
Abstract
Whilst the prevalence of obesity continues to increase at an alarming rate worldwide, the personal and economic burden of obesity-related complications becomes ever more important. Whilst dietary and lifestyle measures remain the fundamental focus of the patient to counter obesity, more frequently pharmacological and/or surgical interventions are required. Nevertheless, these therapies are often limited by weight loss efficacy, side effects, surgical risks and frequently obesity relapse. Currently, only five drug therapies are approved for the specific treatment of obesity. However, our understanding of the pathophysiology of obesity and of gut hormones has developed precipitously over the last 20–30 years. As a result, there has been a recent movement to create and use analogues that manipulate these gut hormones to support weight loss. In this article we review the efficacy of the currently approved drug therapies and discuss future potential drug mechanisms and early clinical trial results exploring these budding avenues. We discuss the use of glucagon-like peptide-1 (GLP-1) analogues as monotherapy and unimolecular dual or triple agonists that exploit the GLP-1 receptor and/or the gastric inhibitory peptide (GIP) receptor and/or the glucagon receptor. We also explore the use of sodium-glucose co-transporter-2 (SGLT-2) inhibitors, amylin mimetics, leptin analogues, ghrelin antagonists and centrally acting agents to suppress appetite [neuropeptide Y (NPY) antagonists, melanocortin-4 receptor (MC4R) agonists and cannabinoid-1 receptor antagonists]. Whilst further evidence is required to support their clinical use, preclinical and early clinical trial results are encouraging.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Obesity is a complex metabolic disorder, with several licensed drug and surgical therapies currently available. |
Current therapies are often associated with inadequate efficacy or complications and side effects, limiting their use. |
Multiple pathophysiological mechanisms of obesity are recognized, though only few of these are manipulated using currently licensed therapies. |
Whilst most drug classes are in early stages of development and/or clinical trials, results are promising for multiple drug targets. |
Introduction
Obesity is the modern epidemic, generally defined as a body mass index (BMI) ≥ 30 kg/m2, though a BMI ≥ 27.5 kg/m2 defines obesity in Asian populations [1]. Currently, 28.7% of people in England are obese with a further 35.6% overweight [2]. Worryingly, the worldwide prevalence of obesity continues to increase and has tripled since 1975, with 1.6 billion overweight adults and 650 million obese adults [3]. Obesity comes with both significant economic and personal costs. One American-based study estimated the additional annual cost associated with obesity in men was US $1152 and in women was US $3613, which given the high prevalence of obesity in the US led to an estimated spending of about $190 billion per year (21% of total healthcare spending) [4]. In the UK, the estimated spend on obesity in 2014/15 was £6.1 billion and is projected to reach £9.7 billion by 2050, with an estimated societal cost of almost £50 billion [5]. Whilst the economic cost of obesity is substantial, the personal cost is often greater with an increased risk of multiple medical and mental health complications. Obesity is associated with increased all-cause mortality and contributed to more than 4 million deaths worldwide in 2015 [6]. Cardiovascular disease accounted for 2.7 million deaths, whilst diabetes was the second leading cause at 0.9 million deaths. Although chronic kidney disease and obesity-associated cancer (hepatic, renal, breast, prostate and colon) each accounted for ≤ 10% of obesity-related deaths, these conditions are associated with major disability and poorer quality of life [6].
There is growing interest in drug therapies that can support weight loss and thereby reduce the prevalence of obesity and obesity-related complications. Intensive lifestyle interventions for people with obesity are associated with significant improvements in weight, amongst other parameters. The Look AHEAD trial observed a 5% reduction in body weight was associated with sustained improvements in key cardiovascular risk factors such as blood pressure and lipid profile [7]. As such, many regulatory bodies use a 5% total weight loss to determine whether a drug induces a meaningful weight loss. Whilst the US Food and Drug Administration (FDA) has approved five drug therapies to treat obesity (orlistat, phentermine/topiramate, lorcaserin, naltrexone/bupropion, liraglutide), the European Medicines Agency (EMA) has approved only three drug therapies (orlistat, bupropion/naltrexone and liraglutide) [8] at the time of writing. Should medical therapies fail to achieve adequate weight loss, bariatric surgery is a final option for some people. Generally, these therapies support weight loss by either enhancing satiety, inhibiting hunger or increasing catabolism. However, with our evolving understanding of the underlying pathophysiology of obesity there are an increasing number of drug targets being identified, and it is anticipated that more medications will be approved by regulatory bodies such as the FDA and EMA to tackle the obesity epidemic.
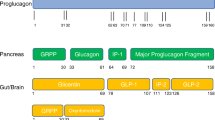
In this article, we discuss the clinical trials investigating pharmacotherapy for people with obesity. However, we do not discuss real-world clinical data related to use of these drugs. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors (Fig. 1).

Summarises the mechanism of action for each class of pharmacotherapy used to treat obesity
Efficacy of Currently Approved Therapies for Obesity
Orlistat
Orlistat is a selective inhibitor of pancreatic lipase, which thereby moderates the intestinal digestion and absorption of fat, approved for use by both the FDA and EMA. It should be taken with meals and is available both by prescription and over the counter in lower doses in the UK [9]. Orlistat is indicated for patients with a BMI ≥ 30 kg/m2 or ≥ 28 kg/m2 in association with other risk factors (e.g., hypertension, diabetes, hyperlipidaemia) [10]. Whilst people with obesity using orlistat lose an extra 2.9–3.4 kg weight over 12 months, gastrointestinal side effects, reduced absorption of fat-soluble vitamins and steatorrhea are commonplace [9]. Indeed, a recent systematic review observed a mean weight loss of 3.1 kg associated with orlistat use [11]. Additional metabolic benefits associated with orlistat use include reduced blood pressure (systolic, 1.15 mmHg; diastolic, 1.07 mmHg) and reduced circulating lipids (total cholesterol, 0.30 mmol/l; low-density lipoprotein (LDL) cholesterol, 0.27 mmol/l; triglycerides, 0.09 mmol/l) [12]. One study found the cost of weight loss per kilogram associated with orlistat was US$546, with a cost of US$71,000 per quality-adjusted life year (QALY) in Europe [13].
Phentermine/Topiramate
Phentermine is a sympathomimetic that stimulates noradrenaline release suppressing appetite, whilst topiramate is an anticonvulsant that augments the weight loss associated with phentermine use though the mechanism of appetite suppression is unclear. It is an effective weight loss agent with studies observing 6.6–8.6 kg weight loss over 12 months [9]. A more recent meta-analysis observed a mean weight loss of 9.8 kg associated with its use in randomised-controlled trials [11]. However, side effects include insomnia, dizziness and paresthesia. Whilst approved by the FDA in 2012, as a result of safety concerns the EMA rejected the use of this therapy. Phentermine/topiramate use was found to cost US $48,340 per QALY, a cost of US $232/kg weight loss [13, 14].
Lorcaserin
Lorcaserin is as an appetite suppressant through activation of hypothalamic 5-HT2C receptors and was approved for use by the FDA in 2012. It is used in people with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with co-morbidity (e.g., hypertension, diabetes, dyslipidaemia) [9]. The use of lorcaserin is associated with an additional annual weight loss of about 3.2–3.6 kg as well as improvements in metabolic parameters including blood pressure (systolic, 0.61 mmHg; diastolic, 0.49 mmHg) and lipids (total cholesterol, 0.35 mmol/l; LDL cholesterol, 0.35 mmol/l [15]. A systematic review of randomised-controlled trials using locaserin noted a mean weight loss of 3.1 kg associated with its use [11] Common side effects associated with lorcaserin use include headache, nausea and dizziness [9, 15]. Studies exploring this therapy report a cost of US$545 per kilogram weight loss attained, with a cost of US$122,640 per QALY gained [13].
Naltrexone/Bupropion
Naltrexone is an opiate antagonist, whilst bupropion is a weak dopamine and noradrenaline reuptake inhibitor. It is approved for use by both the FDA and EMA in the treatment of obesity. In combination, these drugs promote satiety via enhancement of hypothalamic pro-opiomelanocortin (POMC) cell-mediated release of melanocyte-stimulating hormone (MSH) leading to reduced food intake and increased energy expenditure [16]. Like other obesity pharmacotherapy, naltrexone/bupropion is indicated for BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 in association with other obesity-related co-morbidities. An additional annual weight loss of 4.8% total body weight (mean 4.4 kg) has been observed, with side effects including nausea, headache and dizziness [11, 16].
Liraglutide
Liraglutide is a glucagon-like peptide-1 (GLP-1) agonist, originally marketed for the treatment of type 2 diabetes (T2D). It is given as a once daily injection at a dose of 3.0 mg daily (compared with up to 1.8 mg daily for treatment of T2D) [9]. Liraglutide improves weight loss by enhancing satiety via hypothalamic stimulation and delaying gastric emptying, thereby reducing food intake. It is approved by the FDA and EMA to support weight loss in people with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 and obesity-related co-morbidity [9, 17]. More common side effects include nausea/vomiting and pancreatitis. An additional annual weight loss of 5.3–5.9 kg has been observed compared with placebo [11, 17,18,19].
The approved pharmacotherapies for weight loss in people with obesity are presented in Table 1.
Surgical Interventions
Several surgical interventions are approved in the treatment of obesity including devices (e.g., intragastric balloon, endoscopic sleeve gastroplasty, vagal nerve blockade, hydrogels) and surgery [e.g., laparoscopic adjustable gastric banding (LAGB), roux-en-Y gastric bypass (RYGB), biliopancreatic diversion with duodenal switch (BPD-DS)]. Current UK guidelines recommend considering bariatric surgery in people with a BMI ≥ 40 kg/m2 or > 35 kg/m2 with obesity-related co-morbidity [20].
A recent meta-analysis observed that 6 months following bariatric surgery the mean difference in excess weight loss (%) versus the standard of care resulting from BPD-DS, RYGB and sleeve gastrectomy was 38.2%, 32.1% and 32.5%, respectively [21]. Interestingly, LAGB was not superior to the standard of care. Importantly, diabetes remission rates were higher in participants who had undergone BPD-DS, RYGB or sleeve gastrectomy compared with the standard of care [21]. Greater improvements has also been noted in several other metabolic parameters associated with bariatric surgery including blood pressure and lipids compared with medical treatment [22]. Device therapies such as intragastric balloon, endoscopic sleeve gastroplasty or vagal nerve blockade are generally less effective weight loss strategies than surgical intervention, but provide 14–24% excess weight loss when used in the treatment of obesity [23].
Nevertheless, concerns remain about complications in this group of patients who often pose very high surgical risk. Complications include anastomotic leak, anaesthetic complications, bleeding, band slippage or erosion, dumping syndrome, gallstone formation, hernia, luminal stenosis, vitamin deficiencies and venous thromboembolism [24]. Avoiding surgical risks with newer drug therapies would represent major advances in obesity management and improve long-term outcomes for these patients.
Non-approved Therapies for Obesity
Incretin-based Treatments
Since the isolation of the peptide exendin-4 from Heloderma suspectum (Gila Monster) lizard venom in 1992 [25], several drugs that manipulate the incretin axis have been produced and approved for use in people with T2D including exenatide (2005), liraglutide (2009), lixisenatide (2013), albiglutide (2014, withdrawn 2017), dulaglutide (2014) and semaglutide (2017) [26]. The peptide GLP-1 is produced by the enteroendocrine L-cells of the small intestine as a cleavage product of the pre-proglucagon gene within minutes in response to oral glucose or fat. Concentrations are therefore much higher in the fed state, and these peptides are subsequently cleaved by dipeptidyl peptidase4 (DPP-4) enzymes rendering the GLP-1 peptide inactive [26]. The first action of GLP-1 to be characterised was the stimulation of insulin and inhibition of glucagon release from the pancreatic islets [27]. Therefore, this drug class controls dysglycaemia in people with T2D.
Importantly, GLP-1 receptors have been demonstrated in multiple organs throughout the body, including the pancreatic islets, gastrointestinal tract, kidney, lung, heart and centrally within the hypothalamus and pituitary [28]. Therefore, GLP-1 analogues have been found to have several non-glycaemic effects including improved blood pressure through arterial vasodilation and increased renal natriuresis and reduced appetite via delayed gastric emptying and centrally mediated satiety within the hypothalamus. It is these latter actions that lead to reduced appetite and early satiety thereby contributing to weight loss in people who use GLP-1 analogues [26]. Indeed, high-dose liraglutide (3.0 mg daily) is already approved to support weight loss in both Europe and the US. However, other GLP-1 analogues used either as monotherapy or in combination with other drug classes have the potential to encourage additional weight loss in people with obesity [26], but are currently not licensed for this use and are licensed for use in T2D alone.
Whilst the use of DPP-4 inhibitors may theoretically support weight loss in the treatment of obesity through increasing the half-life of endogenous GLP-1, weight loss associated with their use is clinically insignificant (0.16–0.64 kg) [29], and as such their use in obesity is not explored further in this manuscript.
GLP-1 Monotherapy
Exenatide
Exenatide (Byetta, Bydureon) was the first approved GLP-1 analogue in 2005, a synthetic form of the naturally occurring peptide exendin-4 first isolated from Heloderma suspectum (Gila Monster) lizard venom in 1992 [25]. It is available either as a 10 mcg twice daily subcutaneous injection or as a 2 mg once weekly preparation. It is licensed for use in T2D as an add-on therapy to metformin, sulphonylureas, pioglitozone or long-acting insulin only. One systematic review reported that use of exenatide 20 mcg twice daily resulted in an additional 1.4 kg weight loss, whilst exenatide 2 mg once weekly was associated with 1.6 kg additional weight loss compared with placebo [30].
Liraglutide
Liraglutide (Victoza) was approved for use in T2D in 2009 by the EMA at doses of up to 1.8 mg once daily subcutaneous injection for people with T2D as monotherapy in those unable to take metformin or as add-on treatment to oral agents and/or insulin. Subsequently, liraglutide (Saxenda) was approved in 2014 for the treatment of obesity up to a maximum dose of 3.0 mg subcutaneously once daily in people with a BMI ≥ 30 kg/m2, or ≥ 27 kg/m2 and obesity-related co-morbidity [9, 17]. Additional weight loss associated of 1.0 kg and 1.5 kg versus placebo with liraglutide of 1.2 mg and 1.8 mg is reported in one meta-analysis [30]. Absolute weight loss with liraglutide 3.0 mg (Saxenda) is reported up to 5.9 kg over 56 weeks [18, 19].
Lixisenatide
The maximum recommended dose of lixisenatide (Lyxumia) is 20 mcg once daily via subcutaneous injection and is licensed for use in people with T2D as monotherapy in those who cannot take metformin or as an add-on treatment to oral agents and/or insulin [31]. The use of lixisenatide was associated with up to 2 kg additional weight loss over 12 weeks compared with control in the GetGoal-Mono study [32].
Dulaglutide
Dulaglutide (Trulicity) is administered subcutaneously at a maximum dose of 1.5 mg once weekly, and available on license since 2014 for people with T2D as monotherapy in those who cannot take metformin or as add-on treatment in people using oral agents and/or insulin. As monotherapy the use of dulaglutide in people with T2D has been reported to increase weight loss by up to 2.3 kg [33]
Semaglutide
Semaglutide (Ozempic) is the most recent GLP-1 analogue to be approved for use in T2D and is currently available by subcutaneous injection for the treatment of T2D at a dose of up to 1.0 mg once weekly. It is licensed for use as monotherapy in those unable to take metformin or as add-on treatment. The absolute weight loss associated with semaglutide use is up to 4.6 kg and 6.5 kg at doses of 0.5 mg and 1.0 mg respectively over 40 weeks. Indeed, comparative trials have shown that semaglutide 1.0 mg demonstrated greater weight loss efficacy than dulaglutide 1.5 mg (6.5 kg vs. 3.0 kg) [34]. Indeed, a phase II dose-finding trial demonstrated greater body weight reductions associated with daily subcutaneous semaglutide at a dose greater than 0.2 mg compared with liraglutide 3.0 mg daily [35]. The development of oral semaglutide for T2D and/or obesity is of interest, as other GLP-1 analogues are currently available only as injectables often limiting their use. One trial recently reported that oral semaglutide (14 mg once daily) resulted in greater weight loss compared with subcutaneous liraglutide (4.4 kg vs. 3.1 kg) over 26 weeks [36]. Whilst the FDA approved the use of oral semaglutide for the treatment of people with T2D in September 2019 [37], the EMA have not approved its use in T2D at the time of writing.
The effect of currently approved GLP-1 monotherapies for T2D on weight loss are presented in Table 2.
Multi-agonists
A single drug that acts at two or more different receptors may enhance weight loss compared with single receptor agonists and has the advantage of a single pharmacokinetic profile compared with multiple profiles observed with the co-administration of two or more drugs [38]. This has led to the development and investigation of several incretin-related therapies such as unimolecular GLP-1/glucagon receptor dual agonists, GLP-1/gastric inhibitory polypeptide (GIP) dual agonists and GLP-1/GIP/glucagon receptor triple agonists. Whilst these multi-agonists have not yet been licensed for use in people with T2D and/or obesity, preclinical and early clinical trials support a future role for their use [38].
GLP-1/Glucagon Receptor Dual Agonists
The mechanism of GLP-1 receptor stimulation on satiety is discussed above. Glucagon receptor stimulation enhances energy expenditure by increasing oxygen consumption, lipid catabolism and thermogenesis amongst other factors [39]. Oxyntomodulin is an endogenous peptide secreted by the enteroendocrine cells of the intestine in response to oral nutrients, which acts as an agonist at both GLP-1 and glucagon receptors [40]. Indeed, the administration of oxyntomodulin reduces food intake and suppresses appetite [41] by activation of GLP-1 receptors [42] and increases energy expenditure via activation of the glucagon receptor [43]. Thus, drugs that have a dual agonist effect at these receptors ought to potentiate weight loss.
Previous human trials have shown that subcutaneous administration of oxyntomodulin three times daily for 4 weeks resulted in 1.8 kg greater weight loss than controls [44] as a result of increased activity-related energy expenditure (143 kcal/day) and decreased energy intake (128 kcal) [45]. Several clinical trials have observed significant weight loss associated with the use of these drugs from 2.1 to 3.1 kg [46, 47]. Encouragingly, there are several ongoing and unreported preclinical, phase I/II trials investigating drugs utilising this dual mechanism of action [48].
GLP-1/GIP Receptor Dual Agonists
Gastric inhibitory polypeptide (GIP) is a peptide hormone released by K-cells of the small intestine in response to food, which increases pancreatic insulin release. Combination therapy with GIP and GLP-1 enhances the incretin effect to improve dysglycaemia through increased prandial insulin secretion, reducing postprandial glycaemic excursions. However, GLP-1 and GIP demonstrate opposing actions on glucagon secretion. Whilst GIP stimulates glucagon secretion in low glucose environments, GLP-1 inhibits glucagon secretion in euglycaemia or hyperglycaemia [49]. This effect has limited the use and production of GIP analogues as they may potentiate the diabetogenic glucagon effect through augmented gluconeogenesis and glycogenolysis. However, GIP was combined with a GLP-1 receptor agonist to counter the potential obesogenic effect of GIP with the anorectic effect of GLP-1 [38].
Human studies have demonstrated good outcomes for weight reduction in people with (− 2.62 kg) and without (− 5.09 kg) T2D over 4 weeks compared with placebo [50]. Similarly, a weight loss of 1.67% total body weight over 12 weeks was associated with the use of the dual GLP-1/GIP agonist NNC0090-2746 [51]. Promising results have been observed for the GLP-1/GIP agonist LY3298176 in overweight or obese people with T2D, noting a weight loss between 0.9 and 11.3 kg in participants using the dual agent compared with 0.4 kg and 2.7 kg in those using placebo or dulaglutide respectively [52]. Further preclinical and clinical trials utilising these dual agonists are ongoing, though results are unpublished at the time of writing [48].
GLP-1/GIP/Glucagon Receptor Triple Agonists
Unimolecular triple agonists pose the greatest potential with respect to weight loss. A synergistic action in which glucagon receptor activation enhances energy expenditure whilst GLP-1 receptor agonists reduce calorie intake and both GLP-1/GIP potentiate the incretin effect to modulate the diabetogenic effect of glucagon agonists. Two triple agonists are currently in early stages of clinical trials with no yet published clinical trial data [48]. However, preclinical studies of these drugs demonstrate supportive results in rodent and mice models [53,54,55], and we await further clinical trial data with anticipation.
Sodium-Glucose Co-Transporter-2 Inhibitors
Sodium-glucose co-transporter-2 (SGLT-2) inhibitors are currently licensed for the treatment of T2D, though there is a growing body of evidence to support their use in cardiovascular [56] and renal disease [57]. These drugs block the SGLT-2 protein in the proximal convoluted tubule, thereby inducing a glucose-mediated osmotic diuresis and natriuresis, improving several metabolic measures [58]. Interestingly, as a result of the enhanced glycosuria mediated by this class of medications, they are associated with an increased urinary loss of about 60–100 g of glucose (200–300 kcal) per day [59]. One meta-analysis reported a mean weight loss of 2.1–2.7 kg associated with their use from phase III clinical trials in people with T2D [60]. The EMA and FDA have approved several drugs in this class of medication for the treatment of T2D including dapagliflozin, empagliflozin, canagliflozin and ertugliflozin. However, none are yet approved by the EMA or FDA specifically for the treatment of obesity, though trials in obese people without T2D have shown promising results [61].
Dapagliflozin
Dapagliflozin (Forxiga) was the first SGLT-2 inhibitor to go to market, approved by the EMA in 2012 and the FDA in 2014 in the treatment of people with T2D. In people with T2D, dapagliflozin 10 mg daily is associated with a weight loss of 2.7–3.2 kg [62, 63]. In people without diabetes, some trials have explored dapagliflozin as a monotherapy or in combination with other weight loss agents have been explored. One study in people with prediabetes and a mean BMI of 30.3 kg/m2 received either dapagliflozin 10 mg or placebo for 12 weeks. The authors reported dapagliflozin use was associated with a 3.0 kg weight loss as well as improvements in several other metabolic markers [64]. One trial explored the impact of combining dapagliflozin with the GLP-1 analogue exenatide in obese participants without diabetes, finding a mean weight loss 4.5 kg over 24 weeks [65]. Importantly, these weight loss benefits were sustained with a reported weight loss of 5.7 kg over 52 weeks [66].
Canagliflozin
Canagliflozin (Invokana) was approved for use in people with T2D by the EMA and the FDA in 2013. In people with diabetes, canagliflozin induces additional weight loss of 2.5–4.0 kg [67, 68]. Canagliflozin has been trialed in a phase IIb study in overweight and obese subjects without T2D. Over a 12-week study, the authors reported a mean weight loss of 1.9 kg, 2.8 kg and 2.4 kg with the use of 50 mg, 100 mg or 300 mg respectively [69]. Subsequently, a further trial investigated the co-administration of canagliflozin and phentermine in obese patients without T2D. Over 26 weeks, participants using canagliflozin lost 1.9 kg compared with 4.1 kg in those using phentermine and 7.3 kg in participants using both drugs [70].
Empagliflozin
Empagliflozin (Jardiance) was approved for the treatment of people with T2D by the EMA and the FDA in 2014. The use of empagliflozin in addition to metformin or sulphonylurea is associated with weight loss of 2.1–2.5 kg [71, 72]. We are not aware of any trial exploring the effect of empagliflozin on body weight in people without T2D at the time of writing.
Ertugliflozin
Ertugliflozin (Steglatro) is the most recent SGLT-2 inhibitor to come to the market, with approval for use in people with T2D received from the EMA in 2018 and the FDA in 2017. A weight loss of 2.7–3.7 kg over 52 weeks has been associated with its use in people with T2D [73, 74]. To our knowledge, at the time of writing there are no trials exploring weight loss in people without T2D.
Other SGLT-2 Inhibitors
One trial explored the use of the SGLT-2 inhibitors remogliflozin and sergliflozin compared with placebo controls in obese people without co-morbid diabetes over 8 weeks. The authors reported that in people using remogliflozin there was a 7.6 kg weight loss and 6.1 kg weight loss in those using sergliflozin compared with 5.1 kg weight loss in placebo controls [75].
The clinical trials reporting on the impact of SGLT-2 inhibitors for weight loss in people with or without T2D are presented in Table 3.
Other Emerging Therapeutic Targets
Amylin Mimetics
Amylin is a neuroendocrine peptide co-secreted by pancreatic β-cells postprandially with insulin and acts to inhibit glucagon secretion, reduce gastric emptying and centrally induce satiety [76]. The amylin analogue pramlintide was licensed by the FDA in 2005 for patients with insulin-treated diabetes. Early studies reported that pramlintide use in people with insulin-treated diabetes is associated with improved glycaemic control and may support weight loss by reducing food intake [77]. A subsequent trial in obese patients with either non-insulin treated T2D or without T2D found an additional mean weight loss of 3.7 kg versus placebo [78]. Co-administration of pramlintide with either the sympathomimetic sibutramine or phentermine was observed to result in 9.2 kg weight loss compared with placebo over 24 weeks, whereas pramlintide monotherapy resulted in just 1.5 kg additional weight loss [79]. However, development of these combination therapies was discontinued following commercial reassessment. Subsequently, a second-generation amylin analogue, davalintide, was developed and completed phase II trials. However, results were disappointing with weight loss comparable to pramlintide and inferior compared with pramlintide/metreleptin, also resulting in its discontinuation in its development [80]. To further enhance the effect of amylin mimetics, preclinical studies have trialed these drugs in combination with calcitonin-receptor agonists as the amylin receptor consists of calcitonin receptor and activity-modifying proteins, thereby enhancing binding and activation of the amylin receptors [81]. Whilst these preclinical studies show some promise [82], human clinical trials are awaited.
Leptin Analogues
Leptin is a 167 amino acid secreted by white adipose tissue, which promotes satiety and increases energy expenditure via stimulation of hypothalamic POMC neurons and inhibition of neuropeptide Y neurons. Overfeeding and high total body fat stimulate release of leptin, whilst the fasting state and low-fat stores inhibit leptin secretion [83]. People with leptin gene mutations are obese secondary to pronounced hyperphagia [84], and satiety and body weight can be regulated in people with these genetic mutations with leptin treatment [85]. As such, there is a good rationale for leptin analogues in the treatment of obesity.
Metreleptin is a once daily subcutaneously administered leptin analogue approved by the FDA in 2014 for use in people with leptin deficiency or congenital/acquired lipodystrophy, with good clinical results in these conditions [86]. Whilst metreleptin monotherapy supports weight loss in obese individuals, it is not clinically meaningful with a mean 1.5 kg additional weight loss over 24 weeks noted in a previous trial [87]. To enhance the effect of leptin analogues, amylin mimetics such as pramlintide have been used in combination with metreleptin. One study found that use of the pramlintide/metreleptin combination resulted in 11.5 kg weight loss over 20 weeks compared with 7.4 kg and 7.9 kg weight loss respectively in participants receiving either metreleptin or pramlintide monotherapy [88]. Unfortunately, the development of the combination therapy was discontinued in 2011 following commercial reassessment.
Ghrelin Vaccines and Antagonists
Ghrelin is the only known orexigenic peptide hormone and is secreted by the stomach and proximal small intestine. Conversely to the hormone leptin, ghrelin stimulates neuropeptide Y neurons and inhibits hypothalamic POMC neurons [89]. As ghrelin induces hunger, inhibition of the ghrelin receptor represents an attractive therapeutic target in obesity. Ghrelin receptor antagonists and vaccines have shown promise with reduced food intake and body weight in pre-clinical studies [90, 91]. However, results in human trials are less favourable with no difference in weight between obese patients receiving ghrelin immunisation over 20 weeks, with both active and control groups losing 3.6 kg weight [92]. We were unable to identify any current clinical trials exploring ghrelin antagonism under progress, though recently ghrelin receptor inverse agonists have been successfully profiled in humans [93].
Neuropeptide Y Inhibitors
Neuropeptide Y (NPY) neurons inhibit the appetite-suppressing POMC neurons and stimulate the release of appetite-inducing peptides such as orexin and melanin-concentrating hormone, making their inhibition an attractive therapeutic target in obesity. The first clinical trials to explore this mechanism used the NPY-antagonist MK-0557; however, results were disappointing with an additional annual weight loss of just 1.1 kg compared with placebo [94]. Whilst early clinical studies of the NPY antagonist velneperit were encouraging with a reported additional 2.8 kg weight loss compared with placebo over 16 weeks [95], subsequent phase IIb studies were disappointing and studies were discontinued. Further trials exploring the combination of the NPY inhibitor and orlistat were also disappointing and have been discontinued [96].
Melanocortin-4 Receptor Agonists
Melanocortin receptors such as the melanocortin-4 receptor (MC4R) are stimulated by POMC-derived melanocortin peptides within the hypothalamus to suppress appetite. This is typically in response to POMC stimulation by hormones such as leptin. Conversely, POMC neurons are inhibited by NPY or ghrelin as discussed above. Therefore, drugs that enhance MC4R action should modulate food intake centrally to improve weight loss. Whilst mutations of the MC4R are a rare genetic cause of obesity, the treatment of two people with POMC deficiency with the MC4R agonist setmelanotide previously resulted in massive weight loss of 51.0 kg and 20.5 kg over 42 and 12 weeks respectively [97], highlighting the potential of this mechanism to support weight loss in obesity. One phase IIb clinical trial administered setmelanotide to obese participants with heterozygous MC4R deficiency and obese controls over 28 days, observing a placebo-adjusted weight loss of 2.6 kg and 4.0 kg respectively [98]. There are ongoing phase 2 and 3 trials using setmelanotide to further investigate the role of these medications in obesity [99, 100].
Cannabinoid Type-1 Receptor Antagonists
The cannabinoid neurotransmitter system also affects the central control of appetite, with cannabinoid-1 receptor agonists increasing appetite and antagonists suppressing appetite [101]. Therefore, cannabinoid receptor antagonists represent a further therapeutic option in the treatment of obesity. The first cannabanoid-1 receptor antagonist to enter clinical trials was rimonabant, which showed excellent weight loss outcomes with an additional mean weight loss of 4.7 kg [102]. Moreover, rimonabant use was associated with several improved metabolic markers including glycaemic and lipid control [103]. Whilst initially approved in the European market in 2006, the drug was later withdrawn in 2009 because of the increased risk of severe mood disorders and suicide [104]. Whilst there is movement to design cannabanoid-1 receptor antagonists that are less hydrophobic to reduce blood-brain barrier penetration and therefore psychiatric side effects [105], we are not aware of any ongoing clinical trials yet.
Conclusions
As our understanding of obesity and the associated hormonal changes continues to improve, further drug developments and therapies will be introduced. Currently, evidence is lacking to introduce many of these therapies into routine practice, though early results are promising. Within the rapidly developing world of diabetes pharmacotherapy, an increasing number of drug targets have been identified and these drugs are often associated with clinically significant weight loss. In this article we have highlighted important clinical trial data that support the pharmacotherapy of obesity. However, an important limitation is the omission of real-world data, which are often different from clinical trial results, partly because of varying patient populations. As further clinical trials continue, it is anticipated that more drugs will be approved for use in people with obesity to support weight loss before often riskier interventions such as bariatric surgery are required.
References
Purnell JQ. Definitions, classification, and epidemiology of obesity. 2018. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000. https://www.ncbi.nlm.nih.gov/books/NBK279167/. Accessed 31 Mar 2020
UK Parliament. House of Commons Library: Obesity Statistics. 2019. https://researchbriefings.parliament.uk/ResearchBriefing/Summary/SN03336. Accessed 23 Feb 2020.
World Health Organization (WHO). Obesity and Overweight. 2018. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. Accessed 23 Feb 2020.
Cawley J, Meyerhoefer C. The medical care costs of obesity: an instrumental variables approach. J Health Econ. 2012;31(1):219–30.
Public Health England. Health matters: obesity and the food environment. 2017. https://www.gov.uk/government/publications/health-matters-obesity-and-the-food-environment/health-matters-obesity-and-the-food-environment--2. Accessed 23 Feb 2020.
Afshin A, Forouzanfar MH, Reitsma MB, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377(1):13–27.
Look AR, Wing RR. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: four-year results of the Look AHEAD trial. Arch Intern Med. 2010;170(17):1566–75.
Yumuk V, Tsigos C, Fried M, et al. European guidelines for obesity management in adults. Obes Facts. 2015;8(6):402–24.
Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(2):342–62.
NICE. Obesity: identification, assessment and management. Clinical guideline [CG189]. 2014. https://www.nice.org.uk/guidance/cg189/chapter/1-Recommendations#pharmacological-interventions. Accessed 23 Feb 2020.
Singh AK, Singh R. Pharmacotherapy in obesity: a systematic review and meta-analysis of randomized controlled trials of anti-obesity drugs. Expert Rev Clin Pharmacol. 2020;13(1):53–64.
Sahebkar A, Simental-Mendía LE, Reiner Ž, et al. Effect of orlistat on plasma lipids and body weight: a systematic review and meta-analysis of 33 randomized controlled trials. Pharmacol Res. 2017;122:53–655.
Finkelstein EA, Kruger E. Meta- and cost-effectiveness analysis of commercial weight loss strategies. Obesity (Silver Spring). 2014;22(9):1942–51.
Finkelstein EA, Kruger E, Karnawat S. Cost-effectiveness analysis of Qsymia for weight loss. Pharmacoeconomics. 2015;33(7):699–706.
Chan EW, He Y, Chui CS, Wong AY, Lau WC, Wong IC. Efficacy and safety of lorcaserin in obese adults: a meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes Rev. 2013;14(5):383–92.
Sherman MM, Ungureanu S, Rey JA. Naltrexone/bupropion ER (Contrave): newly approved treatment option for chronic weight management in obese adults. Pharm Ther. 2016;41(3):164–72.
Astrup A, Carraro R, Fine N, et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond). 2012;36(6):843–54.
Wadden TA, Hollander P, Klein S, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37:1443–511.
Mehta A, Marso SP, Neeland IJ. Liraglutide for weight management: a critical review of the evidence. Obes Sci Pract. 2017;3(1):3–14.
NICE. Surgery for obese adults: Assessment and offering surgry. 2019. https://pathways.nice.org.uk/pathways/obesity#path=view%3A/pathways/obesity/surgery-for-obese-adults.xml&content=view-node%3Anodes-assessment-and-offering-surgery. Accessed 06 Feb 2020.
Park CH, Nam SJ, Choi HS, et al. Comparative efficacy of bariatric surgery in the treatment of morbid obesity and diabetes mellitus: a systematic review and network meta-analysis. Obes Surg. 2019;29(7):2180–90.
Gloy VL, Briel M, Bhatt DL. Bariatric surgery versus non-surgical treatment for obesity: a systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;347:f5934.
Ruban A, Doshi A, Lam E, Teare JP. Medical devices in obesity treatment. Curr Diabetes Rep. 2019;19(10):90.
Ma IT, Madura JA. Gastrointestinal complications after bariatric surgery. Gastroenterol Hepatol. 2015;11(8):526–35.
Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267(11):7402–5.
Sharma D, Verma S, Vaidya S, Kalia K, Tiwari V. Recent updates on GLP-1 agonists: current advancements and challenges. Biomed Pharmacother. 2018;108:952–62.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–57.
Koliaki C, Doupis J. Incretin-based therapy: a powerful and promising weapon in the treatment of type 2 diabetes mellitus. Diabetes Ther. 2011;2(2):101–21.
Aroda VR, Henry RR, Han J. Efficacy of GLP-1 receptor agonists and DPP-4 inhibitors: meta-analysis and systematic review. Clin Ther. 2012;34(6):1247–1258.e22.
Potts JE, Gray LJ, Brady EM, Khunti K, Davies MJ, Bodicoat DH. The effect of glucagon-like peptide 1 receptor agonists on weight loss in type 2 diabetes: a systematic review and mixed treatment comparison meta-analysis. PLoS One. 2015;10(6):e0126769.
Bain SC. The clinical development program of lixisenatide: a once-daily glucagon-like peptide-1 receptor agonist. Diabetes Ther. 2014;5(2):367–83.
Fonseca VA, Alvarado-Ruiz R, Raccah D, et al. Efficacy and safety of the once-daily GLP-1 receptor agonist lixisenatide in monotherapy: a randomized, double-blind, placebo-controlled trial in patients with type 2 diabetes (GetGoal-Mono). Diabetes Care. 2012;35:1225–311.
Umpierrez G, Tofé Povedano S, Pérez Manghi F, Shurzinske L, Pechtner V. Efficacy and safety of dulaglutide monotherapy versus metformin in type 2 diabetes in a randomized controlled trial (AWARD-3). Diabetes Care. 2014;37:2168–76.
Pratley RE, Aroda VR, Lingvay I, et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018;6(4):275–86.
O'Neil PM, Birkenfeld AL, McGowan B, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392(10148):637–49.
Pratley R, Amod A, Hoff ST, et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394(10192):39–50.
US Food and Drug Administration. 2019. FDA approves first oral GLP-1 treatment for type 2 diabetes. https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-glp-1-treatment-type-2-diabetes. Accessed 31 Mar 2020.
Brandt SJ, Kleinert M, Tschöp MH, Müller TD. Are peptide conjugates the golden therapy against obesity? J Endocrinol. 2018;238(2):R109–R119119.
Sánchez-Garrido MA, Brandt SJ, Clemmensen C, Müller TD, DiMarchi RD, Tschöp MH. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia. 2017;60(10):1851–61.
Shankar SS, Shankar RR, Mixson LA, et al. Native oxyntomodulin has significant glucoregulatory effects independent of weight loss in obese humans with and without type 2 diabetes. Diabetes. 2018;67(6):1105–12.
Cohen MA, Ellis SM, Le Roux CW, et al. Oxyntomodulin suppresses appetite and reduces food intake in humans. J Clin Endocrinol Metab. 2003;88:4696–701.
Dakin CL, Small CJ, Batterham RL, et al. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology. 2004;145(6):2687–95.
Scott R, Minnion J, Tan T, Bloom SR. Oxyntomodulin analogue increases energy expenditure via the glucagon receptor. Peptides. 2018;104:70–7.
Wynne K, Park AJ, Small CJ, et al. Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double-blind, randomized, controlled trial. Diabetes. 2005;54(8):2390–5.
Wynne K, Park AJ, Small CJ, et al. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes (Lond). 2006;30(12):1729–36.
Ambery P, Parker VE, Stumvoll M, et al. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: a randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet. 2018;391(10140):2607–18.
Tillner J, Posch MG, Wagner F, et al. A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: Results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes Metab. 2019;21(1):120–8.
Usui R, Yabe D, Seino Y. Twincretin as a potential therapeutic for the management of type 2 diabetes with obesity. J Diabetes Investig. 2019;10(4):902–5.
Mathiesen DS, Bagger JI, Bergmann NC, et al. The effects of dual GLP-1/GIP receptor agonism on glucagon secretion—a review. Int J Mol Sci. 2019;20(17):4092.
Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018;18:3–14.
Frias JP, Bastyr EJ III, Vignati L, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2. Diabetes Cell Metab. 2017;26(2):343–52 (e2).
Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392(10160):2180–93.
Finan B, Yang B, Ottaway N, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21(1):27–36.
Jall S, Sachs S, Clemmensen C, et al. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol Metab. 2017;6(5):440–6.
Choi IY, Kim JK, Lee JS, et al. Effect of a novel long-acting GLP-1/GIP/glucagon triple agonist (HM15211) in a NASH and fibrosis animal model. Diabetes. 2018;67(Supplement 1):1106-P.
Ali A, Bain S, Hicks D, et al. SGLT2 inhibitors: cardiovascular benefits beyond HbA1c—translating evidence into practice. Diabetes Ther. 2019;10:1595–622.
Williams DM, Nawaz A, Evans M. Renal outcomes in type 2 diabetes: a review of cardiovascular and renal outcome trials. Diabetes Ther. 2020;11:369–86.
Musso G, Gambino R, Cassader M, Pagano G. A novel approach to control hyperglycemia in type 2 diabetes: sodium glucose co-transport (SGLT) inhibitors: systematic review and meta-analysis of randomized trials. Ann Med. 2012;44(4):375–93.
List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32(4):650–7.
Brown E, Wilding JPH, Barber TM, Alam U, Cuthbertson DJ. Weight loss variability with SGLT2 inhibitors and GLP-1 receptor agonists in type 2 diabetes mellitus and obesity: mechanistic possibilities. Obes Rev. 2019;20(6):816–28.
Pereira MJ, Eriksson JW. Emerging role of SGLT-2 inhibitors for the treatment of obesity. Drugs. 2019;79(3):219–30.
Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33(10):2217–24.
Bailey CJ, Gross JL, Hennicken D, Iqbal N, Mansfield TA, List JF. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43.
Ramírez-Rodríguez AM, González-Ortiz M, Martínez-Abundis E. Effect of dapagliflozin on insulin secretion and insulin sensitivity in patients with prediabetes. Exp Clin Endocrinol Diabetes. 2018. https://doi.org/10.1055/a-0664-7583.
Lundkvist P, Sjöström CD, Amini S, Pereira MJ, Johnsson E, Eriksson JW. Dapagliflozin once-daily and exenatide once-weekly dual therapy: A 24-week randomized, placebo-controlled, phase II study examining effects on body weight and prediabetes in obese adults without diabetes. Diabetes Obes Metab. 2017;19(1):49–60.
Lundkvist P, Pereira MJ, Katsogiannos P, Sjöström CD, Johnsson E, Eriksson JW. Dapagliflozin once daily plus exenatide once weekly in obese adults without diabetes: sustained reductions in body weight, glycaemia and blood pressure over 1 year. Diabetes Obes Metab. 2017;19(9):1276–88.
Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15(4):372–82.
Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382(9896):941–50.
Bays HE, Weinstein R, Law G, Canovatchel W. Canagliflozin: effects in overweight and obese subjects without diabetes mellitus. Obesity (Silver Spring). 2014;22(4):1042–9.
Hollander P, Bays HE, Rosenstock J, et al. Coadministration of canagliflozin and phentermine for weight management in overweight and obese individuals without diabetes: a randomized clinical trial. Diabetes Care. 2017;40(5):632–9.
Haring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2014;37(6):1650–9.
Ridderstrale M, Andersen KR, Zeller C, Kim G, Woerle HJ, Broedl UC. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(9):691–700.
Dagogo-Jack S, Liu J, Eldor R, et al. Efficacy and safety of the addition of ertugliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sitagliptin: the VERTIS SITA2 placebo-controlled randomized study. Diabetes Obes Metab. 2018;20(3):530–40.
Pratley RE, Eldor R, Raji A, et al. Ertugliflozin plus sitagliptin versus either individual agent over 52 weeks in patients with type 2 diabetes mellitus inadequately controlled with metformin: the VERTIS FACTORIAL randomized trial. Diabetes Obes Metab. 2018;20(5):1111–20.
Napolitano A, Miller S, Murgatroyd PR, et al. Exploring glycosuria as a mechanism for weight and fat mass reduction. A pilot study with remogliflozin etabonate and sergliflozin etabonate in healthy obese subjects. J Clin Transl Endocrinol. 2013;1(1):e3–e8.
Hay DL, Chen S, Lutz TA, Parkes DG, Roth JD. Amylin: pharmacology, physiology, and clinical. Potential Pharmacol Rev. 2015;67(3):564–600.
Chapman I, Parker B, Doran S, et al. Effect of pramlintide on satiety and food intake in obese subjects and subjects with type 2 diabetes. Diabetologia. 2005;48(5):838–48.
Aronne L, Fujioka K, Aroda V, et al. Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: a phase 2, randomized, placebo-controlled, dose-escalation study. J Clin Endocrinol Metab. 2007;92(8):2977–83.
Aronne LJ, Halseth AE, Burns CM, Miller S, Shen LZ. Enhanced weight loss following coadministration of pramlintide with sibutramine or phentermine in a multicenter trial. Obesity (Silver Spring). 2010;18(9):1739–46.
Hay DL, Garelja ML, Poyner DR, Walker CS. Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br J Pharmacol. 2018;175(1):3–17.
Andreassen KV, Feigh M, Hjuler ST, et al. A novel oral dual amylin and calcitonin receptor agonist (KBP-042) exerts antiobesity and antidiabetic effects in rats. Am J Physiol Endocrinol Metab. 2014;307(1):E24–33.
Gydesen S, Hjuler ST, Freving Z, et al. A novel dual amylin and calcitonin receptor agonist, KBP-089, induces weight loss through a reduction in fat, but not lean mass, while improving food preference. Br J Pharmacol. 2017;174(7):591–602.
Kelesidis T, Kelesidis I, Chou S, Mantzoros CS. Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann Intern Med. 2010;152(2):93–100.
Farooqi IS, Wangensteen T, Collins S, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356(3):237–47.
Farooqi IS, Jebb SA, Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341(12):879–84.
Meehan CA, Cochran E, Kassai A, Brown RJ, Gorden P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev Clin Pharmacol. 2016;9(1):59–68.
Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282(16):1568–75.
Ravussin E, Smith SR, Mitchell JA, et al. Enhanced weight loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity (Silver Spring). 2009;17(9):1736–43.
Ibrahim Abdalla MM. Ghrelin—physiological functions and regulation. Eur Endocrinol. 2015;11(2):90–5.
Esler WP, Rudolph J, Claus TH, et al. Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology. 2007;148(11):5175–85.
Zorrilla EP, Iwasaki S, Moss JA, et al. Vaccination against weight gain. Proc Natl Acad Sci USA. 2006;103(35):13226–31.
Altabas V, Zjačić-Rotkvić V. Anti-ghrelin antibodies in appetite suppression: recent advances in obesity pharmacotherapy. Immunotargets Ther. 2015;4:123–30.
Denney WS, Sonnenberg GE, Carvajal-Gonzalez S, Tuthill T, Jackson VM. Pharmacokinetics and pharmacodynamics of PF-05190457: the first oral ghrelin receptor inverse agonist to be profiled in healthy subjects. Br J Clin Pharmacol. 2017;83(2):326–38.
Erondu N, Gantz I, Musser B, et al. Neuropeptide Y5 receptor antagonism does not induce clinically meaningful weight loss in overweight and obese adults. Cell Metab. 2006;4(4):275–82.
Sargent BJ, Moore NA. New central targets for the treatment of obesity. Br J Clin Pharmacol. 2009;68(6):852–60.
ClinicalTrials.gov. Double-Blind, Multi-Center, Randomized Study to Assess the Efficacy and Safety of Velneperit (S-2367) and Orlistat Administered Individually or Combined with a Reduced Calorie Diet (RCD) in Obese Subjects. 2018. https://clinicaltrials.gov/ct2/show/NCT01126970. Accessed 23 Feb 2020.
Kuhnen P, Clement K, Wiegand S, et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. 2016;375:240–6.
Collet TH, Dubern B, Mokrosinski J, et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol Metab. 2017;6(10):1321–9.
ClinicalTrials.gov. Setmelanotide (RM-493), Melanocortin-4 Receptor (MC4R) Agonist, in Bardet-Biedl Syndrome (BBS) and Alström Syndrome (AS) Patients With Moderate to Severe Obesity. 2020. https://clinicaltrials.gov/ct2/show/NCT03746522. Accessed 23 Feb 2020.
ClinicalTrials.gov. Long Term Extension Trial of Setmelanotide. 2019. https://clinicaltrials.gov/ct2/show/NCT03651765. Accessed 23 Feb 2020.
Richey JM, Woolcott O. Re-visiting the endocannabinoid system and its therapeutic potential in obesity and associated diseases. Curr Diabetes Rep. 2017;17(10):99.
Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007;370(9600):1706–13.
Després JP, Golay A, Sjöström L, Rimonabant in Obesity-Lipids Study Group. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353(20):2121–34.
Sam AH, Salem V, Ghatei MA. Rimonabant: from RIO to Ban. J Obes. 2011;2011:432607.
Yadav MR, Murumkar PR. Advances in patented CB1 receptor antagonists for obesity. Pharm Pat Anal. 2018;7(5):169–73.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work, and have given their approval for this version to be published.
Authorship Contributions
David M. Williams and Asif Nawaz contributed equally.
Disclosures
David M Williams and Asif Nawaz have no conflicts of interest to declare. Marc Evans received financial support for consultancy from Novartis, Merck Sharp & Dohme Corp. and Novo Nordisk and has served on the speaker’s bureau for Novartis, Lilly, Boehringer lngelheim, Merck Sharp & Dohme Corp., Novo Nordisk, Janssen and Takeda. Marc Evans is also the Editor-in-Chief of this journal.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Open Access
This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12063873.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Williams, D.M., Nawaz, A. & Evans, M. Drug Therapy in Obesity: A Review of Current and Emerging Treatments. Diabetes Ther 11, 1199–1216 (2020). https://doi.org/10.1007/s13300-020-00816-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-020-00816-y