168(4):3183–90. https://doi.org/10.1016/j.ijcard.2013.07.236.
Van Bortel LM, Laurent S, Boutouyrie P, et al. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J Hypertens. 2012;30(3):445–8. https://doi.org/10.1097/HJH.0b013e32834fa8b0.
Tomiyama H, Yamashina A. Non-invasive vascular function tests: their pathophysiological background and clinical application. Circ J. 2010;74(1):24–33. https://doi.org/10.1253/circj.cj-09-0534.
Berneis K, Rizzo M, Stettler C, et al. Comparative effects of rosiglitazone and pioglitazone on fasting and postprandial low-density lipoprotein size and subclasses in patients with Type 2 diabetes. Expert Opin Pharmacother. 2008;9:343–9.
Berneis K, Rizzo M, Hersberger M, et al. Atherogenic forms of dyslipidaemia in women with polycystic ovary syndrome. Int J Clin Pract. 2009;63:56–62.
Rizzo M, Berneis K. Should we measure routinely the LDL peak particle size? Int J Cardiol. 2006;107(2):166–70. https://doi.org/10.1016/j.ijcard.2005.02.035.
Rizvi AA, Stoian AP, Janez A, Rizzo M. Lipoproteins and cardiovascular disease: an update on the clinical significance of atherogenic small, dense LDL and new therapeutical options. Biomedicines. 2021;9(11). doi: https://doi.org/10.3390/biomedicines9111579.
Lathief S, Inzucchi SE. Approach to diabetes management in patients with CVD. Trends Cardiovasc Med. 2016;26(2):165–79. https://doi.org/10.1016/j.tcm.2015.05.005.
Thethi TK, Bilal A, Pratley RE. Cardiovascular outcome trials with glucose-lowering drugs. Curr Cardiol Rep. 2021;23(7):75. https://doi.org/10.1007/s11886-021-01505-3.
Maranta F, Cianfanelli L, Rizzo M, Cianflone D. Filling the gap between Guidelines and Real World in the cardiovascular approach to the diabetic patients: the need for a call to action. Int J Cardiol. 2021;329:205–7. https://doi.org/10.1016/j.ijcard.2020.12.074.
Kalra S, Sahay R. A review on semaglutide: an oral glucagon-like peptide 1 receptor agonist in management of type 2 diabetes mellitus. Diabetes Ther. 2020;11(9):1965–82. https://doi.org/10.1007/s13300-020-00894-y.
Cardio-Metabolic Academy Europe East. Adoption of the ADA/EASD guidelines in 10 Eastern and Southern European countries: Physician survey and good clinical practice recommendations from an international expert panel. Diabetes Res Clin Pract. 2021;172:108535. https://doi.org/10.1016/j.diabres.2020.108535
Husain M, Birkenfeld AL, Donsmark M, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2019;381(9):841–51. https://doi.org/10.1056/NEJMoa1901118.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19):1834–44. https://doi.org/10.1056/NEJMoa1607141.
Rizzo M, Berneis K. Who needs to care about small, dense low-density lipoproteins? Int J Clin Pract. 2007;61(11):1949–56. https://doi.org/10.1111/j.1742-1241.2007.01596.x.
Acknowledgements
Funding
Sponsorship for this study is provided by University Medical Centre Ljubljana, Ljubljana, Slovenia, as a part of the tertiary project of the University Medical Centre Ljubljana (Grant No. 20210043). No funding or sponsorship was received for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal editors (ICMJE) criteria for authorship of this article, take responsibility for integrity of the work, and have given their approval for this version to be published.
Author Contributions
All authors contributed to the study conception and design. The first draft of the manuscript was written by Miodrag Janić, Manfredi Rizzo, Mojca Lunder and Andrej Janež. All authors commented on previous versions of the manuscript. All authors have read and approved the final version of the manuscript.
Disclosures
All the authors declare that they have no conflict of interests regarding this work. Miodrag Janić has received lecture fees from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Eli Lilly, Merck Sharp & Dohme, Novartis, Novo Nordisk, Pfizer, Sanofi and Viatris. Manfredi Rizzo is Editorial Board Member of Diabetes Therapy and former Director of the Clinical Medical & Regulatory Department, Novo Nordisk Europe East and South; he has given lectures, received honoraria and research support and participated in conferences, advisory boards and clinical trials sponsored by many pharmaceutical companies, including Amgen, Astra Zeneca, Boehringer Ingelheim, Kowa, Eli Lilly, Meda, Mylan, Merck Sharp & Dohme, Novo Nordisk, Novartis, Roche Diagnostics, Sanofi and Servier. Francesco Cosentino reports personal fees from AstraZeneca, Bayer, Boehringer-Ingelheim, Bristol-Myers Squibb, Merck Sharp & Dohme, Novo Nordisk and Pfizer; and grants from the Swedish Research Council, Swedish Heart & Lung Foundation and the King Gustav V and Queen Victoria Foundation. Anca Pantea Stoian has participated in scientific advisory boards and received consulting fees from Astra Zeneca, Amgen, Boehringer Ingelheim, Coca-Cola, Eli Lilly, Merck, Medtronic, Roche Diabetes, Novo Nordisk and Sanofi; and she is currently the Vice President of the National Committee of Diabetes. Mojca Lunder has received lecture fees from Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Merck Sharp & Dohme, Novo Nordisk and Sanofi. Mišo Šabović has received honoraria for participation on advisory boards for Amgen and Boehringer Ingelheim, and as a speaker for Amgen, Boehringer Ingelheim, Novartis, Pfizer, Eli Lilly and Janssen. Andrej Janež has served as a consultant and is on speakers’ bureaus for AstraZeneca, Boehringer Ingelheim, Eli Lilly, Merck Sharp & Dohme, Novo Nordisk and Sanofi. None of the above-mentioned pharmaceutical companies played any role in this article, which has been written independently, without any financial or professional help, and reflects only the opinion of the authors, without any role of the industry.
Compliance with Ethics Guidelines
The study was approved on 23 August 2021, by the Slovenian National Medical Ethics Committee (Approval No. 0120–245/2021/3) and will be conducted in accordance with the Declaration of Helsinki and Good Clinical practice guidelines. The study is supported by a grant of the University Medical Centre Ljubljana, Ljubljana, Slovenia (Grant No. 20210043). Participants will have to sign informed consent forms to participate.
Data Availability
The datasets generated and/or analysed during the study will be available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Janić, M., Rizzo, M., Cosentino, F. et al. Effect of Oral Semaglutide on Cardiovascular Parameters and Their Mechanisms in Patients with Type 2 Diabetes: Rationale and Design of the Semaglutide Anti-Atherosclerotic Mechanisms of Action Study (SAMAS). Diabetes Ther 13, 795–810 (2022). https://doi.org/10.1007/s13300-022-01226-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-022-01226-y