
Abstract
Maturity-onset diabetes of the young (MODY) is an unusual form of diabetes with specific features that distinguish it from type 1 and type 2 diabetes. There are 14 known subtypes of MODY, and mutations in three genes (HNF1A, HNF4A, GCK) account for about 95% of all MODY cases. Diagnosis usually occurs before the age of 25 years, although less frequent forms may occur more often—but not necessarily—later in life. The molecular diagnosis may tailor the choice of the most appropriate treatment, with the aim to optimize blood glucose control, reduce the risk of hypoglycemic events and long-term complications, and enable proper genetic counseling. Treatment is usually unnecessary for patients with mutations in the GCK gene, while oral hypoglycemic agents (generally sulphonylureas) are recommended for patients with mutations in the HNF4A and HNF1A genes. More recent data show that other glucose-lowering agents can be effective in the latter patients, and additional and alternative therapies have been proposed. Proper management guidelines during pregnancy have been developed for carriers of GCK gene mutations, but such guidelines are still a subject of debate in other cases, although some recommendations are available. The other subtypes of MODY are even more rare, and very little data are available in the literature. In this review we summarize the most pertinent findings and recommendations on the treatment of patients with the different subtypes of MODY. Our aim is to provide the reader with an easy-to-read update that can be used to drive the clinician’s therapeutical approach to these patients after the molecular diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Genetic testing plays a pivotal role in the management of maturity onset diabetes of the young (MODY), driving the choice of the treatment, the assessment of the risk for complications, and the need for genetic counseling. |
Patients with glucokinase (GCK)-MODY should not be treated, except during pregnancy and when the fetus is healthy. |
Sulphonylureas are effective in managing patients with HNF1A- and HNF4A–MODY, while additional treatment with other oral hypoglycemic agents may improve blood glucose control. |
Avoid treating carriers of HNF1A and HNF4A mutations with sulphonylureas during pregnancy. |
Introduction
Maturity onset diabetes of the young (MODY; MIM# 606391) is an uncommon form of diabetes mellitus caused by a defect in a single gene and characterized by impaired insulin secretion. MODY is a genetically and clinically heterogeneous disease affecting 1–6% of patients with diabetes [1, 2]. It is an autosomal dominant disorder, but a de novo mutation should be taken into consideration in those patients without a familial history of diabetes but with clinical and biochemical findings highly suggestive of MODY. The disease is characterized by three main features: (1) mild hyperglycemia or overt diabetes in at least three consecutive generations; (2) onset usually before the age of 25 years; (3) absence of islet autoantibodies and lack of characteristics of type 2 diabetes (i.e., insulin resistance, obesity).
To date, 14 subtypes of MODY have been identified and characterized; these differ in terms of gene mutation, age at onset, treatment, and pattern of hyperglycemia (Table 1). Some extra-pancreatic features can be used as markers of specific subtypes of MODY, such as the presence of macrosomia and neonatal hypoglycemia in subtype HNF4A–MODY or renal cysts in subtype HNF1B-MODY (Table 2). Some subtypes of MODY are characterized by stable levels of blood glucose throughout the patient’s lifespan, others by a progressive worsening of insulin secretion and glucose control, and still others are associated to extra-pancreatic features. Furthermore, patients with some subtypes of MODY are prone to develop micro- and macrovascular complications while those with other subtypes do not, and it is this characteristic which drives the decision to treat or not as early as childhood. Interestingly, the “glycemic” phenotype can vary among carriers of the same mutations and even within the same pedigree. Achievement of target glycemic control reduces the risk of onset and progression of microvascular complications, which can be detected early in adolescents with diabetes mellitus [3, 4].
Most cases of MODY are due to mutations in three genes (HNF1A, GCK, and HNF4A), with a detection rate that varies among different study populations. Progress in genetic testing, facilitated by the development of new techniques (i.e., Next Generation Sequencing) and increased accessibility to genetic testing facilities, enables clinicians to establish the correct molecular diagnosis, thereby avoiding misdiagnosis with type 1 or type 2 diabetes.
A correct diagnosis is closely linked to important treatment benefits, such as a more accurate prognosis of the risk of complications, avoidance of stigma and limitations to the patients, and appropriate genetic counseling for relatives and offsprings, but most importantly it drives the choice of the best treatment. The tailoring of medical treatment to the characteristics of each patient has been designated “Precision Medicine” [5]. Precision medicine refers to the use of combined knowledge of a person to predict: (1) susceptibility to a particular disease; (2) etiologic mechanisms and prognosis of the disease, (3) response to a specific treatment. Being able to determine with some degree of accuracy the most appropriate treatment is associated with cost-saving and the avoidance of ineffective therapy and its possible side effects. Specifically for diabetic patients, precision medicine refers to determining the most appropriate method for self-monitoring blood glucose and avoiding the burden of insulin injections when unnecessary.
Genetic testing can be not only expensive, but also time- and money-consuming. Diabetologists need to increase their expertise in this area, and suspected cases should be referred to a specialist in monogenic diabetes or a clinical genetist working in this field to maximize the detection rate. Results from recent simulation modeling suggest that testing for MODY genes is cost-effective in selected patients [6]. An online calculator to assess the probability of MODY is currently available [7].
We have reviewed the pertinent literature on therapeutical approaches to MODY patients with the aim to provide the reader with a quick and easy-to-use guide for clinical practice.
Methods
A systematic literature review of the PubMed database up to 15 April 2020 was performed independently by two of the authors (CP and MD) to identify relevant papers. The search terms were “MODY” OR “maturity-onset diabetes of the young” AND “treatment” or “therapy.” Only papers on the 14 subtypes of MODY were considered for inclusion in this review; any other genetic form of diabetes was not considered. The search included guidelines, reviews, clinical trials, observational studies, and case reports. Potentially relevant papers were initially evaluated by checking the title and abstract, and all eligible studies were retrieved. For the purpose of this review, we considered eligible all papers that clearly stated that the diagnosis of MODY was genetically confirmed, defined the treatment, and reported data about glucose control and outcomes. Additional papers were identified by a manual search of the references from the retrieved articles. Non-English language manuscripts were not included in the search and review.
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
HNF4A–MODY (MIM# 125850) or MODY1
Genetic Defect and Pathophysiological Effects
Hepatocyte nuclear factor-4-alpha (HNF4A) is a member of the nuclear receptor family of transcription factors and is encoded by the HNF4A gene. It is a DNA-binding protein that is found in abundance in the liver, where it regulates genes involved in lipid metabolism and hepatic gluconeogenesis. It controls the expression of transcription factor HNF1. Mutations in the HNF4A gene cause a reduction in insulin secretion.
Clinical Presentation
MODY1 is characterized by a steady increase of blood glucose over time. The beta-cell response to glucose stimulus is decreased as the insulin secretion is impaired by the defect in the transcription factor. Glucagon secretion is also impaired. Due to progressive worsening of blood glucose control, these patients may present the full spectrum of diabetes complications. Microvascular complications, in particular those involving the retina and kidneys, are as common as in patients with type 1 or type 2 diabetes and are related to overall glycemic control [1, 8].
Extra-Pancreatic Associations
One-half of patients with MODY1 are macrosomic at birth, and some present diazoxide-responsive neonatal hyperinsulinemic hypoglycemia [9]. A deficiency of HNF4A also affects the biosynthesis of apolipoproteins and triglycerides, with the result being lowered serum concentrations [10]. An atypical form of Fanconi syndrome, including hypercalciuria and nephrocalcinosis, is present in carriers of the p.Arg76Trp (R76W) mutation of HNF4A [11].
Management Plan
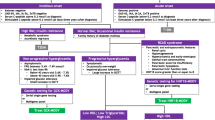
Patients with MODY1 display a relatively normal glucose tolerance during the first decade of life, with testing, and subsequent diagnosis, generally based on familial screening or incidental hyperglycemia during intercurrent illness. Glucose intolerance generally becomes evident during adolescence or early adulthood with hormonal changes causing insulin resistance and derangement of glucose control. Clinical heterogeneity is reported even within the same pedigree [12]. At diagnosis and at the first stage of the disease, when the blood glucose and the glycated hemoglobin (HbA1c) levels are still in the “non-diabetic” range, a reasonable strategy is to manage these patients by placing them on a low carbohydrate diet. These patients are prone to develop marked hyperglycemia after oral glucose load [13], and thus a low carbohydrate diet may reduce the postprandial blood glucose level. Glucose control shows a progressive deterioration with increasing HbA1c. This worsening may last several years, and there is no a common agreement among clinicians on when to switch the patient from management with diet to oral hypoglycemic agents (OHAs). Kyithar et al. [14] reported optimal blood glucose control in two patients on metformin after 15.5 years of diabetes (HbA1c 6.15%), but these authors did not report the dose or any data on the age of the patients. In the same article, the authors describe seven other patients with HNF1A–MODY, four of whom were newly diagnosed (HbA1c 5.23%) and managed only with diet, one whose disease did not respond to the maximum dose of metformin and sulphonylurea and was switched to insulin injections, and two who were on insulin before the molecular diagnosis and continued on insulin by choice (HbA1c 7.2%). Based on our experience, diet is a reasonable treatment approach in children and adolescents with HNF4A–MODY [15]. When the diet appears to be ineffective, switching to sulfonylureas, which achieves better glycemic control than insulin, is suggested [1].
Sulfonylureas bind to the sulphonylurea receptor 1 (SUR1), a subunit of the ATP-dependent potassium channel of the pancreatic beta-cell, closing the channel which leads to depolarization of the membrane. This change triggers the opening of the voltage-dependent calcium channels, leading to an increased influx of calcium, which mediates the fusion of the vesicles of insulin with the cell membrane, yielding the release of the contents [16, 17]. Through this mechanism, sulphonylurea derivatives bypass the dysfunction due to the defect in the gene and restore the beta-cell response to glucose stimulus.
The first aim of the treatment is to avoid hypoglycemia; thus, an initial dose of one-quarter that of the normal initial dose in adults, to be progressively increased on the basis of blood glucose control, is suggested. To further reduce the risk of hypoglycemia, a slow-release preparation may be prescribed and, when OHAs fail, insulin injections [1].
When to Suspect
HNF4A–MODY should be suspected with presentation of familial symptomatic diabetes, although it is not the most frequent cause. Large-for-gestational age with neonatal hyperinsulinemic hypoglycemia and low levels of apolipoproteins and triglycerides may drive the choice for genetic testing. Glucose intolerance usually becomes evident during adolescence or early adulthood.
Pregnancy
In healthy pregnant women, inheritance of a paternal HNF4A gene mutation results in increased birth weight (790 g more than unaffected siblings) with macrosomia-related risks [18]. Regular monitoring of fetal growth with ultrasonography is recommended after 28 weeks of gestation, and early delivery should be considered [9] (Table 3).
The appropriate treatment for pregnant women with HNF4A–MODY should be carefully evaluated. Glibenclamide, a sulfonylurea, crosses the placenta, and its use in pregnancy increases the risk of large-for-gestational-age neonates and neonatal hypoglycemia. No teratogenic effects have been reported for this medication, but it is contraindicated during pregnancy and breastfeeding as well. It should be noted that achieving optimal glycemic control during the first trimester to avoid fetal malformations is the main treatment goal for all women with diabetes. It has been suggested that in the pre-pregnancy phase, these patients could be switched to insulin, which can be continued during pregnancy. Alternatively, the switch to insulin could be made at the end of the second trimester of pregnancy, even if titration of the insulin dose is more difficult at this stage of pregnancy. Fetal growth should be monitored at 2-week intervals after 28 weeks of gestation with ultrasound. Elective caesarean section or induction of labor should be planned between 35 and 38 weeks [9]. The mother can be switched to her previous sulphonylurea medication once she stops breastfeeding.
GCK–MODY (MIM# 125851) or MODY2
Genetic Defect and Pathophysiological Effects
Glucokinase (GCK) is an hexokinase that regulates the phosphorylation of glucose at the sixth carbon position, the first step in glycolysis, converting glucose into glucose-6 phosphate. This enzyme has unique functional characteristics, and plays a pivotal role in the regulation of glucose metabolism.
Clinical Presentation
GCK–MODY is characterized by mild non-progressive hyperglycemia. In persons with GCK–MODY, the regulation of insulin secretion occurs at a higher set point than in the normal population, resulting in impaired fasting glucose and impaired glucose tolerance. HbA1c is usually below 7.3–7.5% [19, 20]. Patients are almost asymptomatic as blood glucose is below the level that causes osmotic symptoms. Diagnosis is often made after an incidental finding of hyperglycemia or with familial screening. In adulthood, GCK–MODY is often misdiagnosed as type 2 diabetes and represents a possible cause of “gestational-like” diabetes. The use of a diagnostic flow-chart may significantly increase the detection rate of GCK mutations [7, 21].
Diabetes-Related Complications
Carriers of GCK mutations are characterized by a particularly low rate of vascular complications, substantially less than those associated with other forms of diabetes [22, 23]. Steele et al. [23] reported that the prevalence of clinically significant microvascular complications (i.e., a grade of severity higher than background retinopathy or persistent microalbuminuria or proteinuria) was 1% (1 subject with persistent microalbuminuria) among the 99 GCK–MODY patients (median HbA1c 6.9%, median age 48.6 years) in their study, which did not differ from the percentage detected in the 89 healthy controls (2 subjects with persistent microalbuminuria or 2%; median HbA1c 5.8%). In this same study, 30% of GCK patients had background retinopathy (27/90, 22 with 1–4 microaneurisms based on digital retinal imaging) as compared to 14% of the healthy controls (all with 1–4 microaneurisms). None of the patients with GCK mutations had severe eye complications (i.e., preproliferative/proliferative retinopathy or maculopathy) compared to 21.7% of patients with type 2 diabetes. Microalbuminuria and neuropathies were equally frequent (1 and 2%, respectively) as in the control population (2 and 0%), and the rate of macrovascular complications was 4% (11% in controls) [23].
Extra-Pancreatic Associations
No extra-pancreatic associations have been reported.
Management Plan
Taking into account data currently available on the outcomes, the treatment of GCK–MODY is usually unnecessary, as the long-term outcomes are similar to those of healthy persons. The findings by Stride et al. [24] support this statement, as they pointed out that at referral for genetic testing 13.5 and 7.5% of 799 patients were on OHAs and insulin, respectively, without any difference in HbA1c levels between treated (6.5%) and untreated (6.4%) patients. In a small subgroup of six patients on OHAs and ten on insulin, the treatment was stopped without any change in HbA1c levels after 3 months. More recently, Hohendorff et al [25] evaluated the efficacy of SGLT-2 medications and showed that a single dose of dapagliflozin (10 mg) induces higher glycosuria in patients with GCK–MODY than in those with type 2 diabetes.
Data from the literature support the clinical evidence that the molecular diagnosis of GCK–MODY is not needed to decide upon the best treatment, but it is needed to stop treatment in patients on insulin or OHAs or to support the decision not to start any treatment at all [1]. In all of these cases, the result is a decrease of medical surveillance.
When to Suspect
GCK–MODY should be suspected in patients presenting with incidental asymptomatic mild hyperglycemia without pancreatic autoimmunity. In adulthood, it is often misdiagnosed as type 2 diabetes and could cause “gestational-like” diabetes.
Pregnancy
It is important that GCK–MODY be correctly diagnosed in pregnant women for correct management. Its prevalence is quite variable, ranging from 2 to 6% of all cases of gestational diabetes mellitus [26, 27], but depends on the screening criteria. In the “Atlantic DIP” study [26], the estimated prevalence in pregnant women with gestational diabetes mellitus was < 1%, but in a more selected population it was up to 80% [28]. A correct diagnosis during pregnancy is very important in terms of defining the most appropriate treatment for fetus health.
If both the mother and the fetus carry a GCK mutation, both will have the same glucose set point, and the result will be a normal birth weight if maternal hyperglycemia is not treated [29, 30]. Insulin treatment is not recommended in this case since it can potentially reduce fetal growth [31]. If the fetus is healthy (i.e., no GCK mutation) and the woman carries a GCK mutation, the fetus will be exposed to maternal hyperglycemia and will present the typical features of an infant of diabetic mother (i.e., large for gestational age, minor malformations, neonatal hypoglycemia). In this case, a large dose (> 1 U/kg per day) of insulin to lower maternal blood glucose is recommended [31, 32], and induction at 38 weeks with monitoring for neonatal glycemia should be considered [33].
HNF1A–MODY (MIM# 600496) or MODY3
Genetic Defect and Pathophysiological Effects
Hepatocyte nuclear factor-1-alpha (HNF1A) is a homodiameric transcription factor that is required for the expression of several liver-specific genes. Mutations in the HNF1A gene can result in impaired dimerization, causing a deregulation of the molecular mechanisms and leading to diabetes mellitus. The pathophysiologic mechanisms are very similar to those of HNF4A–MODY since HNF4A regulates the expression of HNF1A.
Clinical Presentation
The glycemic pattern of HNF1A–MODY is characterized by mild fasting hyperglycemia and very high glucose concentrations following glucose administration. In patients with HNF1A–MODY, insulin secretion progressively decreases and glucose control tends to worsen over time, resulting in the need for treatment, with 63% of patients developing diabetes before 25 years of age, 79% before age 35 years, and 96% before age 55 years [34]. The renal glucose threshold is lower in patients with HNF1A–MODY than in the healthy population. The location of the mutation determines the age at diagnosis; for example, patients with mutations of terminal exons 8–10 are diagnosed with HNF1A–MODY 8 years later than those with mutations in exons 1–6 [1].
Diabetes-Related Complications
Patients with HNF1A–MODY may have the full spectrum of micro- and macrovascular complications, involving both the retina and kidneys, at a similar frequency as in patients with type 1 and type 2 diabetes [1]. The development of such complications is related to the degree of metabolic control [35].
Extra-Pancreatic Associations
In HNF1A–MODY, the renal tubular transport of glucose is impaired, causing a low renal threshold for glucose reabsorption. Patients with MODY3 present with glycosuria before developing significant hyperglycemia [36].
Management Plan
Treatment of patients with HNF1A-MODY depends on age and HbA1c level. If HbA1c is < 6.5%, the first-line therapy consists of a diet without saccharides; otherwise, sulphonylureas may be considered [37].
The glucose-stimulated insulin secretion is impaired due to reduced beta-cell glucose uptake that decreases the intracellular ATP levels [16, 24]. In turn, ATP-sensitive potassium channels are open and, consequently, cell-membrane depolarization and insulin release are prevented [38]. The International Society for Pediatric and Adolescent Diabetes/International Diabetes Federation [1] and the American Diabetes Association [2] guidelines recommend sulphonylureas as first-line treatment since they stimulate insulin release from beta-cells, thereby improving glycemic control [39, 40].
In a randomized crossover study conducted nearly 20 years ago, Pearson et al. [41] showed that patients with HNF1A-MODY have about a fivefold greater response to gliclazide (80 mg twice daily) than to metformin (1 g twice daily) and about a fourfold greater response to gliclazide than patients with type 2 diabetes. In addition, the insulin secretory response to intravenous tolbutamide was found to be higher than that to intravenous glucose, with a better response, even compared to that in patients with type 2 diabetes. Taking these data into account, Shepherd et al. [42] switched eight patients (age 34 years; median time on insulin 20 years) from 0.5 U/kg per day of insulin to a median dose of 80 mg/day of gliclazide, and achieved a median reduction of HbA1c of 0.8%.
Regarding sulphonylurea derivatives, their known safety and long-term efficacy, associated improvement in quality of life, and better patient compliance are well established [43]. Thus, trial therapy with a sulphonylurea in hyperglycemic patients who are carriers of HNF1A mutations is mandatory. Factors influencing the success of sulphonylurea therapy include the duration of diabetes, initial HbA1c value, and weight gain [8]. Similarly to HNF4A–MODY, the first aim of the therapeutic regimen is to avoid hypoglycemia; thus, an initial dose of one-quarter of the starting dose in adults, to be progressively increased on the basis of blood glucose control, is suggested. A slow-release preparation may be suggested to further reduce the risk of hypoglycemia, and insulin injections should be started when glycemic control is not achieved with OHAs [1].
The insulinotropic effect of sulfonylureas and the normal to high insulin sensitivity typical of these patients [16, 44] exert a glucose-lowering effect [41, 45]. It has been hypothesized, but not confirmed [46], that the hepatic clearance of some sulphonylurea derivatives in patients with mutations in the HNF1A and HNF4A genes decreases, with the result that their serum level increases. The elevated circulating levels of these medications would then account for their enhanced efficacy [45, 47].
Primary failure of sulphonylurea medications is almost rare and has actually been detected in only a few patients carrying the c.618G->A mutation [48]. The development of absolute insulinopenia due to secondary failure has been reported after 3–25 years [49], with a decrease in insulin secretion of about 1% per year of treatment.
There are some limitations to the use of sulfonylureas, such as the need for additional glucose-lowering treatment and overall increased risk of hypoglycemia [50, 51]. This has led to to evaluations of the efficacy of other OHAs, with the aim to reduce the risk of hypoglycemic events and improve blood glucose control and variability. Tuomi et al. [50] showed that in adult patients meglitinide analogues, a class of oral antidiabetic agents, reduce the postprandial glucose levels and the risk of hypoglycemic episodes compared to sulphonylureas. A case report which described the use of meglitinides in three adolescents also emphasized the underlying efficacy and safety of this therapy [52]. However, in pediatric patients, the use of meglitinides has not yet been reviewed, and the administration of any secretagogues is still off-label.
Noting the impaired effect of incretin, a gut hormonal factor [51], various research groups have studied the efficacy of the GLP-1 receptor agonist (GLP-1 RA). Docena et al. [53] showed in three patients belonging to the same family that the addition of once-daily liraglutide therapy improved glycemic control, reducing the risk of hypoglycemia and weight gain. More recently, the switch from sulphonylurea to once-weekly GLP-1 RA as monotherapy in a 27-year old patient with inadequate glucose control was described in a case report; the effect of this switch was optimal glycemic control without hypoglycemia for more than 1 year [54]. The effectiveness of GLP-1 RAs has also been evaluated in 16 patients (mean age 39 years, HbA1c 6.46%) in a 6-week double-blind, randomized, crossover trial [55]. The authors of this trial showed that the sulfonylurea glimepiride (up-titrated once weekly in a treat-to-target manner) was more effective in reducing fasting plasma glucose and postprandial glucose excursions than the GLP-1RA liraglutide (up-titrated once weekly to 1.8 once a day), but that the former treatment was associated with a greater risk for hypoglycemic events [55]. A trial with exogenous gastric inhibitory polypeptide (GIP) and GLP-1 infusions showed a potentiation of the sulfonylurea-induced insulin secretion [56]. Interesting and effective data might be provided by an ongoing randomized, double-blinded, placebo-controlled, crossover trial with a combination of the sulfonylurea glimepiride and the dipeptidyl peptidase 4 (DPP-4) inhibitor linagliptin versus glimepiride monotherapy [57].
Sodium-glucose co-transporter-2 (SGLT-2) inhibitors have also been tested. It has been proven that a single dose of the SGLT-2 inhibitor dapagliflozin (10 mg) as adjuvent in standard treatment induces higher glycosuria in patients with HNF1A–MODY than in those with type 2 diabetes. Its possible role as an additional therapeutic long-term option has not been evaluated to date [25].
When to Suspect
This subtype of MODY is the most frequent cause of familial symptomatic diabetes, and is about tenfold more frequent than HNF4A–MODY. HNF1A–MODY is more frequent in adulthood than in childhood [58]. Euglycemic glycosuria should prompt a suspicion of this subtype of diabetes, which usually becomes evident during adolescence or early adulthood [1].
Pregnancy
Neonates with mutations in the HNF1A gene do not differ with unaffected neonates in terms of any difference in birth weight and frequency of neonatal hypoglycemic, suggesting that insulin secretion is not affected in fetuses carrying HNF1A gene mutations [9]. Similarly to HNF4A–MODY, there is inconclusive data on treatment during pregnancy. The suggested management of these cases is the same as for HNF4A–MODY, with elective caesarean section or induction of labor to be planned between 35 and 38 weeks of gestation [18].
PDX1–MODY (MIM# 606392) or MODY4
PDX1–MODY is a rare mild form of monogenic diabetes caused by heterozygous mutations in the PDX1 gene, which encodes the transcription factor PDX1 (also known as insulin promoter factor-1 [IPF-1]) which is crucial for pancreatic and beta-cell development and function [59]. Mutations in this gene seem to exert a dominant-negative inhibition of transcription of the insulin gene, leading to diabetes. Patients with PDX1–MODY present an early-onset type 2 diabetes mellitus without extra-pancreatic associations. PDX1 plays an important role in glucose-stimulated insulin release, particularly in postprandial hyperglycemia [60] and indirectly impairs the incretin pathway. OHAs have been suggested as therapy [61]. The effectiveness of metformin [62] and DPP-4 inhibitors [63] has been reported in case reports.
HNF1B–MODY (MIM# 606391) or MODY5
Genetic Defect and Pathophysiological Effects
The transcription factor HNF1B is a member of the homeodomain-containing superfamily of transcription factors and is expressed in different tissues. It acts either as a homodimer or as a heterodimer with HNF1A.
Clinical Presentation
HNF1B–MODY, also known as renal cysts and diabetes syndrome (RCAD), occurs more frequently in isolated cases rather than in the context of familial diabetes due to the de novo origin of the mutations. Differently from the other subtypes of monogenic diabetes, which are associated to point mutations, large genomic rearrangements and gene deletions account for about one-third to two-thirds of HNF1B–MODY cases [64, 65]. Onset occurs typically during adolescence or early adulthood. Neonatal onset has been reported.
Diabetes-Related Complications
There is little mention of chronic complications in literature but they may as common as in other types of diabetes and related to glycemic control [66]. However, long-term data are missing.
Extra-Pancreatic Associations
HNF1B–MODY shows involvement of the pancreas, which takes on a hypoplastic appearance with insulin deficiency, and of the genitourinary tract, with renal cysts or renal dysplasia; uterine malformations are also present. Increased uric acid levels and liver involvement with higher liver enzyme levels have also been described. The penetrance is incomplete, and thus the phenotype varies broadly [1].
Management Plan
Patients with HNF1B–MODY present some degree of hepatic insulin resistance [67], thus the response to sulfonylureas is not satisfactory and early insulin therapy may be required [1]. In a cohort of 201 patients, Dubois-Laforgue et al. [66] reported that 49% started insulin at diabetes onset and 79% at follow-up due to worsening of glucose control on a diet or OHAs. In a cohort of patients, treatment with a sulphonylurea or the meglitinide repaglinide was started shortly after diagnosis (8 months) and was successful in 57% of patients, lowering the HbA1c from 7.1 to 6.1% and improving the residual beta-cell function after 5 years; however, the switch from insulin to a sulphonylurea or repaglinide was successful only in three of ten patients. Carrillo et al. [68] reported on a patient who initiated treatment with insulin and then switched to OHAs after reaching an excellent metabolic control (HbA1c 6.3–6.5%); the switch was to glimepiride followed by sitagliptin as add-on, and very good metabolic control (HbA1c about 6.5%) was achieved for 6 years. Thereafter, HbA1c increased and insulin treatment was started again.
When to Suspect
Patients with HNF1B-MODY rarely present with only diabetes [69]; rather, extra-pancreatic features associated with the renal developmental disorders (overall renal cysts and renal dysplasia) are present in almost all patients and are the main evidence in children, even if blood glucose is normal [64]. This diagnosis should be suspected in the case of renal cysts and diabetes syndrome, also when a familial history is absent.
Pregnancy
No data on care during pregnancy are available in literature, but insulin treatment could be a reasonable option.
NEUROD1–MODY (MIM# 606394) or MODY6
NEUROD1–MODY is very rare. The transcription factor Neurogenic differentiation 1 (NEUROD1) is involved in the activation of insulin transcription, binding and activating the promoter of SUR1, GCK, the glucose-6-phosphatase catalytic subunit-related protein, and PAX6. Disease severity may be different within the same pedigree and require different treatment. Age at onset of diabetes is variable (from adolescence to 60 years s), and there are both obese and non-obese patients. A defect in the early phase of insulin secretion, without insulin dependence, leading to diabetic ketoacidosis or ketosis may be the only defect present [70].
Insulin therapy is commonly used, but MODY6 shows incomplete penetrance of diabetes; thus, management with OHAs and diet only appear to be effective in half of the MODY6 patients described in the literature [71]. Abreu et al. [72] described a patient treated with insulin and then with glimepiride and metformin, with satisfactory glycemic control.
Patients with NEUROD1–MODY may develop microvascular complications, whose severity ranges from mild to serious (proliferative retinopathy and renal failure) [71].
KLF11–MODY (MIM# 610508) or MODY7
CEL–MODY (MIM# 609812) or MODY8
CEL-MODY is caused by heterozygous mutations in the CEL gene and involves endocrine and exocrine pancreatic function [75]. In the absence of evidence on other pharmacological approaches, insulin seems to be the most reasonable treatment.
PAX4–MODY (MIM# 612225) or MODY9
PAX4–MODY is caused by mutations in the PAX4 gene, which encodes a transcription factor that influences the generation, differentiation, development, and survival of insulin-producing beta-cells. Plengvidhya et al. [76] were the first to describe patients with PAX4 mutations. At the time of molecular diagnosis these patients were treated with diet or OHAs [76]. In a later case report, affected patients required insulin following diagnosis [77].
BLK–MODY (MIM# 191305) or MODY11
BLK–MODY has incomplete penetrance, thus not all the carriers present with diabetes. It has been suggested that environmental and genetic modifiers play a significant role in BLK–MODY and that body weight is the most important factor that triggers hyperglycemia [80]. Pregnancy is also likely to exert a role in causing hyperglycemia [81]. Mostf patients require insulin, but some of them may be treated with diet or OHAs [82].
ABCC8–MODY or MODY12
ABCC8–MODY is caused by mutations in the ATP-sensitive potassium channel, which regulates insulin secretion. While mutations in the ABCC8 gene are more often associated with neonatal diabetes, some gain-of-function and loss-of-function mutations can cause transient neonatal hyperinsulinism with diabetes later in life [83, 84]. Most of these patients are misdiagnosed as having other types of diabetes and erroneously treated with insulin, which may result in poor control and episodes of hypoglycemia. Rafiq et al. [85] suggested that all carriers of ABCC8 mutations could be switched to sulphonylureas, also in adulthood. More recently, Reilly et al. [86] described ten patients with a mean age of 33.8 years at diagnosis treated with insulin or diet or metformin at recruitment. The HbA1c value was 6.9% after the molecular diagnosis and 6.2% after the switch to the anti-diabetic medication gliclazide (mean daily dose 36.87 ± 32.2 mg). Three patients stopped insulin (mean duration of diabetes 10.6 years) and started on therapy with a sulphonylurea, achieving a reduction in HbA1c.
KCNJ11–MODY (MIM# 616329) or MODY13
KCNJ11–MODY is caused by an impaired function of the potassium inwardly-rectifying channel. Mutations in the KCNJ11 gene are typically associated to permanent neonatal diabetes, and patients can be safely and effectively treated with high-dose sulphonylurea for a long time [87]. Patients with KCNJ11–MODY carry the p.Glu227Lys mutation, and diabetes occurs from the second decade of life onwards. These patients can be ideally treated with sulphonylurea [88].
APPL1–MODY (MIM# 604299) or MODY14
APPL1–MODY has been reported just in one paper describing two families with 24 carriers, of whom seven were on insulin, four were following a diet, and two were on OHAs. APPL1–MODY mostly develops after 40 years of age [89].
Conclusion
Maturity-onset diabetes of the young is a subtype of monogenic diabetes caused by mutations in at least 14 known genes. The phenotype may feature mild non-progressive hyperglycemia, progressive hyperglycemia, and hyperglycemia with extra-pancreatic features. In patients with mild hyperglycemia at diagnosis, diet seems to be a reasonable and effective therapeutic strategy in most cases; however, in the event of progressive hyperglycemia a pharmacological approach should be attempted.
Molecular diagnosis is pivotal to choosing the best treatment for most MODY patients, while for carriers of GCK mutations, molecular diagnosis is fundamental to the choice of not treating patients without any negative effect on prognosis and decreasinge medical surveillance.
Oral hypoglycemic agents and, in particular, sulphonylureas which bypass the molecular defect and activate the ATP-sensitive potassium channel, are effective in most patients with MODY, although not in patients with INS–MODY. HNF1A–MODY and HNF4A–MODY are particular responsive to sulphonylurea, which provides optimal blood glucose control. In patients with HNF1A–MODY, recent data suggest that other OHAs may be more effective in reducing the blood glucose variability. In the other MODY subtypes, different OHAs have been used, but the limited number of treated patients does not allow definitive recommendations to be made.
References
Hattersley AT, Greeley SAW, Polak M, et al. ISPAD Clinical Practice Consensus Guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19(Suppl 27):47–63.
American Diabetes Association. Classification and diagnosis of diabetes: standards of medical care in diabetes—2020. Diabetes Care. 2020;43(Suppl 1):S14–31.
El Samahy MH, Matter RM, Youssef OI, et al. Relation between carotid intima media thickness and oxidative stress markers in type 1 diabetic children and adolescents. J Diabetes Metab Disord. 2013;12(1):50.
Faienza MF, Acquafredda A, Tesse R, et al. Risk factors for subclinical atherosclerosis in diabetic and obese children. Int J Med Sci. 2013;10(3):338–43.
Hattersley AT, Patel K. Precision diabetes: learning from monogenic diabetes. Diabetologia. 2017;60(5):769–77.
Naylor RN, John PM, Winn AN, et al. Cost-effectiveness of MODY genetic testing: translating genomic advances into practical health applications. Diabetes Care. 2014;37(1):202–9.
Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia. 2012;55(5):1265–72.
Bacon S, Kyithar MP, Rizvi SR, et al. Successful maintenance on sulphonylurea therapy and low diabetes complication rates in a HNF1A-MODY cohort. Diabet Med. 2016;33(7):976–84.
Pearson ER, Boj SF, Steele AM, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PloS Med. 2007;4:e118.
Pearson ER, Pruhova S, Tack CJ, et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia. 2005;48(5):878–85.
Hamilton AJ, Bingham C, McDonald TJ, et al. The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a beta cell phenotype. J Med Genet. 2014;51(3):165–9.
Delvecchio M, Di Paola R, Mangiacotti D, et al. Clinical heterogeneity of abnormal glucose homeostasis associated with the HNF4A R311H mutation. Ital J Pediatr. 2014;40:58.
Stride A, Hattersley AT. Different genes, different diabetes: lessons from maturity-onset diabetes of the young. Ann Med. 2002;34(3):207–16.
Kyithar MP, Bacon S, Pannu KK, et al. Identification of HNF1A-MODY and HNF4A–MODY in Irish families: phenotypic characteristics and therapeutic implications. Diabetes Metab. 2011;37(6):512–9.
Delvecchio M, Mozzillo E, Salzano G, et al. Monogenic diabetes accounts for 6.3% of cases referred to 15 Italian pediatric diabetes centers during 2007 to 2012. J Clin Endocrinol Metab. 2017;102(6):1826–34.
Pearson ER, Velho G, Clark P, et al. Beta-cell genes and diabetes: quantitative and qualitative differences in the pathophysiology of hepatic nuclear factor-1alpha and glucokinase mutations. Diabetes. 2001;50(Suppl 1):S101–7.
Hansen T, Eiberg H, Rouard M, et al. Novel MODY3 mutations in the hepatocyte nuclear factor-1α gene: evidence for a hyperexcitability of pancreatic β-cells to intravenous secretagogues in a glucose-tolerant carrier of a P447L mutation. Diabetes. 1997;46(4):726–30.
Shepherd M, Brook AJ, Chakera AJ, Hattersley AT. Management of sulfonylurea-treated monogenic diabetes in pregnancy: implications of placental glibenclamide transfer. Diabet Med. 2017;34(10):1332–9.
Steele AM, Wensley KJ, Ellard S, et al. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS One. 2013;148(6):e65326.
Delvecchio M, Salzano G, Bonura C, et al. Can HbA1c combined with fasting plasma glucose help to assess priority for GCK–MODY vs HNF1A-MODY genetic testing? Acta Diabetol. 2018;55(9):981–3.
Pinelli M, Acquaviva F, Barbetti F, et al. Identification of candidate children for maturity-onset diabetes of the young type 2 (MODY2) gene testing: a seven-item clinical flowchart (7-iF). PLoS One. 2013;8(11):e79933.
Velho G, Blanché H, Vaxillaire M, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia. 1997;40:217–24.
Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311(3):279–86.
Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2014;45(3):427–35.
Hohendorff J, Szopa M, Skupien J, et al. A single dose of dapagliflozin, an SGLT-2 inhibitor, induces higher glycosuria in GCK- and HNF1A-MODY than in type 2 diabetes mellitus. Endocrine. 2017;57(2):272–9.
Chakera AJ, Spyer G, Vincent N, Ellard S, Hattersley AT, Dunne FP. The 0.1% of the population with glucokinase monogenic diabetes can be recognized by clinical characteristics in pregnancy: the Atlantic Diabetes in Pregnancy cohort. Diabetes Care. 2014;37(5):1230–6.
Rudland VL, Hinchcliffe M, Pinner J, et al. Identifying glucokinase monogenic diabetes in a multiethnic gestational diabetes mellitus cohort: new pregnancy screening criteria and utility of HbA1c. Diabetes Care. 2016;39(1):50–2.
Ellard S, Beards F, Allen LI, et al. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia. 2000;43(2):250–3.
Hattersley AT, Beards F, Ballantyne E, et al. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet. 1998;19:268–70.
Spyer G, Macleod KM, Shepherd M, et al. Pregnancy outcome in patients with raised blood glucose due to a heterozygous glucokinase gene mutation. Diabet Med. 2009;26:14–8.
Spyer G, Hattersley AT, Sykes JE, Sturley RH, MacLeod KM. Influence of maternal and fetal glucokinase mutations in gestational diabetes. Am J Obstet Gynecol. 2001;185:240–1.
Hattersley AT, Pearson ER. Minireview: pharmacogenetics and beyond: the interaction of therapeutic response, beta-cell physiology, and genetics in diabetes. Endocrinology. 2006;147:2657–63.
Chakera AJ, Steele AM, Gloyn AL, et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care. 2015;38(7):1383–92.
Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200–13.
Isomaa B, Henricsson M, Lehto M, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia. 1998;41(4):467–73.
Stride A, Ellard S, Clark P, et al. Beta-cell dysfunction, insulin sensitivity, and glycosuria precede diabetes in hepatocyte nuclear factor-1alpha mutation carriers. Diabetes Care. 2005;28(7):1751–6.
Valkovicova T, Skopkova M, Stanik J, Gasperikova D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr Regul. 2019;53(2):110–34.
Wollheim CB. Beta-cell mitochondria in the regulation of insulin secretion: a new culprit in type II diabetes. Diabetologia. 2000;43(3):265–77.
Raile K, Schober E, Konrad K, et al. Treatment of young patients with HNF1A mutations (HNF1A-MODY). Diabet Med. 2015;32(4):526–30.
Khelifa SB, Dendana A, Barboura I, et al. Successful switch from insulin to oral sulfonylurea therapy in HNF1A-MODY Tunisian patient with the P291fsinsC mutation. Diabetes Res Clin Pract. 2016;115:133–6.
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362(9392):1275–81.
Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, Hattersley AT. No deterioration in glycemic control in HNF-1alpha maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care. 2003;26(11):3191–2.
Brunerova L, Rahelić D, Ceriello A, Broz J. Use of oral antidiabetic drugs in the treatment of maturity-onset diabetes of the young: a mini review. Diabetes Metab Res Rev. 2018;34(1):e2940.
Tripathy D, Carlsson Å-L, Lehto M, Isomaa B, Tuomi T, Groop L. Insulin secretion and insulin sensitivity in diabetic subgroups: studies in the prediabetic and diabetic state. Diabetologia. 2000;43(12):1476–83.
Pearson E, Liddell W, Shepherd M, Corrall R, Hattersley A. Sensitivity to sulphonylureas in patients with HNF-1α gene mutations: evidence for pharmacogenetics in diabetes. Diabet Med. 2000;17:543–5.
Urbanova J, Andel M, Potockova J, et al. Half-life of sulfonylureas in HNF1A and HNF4A human MODY patients is not prolonged as suggested by the mouse Hnf1a(−/−) model. Curr Pharm Des. 2015;21(39):5736–48.
Boileau P, Wolfrum C, Shih D, Stoffel M. Decreased glibenclamide uptake in hepatocytes of HNF-1α defficient mice. Diabetes. 2002;51(3):S343–8.
Demol S, Lebenthal Y, Bar-Meisels M, Phillip M, Gat-Yablonski G, Gozlan Y. A family with a novel termination mutation in hepatic nuclear factor 1α in maturity-onset diabetes of the young type 3 which is unresponsive to sulphonylurea therapy. Horm Res Paediatr. 2014;81(4):280–4.
Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med. 2009;26:437–41.
Tuomi T, Honkanen EH, Isomaa B, Sarelin L, Groop LC. Improved prandial glucose control with lower risk of hypoglycemia with nateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care. 2006;29(2):189–94.
Østoft SH, Bagger JI, Hansen T, et al. Incretin effect and glucagon responses to oral and intravenous glucose in patients with maturity-onset diabetes of the young—type 2 and type 3. Diabetes. 2014;63(8):2838–44.
Becker M, Galler A, Raile K. Meglitinide analogues in adolescent patients with HNF1A-MODY (MODY 3). Pediatrics. 2014;133(3):e775–9.
Docena MK, Faiman C, Stanley CM, Pantalone KM. Mody-3: novel HNF1A mutation and the utility of glucagon-like peptide (GLP)-1 receptor agonist therapy. Endocr Pract. 2014;20(2):107–11.
Fantasia KL, Steenkamp DW. Optimal glycemic control in a patient with HNF1A MODY with GLP-1 RA monotherapy: implications for future therapy. J Endocr Soc. 2019;3(12):2286–9.
Østoft SH, Bagger JI, Hansen T, et al. Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial. Diabetes Care. 2014;37(7):1797–805.
Christensen AS, Haedersdal S, Storgaard H, et al. 981-P:GIP and GLP-1 potentiate sulfonylurea-induced insulin secretion in HNF1A diabetes. Diabetes. 2019;68(Supp 1):981-P.
Christensen AS, Storgaard H, Hædersdal S, Hansen T, Knop FK, Vilsbøll T. Glimepiride monotherapy versus combination of glimepiride and linagliptin therapy in patients with HNF1A-diabetes: a protocol for a randomised, double-blinded, placebo-controlled trial. BMJ Open. 2018;8(10):e022517.
Delvecchio M, Ludovico O, Menzaghi C, et al. Low prevalence of HNF1A mutations after molecular screening of multiple MODY genes in 58 Italian families recruited in the pediatric or adult diabetes clinic from a single Italian hospital. Diabetes Care. 2014;37(12):e258–60.
Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the betacell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–8.
Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57(9):2280–7.
Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345(13):971–80.
Deng M, Xiao X, Zhou L, Wang T. First case report of maturity-onset diabetes of the young type 4 pedigree in a Chinese family. Front Endocrinol (Lausanne). 2019;10:406.
Mangrum C, Rush E, Shivaswamy V. Genetically targeted dipeptidyl peptidase-4 inhibitor use in a patient with a novel mutation of MODY type 4. Clin Med Insights Endocrinol Diabetes. 2015;8:83–6.
Ulinski T, Lescure S, Beaufils S, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17(2):497–503.
Bellanné-Chantelot C, Clauin S, Chauveau D, et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes. 2005;54(11):3126–32.
Dubois-Laforgue D, Cornu E, Saint-Martin C, et al. Diabetes, associated clinical spectrum, long-term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Care. 2017;40(11):1436–43.
Pearson ER, Badman MK, Lockwood CR, et al. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1alpha and-1beta mutations. Diabetes Care. 2004;27(5):1102–7.
Carrillo E, Lomas A, Pinés PJ, Lamas C. Long-lasting response to oral therapy in a young male with monogenic diabetes as part of HNF1B-related disease. Endocrinol Diabetes Metab Case Rep. 2017;17-0052. https://doi.org/10.1530/EDM-17-0052.
Bingham C, Hattersley AT. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol Dial Transplant. 2004;19(11):2703–8.
Horikawa Y, Enya M, Mabe H, et al. NEUROD1-deficient diabetes (MODY6): identification of the first cases in Japanese and the clinical features. Pediatr Diabetes. 2018;19:236–42.
Horikawa Y, Enya M. Genetic dissection and clinical features of MODY6 (NEUROD1-MODY). Curr Diab Rep. 2019;19(3):12.
Abreu GM, Tarantino RM, Cabello PH, et al. The first case of NEUROD1-MODY reported in Latin America. Mol Genet Genom Med. 2019;7(12):e989.
Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci USA. 2005;102(13):4807–12.
Ushijima K, Narumi S, Ogata T, et al. KLF11 variant in a family clinically diagnosed with early childhood-onset type 1B diabetes. Pediatr Diabetes. 2019;20:712–9.
Raeder H, Johansson S, Holm PI, et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. 2006;38:54–62.
Plengvidhya N, Kooptiwut S, Songtawee N, et al. PAX4 mutations in Thais with maturity-onset diabetes of the young. J Clin Endocrinol Metab. 2007;92(7):2821–6.
Sujjitjoon J, Kooptiwut S, Chongjaroen N, Tangjittipokin W, Plengvidhya N, Yenchitsomanus PT. Aberrant mRNA splicing of paired box 4 (PAX4) IVS7-1G>A mutation causing maturity-onset diabetes of the young, type 9. Acta Diabetol. 2016;53(2):205–16.
Molven A, Ringdal M, Nordbø AM, et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes. 2008;57(4):1131–5.
Edghill EL, Flanagan SE, Patch AM, et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57(4):1034–42.
Bonnefond A, Yengo L, Philippe J, et al. Reassessment of the putative role of BLK-p.A71T loss-of-function mutation in MODY and type 2 diabetes. Diabetologia. 2013;56(3):492–6.
Mruthyunjaya MD, Chapla A, Shyamasunder AH, et al. Comprehensive maturity onset diabetes of the young (MODY) gene screening in pregnant women with diabetes in India. PLoS One. 2017;12(1):e0168656.
Borowiec M, Liew CW, Thompson R, et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and beta-cell dysfunction. Proc Natl Acad Sci USA. 2009;106(34):14460–5.
Kapoor RR, Flanagan SE, James CT, et al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia. 2011;54:2575–83.
Occal G, Flanagan SE, Hacihamdioglu B, et al. Clinical characteristics of recessive and dominant congenital hyperinsulinism due to mutation(s) in the ABCC8/KCNJ11 genes encoding the ATP-sensitive potassium channel in the pancreatic beta cell. J Pediatr Endocrinol Metab. 2011;24:1019–23.
Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT. Neonatal Diabetes International Collaborative Group. Effective treatment with oral sulfonylureas in peoples with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31:204–9.
Reilly F, Sanchez-Lechuga B, Clinton S, et al. Phenotype, genotype and glycaemic variability in people with activating mutations in the ABCC8 gene: response to appropriate therapy. Diabet Med. 2020;37(5):876–84.
Bowman P, Sulen Å, Barbetti F, et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol. 2018;6(8):637–46.
Bonnefond A, Philippe J, Durand E, et al. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS One. 2012;7(6):e37423.
Prudente S, Jungtrakoon P, Marucci A, et al. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet. 2015;97:177–85.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Maurizio Delvecchio, Carmela Pastore, and Paola Giordano have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Open Access
This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12465173.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Delvecchio, M., Pastore, C. & Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther 11, 1667–1685 (2020). https://doi.org/10.1007/s13300-020-00864-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-020-00864-4