
Abstract
Introduction
This study compared the efficacy and safety of similar U-100 insulin glargine products, namely, Lilly insulin glargine (LY IGlar; Basaglar®) and the reference insulin glargine product (IGlar; Lantus®), used once daily in combination with oral antihyperglycemic medications (OAMs) in adults with type 2 diabetes (T2D).
Methods
ELEMENT 5 was a phase III, randomized, multinational, open-label, treat-to-target, 24-week trial. Participants were insulin naïve (glycated hemoglobin [HbA1c] ≥ 7.0% to ≤ 11.0%) or on basal insulin (IGlar, neutral protamine Hagedorn or insulin detemir; HbA1c ≤ 11.0%) and taking ≥ 2 OAMs. The primary objective was to show that LY IGlar is noninferior to IGlar in terms of HbA1c reduction (0.4% noninferiority margin).
Results
The study population (N = 493) was predominantly Asian (48%) or White (46%), with similar baseline characteristics between arms (P > 0.05). At 24 weeks, LY IGlar was noninferior to IGlar in terms of change in HbA1c level from baseline (− 1.25 vs. − 1.22%, respectively; least squares mean difference − 0.04%; 95% confidence interval − 0.22%, 0.15%). Other 24-week efficacy and safety results were also similar between treatments (P > 0.05), including insulin dose; percentage of patients having HbA1c of < 7% and ≤ 6.5%; overall rate and incidence of total, nocturnal, and severe hypoglycemia; adverse events; insulin antibody response; and weight gain. Daily mean 7-point self-monitored blood glucose reduction was similar between treatments at 24 weeks, with no differences at any time point except premorning-meal (fasting) blood glucose (LY IGlar − 2.37 mmol/L; IGlar − 2.69 mmol/L; P = 0.007).
Conclusion
Overall, LY IGlar and IGlar combined with OAMs provided similar glucose control and safety findings in this T2D population, which included a greater proportion of Asian patients and had broader background basal insulin experience than a previously studied T2D population.
Trial Registration
ClinicalTrials.gov identifier, NCT02302716.
Funding
Eli Lilly and Company and Boehringer Ingelheim.
Plain Language Summary
Plain language summary available for this article.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Plain Language Summary
The phase III clinical trial reported here compared two similar basal insulin products, namely, Lilly insulin glargine (Basaglar®) and a reference insulin glargine product (Lantus®), used to treat patients with type 2 diabetes. Some patients were using basal insulin prior to entry, and others were insulin naïve. The main racial groups were Asian (48% of the study population), White (46%), and African American (6%). Patients were recruited from India, Korea, Taiwan, Russia, Turkey, the USA, and Puerto Rico. Efficacy and safety measures were acquired over 24 weeks. The main efficacy measures evaluated blood glucose, including glycated hemoglobin, an indicator of blood glucose levels over approximately 3 months, and self-monitored blood glucose taken at several time points over the course of the day. Safety measures included incidence and rate of low blood sugar, incidence of adverse events, and incidence and levels of insulin antibodies. Dose and weight assessments were also obtained. In general, the two treatments provided similar results for these measures. Only one result showed a statistically significant difference between treatments, namely, the reduction in premorning meal self-monitored blood glucose was greater in patients on Lantus® at 24 weeks. To further evaluate this finding, we analyzed treatment effects on this measure across different sets of patient subgroups. No significant differential treatment effects were observed. Considering this and other results, the difference was not expected to be clinically meaningful. Findings from this study extend the body of evidence from other trials supporting the similarity of these two insulin glargine products.
Introduction
Clinical guidelines recommend introduction of a basal insulin as an effective strategy for initiating insulin therapy in type 2 diabetes (T2D) when patients fail to meet glycemic goals using non-insulin antihyperglycemic agents [1,2,3]. Basal insulins should provide relatively constant blood levels sufficient to suppress hepatic glucose output between meals and during the night [4]. Long-acting basal insulin analogs were developed several years ago to overcome the deficiencies of conventional basal insulin therapy and more closely mimic endogenous basal insulin secretion [4]. Insulin glargine (IGlar; Lantus®, Sanofi-Aventis, Paris, France), the first long-acting basal insulin analog, became available in 2000 and continues to be an important treatment option for people with diabetes [4]. Compared with neutral protamine Hagedorn (NPH), IGlar has demonstrated less variability and a more prolonged duration of action (up to 24 h) with a more even concentration–time profile [4,5,6]. Clinically, this has allowed once-daily (QD) dosing in most patients and resulted in a lower risk of hypoglycemia [7,8,9,10,11,12].
In 2014, Lilly insulin glargine (LY IGlar; Basaglar®, Eli Lilly and Company, Indianapolis, IN, USA and Boehringer Ingelheim, Ingelheim am Rheim, Germany) received marketing authorization in the European Union as the first biosimilar insulin product [13], which was followed by approvals in Japan [14] and the USA [15] under the appropriate pathways. Protein-derived biosimilars or follow-on biologics, such as insulins, are highly similar versions of previously approved biopharmaceuticals with an amino acid sequence (primary protein structure) that is identical to that of the corresponding reference product. Their development may reduce costs and increase patient access to such therapies. Because of the nature of protein drugs, complex manufacturing processes are utilized, and differences in these processes can affect the final products [16, 17]. Thus, nonclinical and clinical studies comparing similar biologics with their reference products must demonstrate similar structural and functional characteristics and clinically similar outcomes, as required by regulatory agencies [18,19,20,21,22,23]. Several such studies were conducted as part of the LY IGlar development program [24,25,26,27,28,29,30,31,32,33]. Assessment of outcome effects across different racial backgrounds and clinical characteristics, such as prior insulin exposure, is also important as further characterization of the extent of effect comparability. Herein we report results from a phase III clinical trial (ELEMENT 5) that compares the efficacy and safety of LY IGlar and IGlar in a T2D population with expanded Asian participation and on additional basal insulin background therapies compared with that from an earlier study [26].
Methods
Study Design and Patients
ELEMENT 5 was a phase III, prospective, multinational, multicenter, two-arm, active-control, open-label, parallel assignment, randomized controlled trial (RCT) in patients with T2D, which was conducted from December 2014 to July 2016 in India, Korea, Taiwan, Russia, Turkey, the USA, and Puerto Rico. The primary objective was to determine whether LY IGlar is noninferior to IGlar (both administered QD in combination with oral antihyperglycemic medications [OAMs]), as measured by change in glycated hemoglobin A1c (HbA1c) over the treatment period. The trial consisted of a screening visit (approximately 2 weeks prior to randomization), randomization visit (week 0), 24-week treatment period (clinic visits at 2, 4, 8, 12, 16, 20, and 24 weeks), and a post-treatment-period telephone follow-up visit after approximately 4 weeks (Electronic Supplementary Material [ESM] Fig. S1). The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki [34]. The protocol was approved by the appropriate ethics review boards, and all patients provided written informed consent.
Eligible participants had T2D and were ≥ 18 years of age with a body mass index (BMI) of ≤ 45 kg/m2. The study included both insulin-naïve patients (HbA1c ≥ 7% and ≤ 11%) and patients on basal insulin (HbA1c ≤ 11%), all taking ≥ 2 OAMs. Allowed prior basal insulin therapy included NPH or insulin detemir (IDet), administered either QD or twice daily (BID), or IGlar, administered QD. Recent use of insulins other than those mentioned, pramlintide, or chronic systemic glucocorticoid therapy (within approximately 30 days) or of glucagon-like peptide-1 receptor agonists or biosimilar IGlar products (within 90 days) was not allowed. Additionally, patients with significant cardiac or gastrointestinal disease, a history or diagnosis of human immunodeficiency virus infection, active/recent (within 5 years) cancer (excluding basal cell carcinoma or carcinoma in situ), or evidence of liver disease were excluded. Other exclusion criteria included excessive insulin resistance (i.e., insulin dose ≥ 1.5 units/kg/day), > 1 episode of severe hypoglycemia in the previous 6 months, and hypersensitivity or allergy to IGlar or its excipients.
Randomization to LY IGlar or IGlar treatment was stratified by country, HbA1c level at screening (< 8.5%, ≥ 8.5%), sulfonylurea (SU) use (yes, no), and prestudy basal insulin use (yes, no). Treatment assignments were managed using an interactive web response system. Treatments were administered QD (a.m. or p.m.) with continued administration at the same time each day. The starting dose for insulin-naïve patients was 10 units/day. For patients on prestudy basal insulin, the starting dose was the same as the QD basal insulin dose or 80% of the BID dose. Patients self-titrated with insulin thereafter, adding 1 unit/day until fasting plasma glucose (FPG) levels reached ≤ 5.6 mmol/L [35]. Most insulin dose adjustments were expected to occur during the first 12 weeks of the study (titration period). Prestudy OAMs were continued as prescribed through week 24. The insulin or SU dose could be adjusted for safety concerns.
Outcomes
The primary outcome was the change in HbA1c from baseline to 24 weeks of treatment. The change in HbA1c from baseline to weeks 4, 8, 12, 16, and 20 was a secondary outcome. HbA1c analyses were performed by Covance (Princeton, NJ, USA) at regional centers. Other secondary efficacy measures included the percentage of patients achieving HbA1c target levels of < 7% and ≤ 6.5% and 7-point self-monitored blood glucose (SMBG) assessments (premeal for each meal, 2-h postmeal for morning/mid-day meals, bedtime, and 3 a.m.). Commercially available glucometers (Roche Accu-Chek® Performa; Roche, Indianapolis, IN, USA) were provided for SMBG measurements. Other secondary outcomes included basal insulin dose (units/day and units/kg/day), weight, BMI, and intrapatient variability (measured as FPG standard deviation [SD]). A health outcomes assessment of patient satisfaction was conducted using the Insulin Treatment Satisfaction Questionnaire (ITSQ) [36].
Clinical chemistry and hematology assessments were performed at screening and week 24. Adverse events (AEs) were recorded at every visit and summarized as treatment-emergent AEs, defined as postrandomization events either newly reported or increasing in severity. Their relatedness to the study drug was determined by the investigator. Injection site AEs were evaluated through patient responses to the Skin Evaluation Questionnaire and Insulin Questionnaire: Injection Sites. These AEs were assessed for factors associated with the injection (pain, pruritus, and rash) and injection site characteristics, including lipohypertrophy, hemorrhage, or induration. Allergic events were characterized according to a prespecified list of allergic reaction terms by blinded review of reported episodes. Immunogenicity was determined by the proportion of patients with measurable anti-insulin antibodies and by antibody levels (percentage binding). Immunogenicity samples were analyzed at Eurofins Pharma Bioanalytics (St. Charles, MO, USA).
Categories of hypoglycemia were assessed as incidences (number/proportion of patients with ≥ 1 event) and annualized rates (events/person/year). A hypoglycemic episode was defined as any event associated with signs or symptoms of hypoglycemia or measured blood glucose (BG) ≤ 3.9 mmol/L. Total hypoglycemia included all such events. Nocturnal hypoglycemia was defined as any hypoglycemic episode, as defined above, that occurred after bedtime and prior to the first meal upon waking. Documented symptomatic hypoglycemia was any confirmed event (BG ≤ 3.9 mmol/L) accompanied by signs/symptoms of hypoglycemia. Severe hypoglycemia was defined as any hypoglycemic event accompanied by neurologic (cognitive) impairment requiring assistance from another person (with or without a BG measurement). Severe hypoglycemia episodes were reported as serious AEs (SAEs).
Statistical Analyses
Calculation of the sample size was based on the primary outcome, namely, HbA1c change from baseline at 24 weeks. An estimated 209 completers per arm (418 total) were needed to show noninferiority of LY IGlar to IGlar at the 0.4% noninferiority margin (NIM), assuming no treatment difference between comparators, an SD of 1.1%, a two-sided significance level of 0.05, and 95% power. With an estimated 15% dropout rate at 24 weeks, the study required 492 randomized patients (246 per arm).
All analyses were conducted using SAS® version 9.2 (SAS Institute, Cary, NC, USA). Efficacy and safety analyses were conducted on the full analysis set (FAS), a modified intent-to-treat population comprising all randomized patients who took ≥ 1 dose of the assigned study drug. Unless otherwise stated, all analyses of treatment effects used the two-sided alpha level of 0.05 and corresponding 95% confidence intervals (CIs).
The primary endpoint was evaluated using the mixed model repeated measures (MMRM) method with change from baseline in HbA1c as the dependent variable and treatment (LY IGlar, IGlar), pooled country, basal insulin at entry (yes/no), SU use (yes/no), visit, and interaction between visit and treatment as fixed effects. The baseline HbA1c level was a covariate, and patient was a random effect. Noninferiority of LY IGlar to IGlar, the primary objective, was demonstrated if the upper limit of the 95% CI for the difference (LY IGlar − IGlar) in HbA1c change from baseline to 24 weeks was below + 0.4% (NIM). A key secondary analysis assessed noninferiority of IGlar to LY IGlar at the NIM of − 0.4%. If both noninferiority criteria were met, LY IGlar was considered to have equivalent efficacy to IGlar. Noninferiority of LY IGlar to IGlar for the same measure was then further analyzed by a second gated test within the subgroup of patients on prestudy IGlar. The family-wise Type 1 error rate was controlled at a one-sided 0.025 level using this gate-keeping procedure.
Analyses of other continuous secondary outcomes, including SMBG values, weight, insulin dose, and ITSQ results, used the same MMRM model that was used for the primary analysis with the baseline value of the response variable added as a covariate (FAS population). Continuous laboratory measures were analyzed using the analysis of covariance (ANCOVA) model. Comparisons of categorical variables used either Fisher’s exact test or Pearson’s Chi-square test. The rate of hypoglycemic episodes per person per year (total, nocturnal, documented symptomatic, and severe) was analyzed using the Wilcoxon rank-sum test. The percentage insulin antibody binding analysis also used the Wilcoxon rank-sum test, and analyses of the relationship between insulin antibody levels and clinical outcomes used a last-observation-carried-forward ANCOVA method whereby a significant treatment-by-insulin antibody interaction was used to indicate a potential differential treatment effect (ESM File S1). Post hoc analyses of treatment effects on FPG across several distinct sets of subgroups defined by prespecified baseline characteristics used an MMRM model with treatment, pooled country, basal insulin at entry (yes/no), SU use (yes, no), visit, subgroup, subgroup-by-treatment interaction, subgroup-by-visit interaction, treatment-by-visit interaction, and visit-by-subgroup-by-treatment interaction as fixed effects; the baseline values of FPG and HbA1c as covariates; and a random effect for patient for the FAS population.
Results
Patients
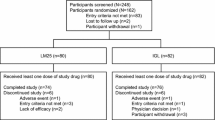
More than 90% of randomized patients (455/493) completed the 24-week treatment period; patient decision was the most common reason for discontinuation (ESM Fig. S2). Baseline demographics and clinical characteristics were balanced between arms (Table 1). The population was predominantly Asian (48%) or White (46%), in contrast to the earlier ELEMENT 2 T2D trial (Asian, 9% and White, 78%) [26]. The mean duration of diabetes was 12 years. Of the patients on prestudy basal insulin, 63% were taking IGlar (all QD), which was the only prestudy insulin used in the ELEMENT 2 trial. Other prestudy insulins included IDet (22%) and NPH (15%), which were primarily administered QD (< 10% administered BID). A minority of patients were insulin naïve (45%), and a majority (84%) were taking a SU in combination with one or more additional OAMs. Mean age, HbA1c level, and BMI were 57.4 years, 8.6%, and 29.0 kg/m2, respectively.
Glycemic Responses
Both the primary objective, noninferiority of LY IGlar to IGlar, and the key secondary efficacy objective, noninferiority of IGlar to LY IGlar, were achieved (NIM ± 0.4%; Table 2; Fig. 1a). Consequently, LY IGlar and IGlar were considered to have equivalent efficacy. Least squares (LS) mean reductions in HbA1c at 24 weeks were − 1.25% for LY IGlar and − 1.22% for IGlar (difference − 0.04%; 95% CI − 0.22%, 0.15%). Both treatment groups demonstrated statistically significant HbA1c reductions from baseline, beginning at week 4 and continuing through week 24 (P< 0.001; Fig. 1b). In a gated analysis used to control the Type 1 error rate, LY IGlar and IGlar also showed similar LS mean HbA1c reductions from baseline at 24 weeks in a subgroup of patients on prior IGlar therapy (LY IGlar − 0.91% [N = 81] and IGlar − 0.81% [N = 80]; P = 0.46). The same was true of the other subgroups associated with this analysis, including a subgroup of patients on insulins other than IGlar at baseline and an insulin-naïve subgroup (24-week LS mean HbA1c reductions for LY IGlar and IGlar, respectively: − 0.93% [N = 43] and − 1.03% [N = 47], P = 0.61 [non-IGlar insulin treated]; − 1.67% [N = 102] and − 1.64% [N = 96], P = 0.84 [insulin naïve]). No differential treatment effects were observed across these subgroups (i.e., treatment-by-subgroup interactions). Additionally, the proportions of patients who achieved the HbA1c targets of < 7% or ≤ 6.5% at 24 weeks were similar (P > 0.05) between treatment groups (Table 2).

a Change in glycated hemoglobin A1c (HbA1c) from baseline at 24 weeks, b HbA1c from baseline to 24 weeks, c 7-point self-monitored blood glucose (SMBG) at baseline and 24 weeks (reported as plasma-equivalent glucose values), d fasting plasma glucose (FPG) over 24 weeks. Data are shown as the least squares mean (LSM) with the standard error. Analyses are based on a mixed model repeated measures. CI Confidence interval, IGlar insulin glargine (Lantus), LSM Diff least squares mean difference, LY IGlar Lilly insulin glargine, PPG postprandial glucose. Asterisk indicates P< 0.05 for between-treatment difference
Both treatment groups showed improved LS mean SMBG levels for all seven time points over the 24-h period at 24 weeks (Fig. 1c). These results, including the SMBG daily mean, showed no statistically significant differences between treatments, with the exception of the LS mean difference in the premorning-meal SMBG (FPG) change from baseline (0.32 mmol/L; 95% CI 0.09, 0.55; P = 0.007; actual LS mean [standard error {SE}] FPG values: LY IGlar 5.96 [0.08] mmol/L and IGlar 5.65 [0.08] mmol/L; Fig. 1d). No significant differences in FPG results were observed at other visits over the course of the study. Post hoc analyses were conducted to compare LY IGlar and IGlar treatment effects on FPG change from baseline at 24 weeks across several distinct sets of patient subgroups to further probe this finding. Results from these analyses showed no significant differential treatment effects (ESM Table S1). Additionally, at 24 weeks, no differences were observed between treatment groups in premorning-meal intrapatient BG variability, as assessed by the standard deviation (Table 2).
Basal Insulin Dose and Body Weight
Significant increases from baseline in basal insulin dose were observed for both treatment groups at 24 weeks (LS mean [SE]: LY IGlar 29.0 [2.2] units/day; IGlar 28.9 [2.2] units/day; P < 0.001, both arms), and these increases were similar between arms (P = 0.99). Additionally, similar increases in LS mean weight for LY IGlar and IGlar were seen at 24 weeks (Table 2).
Hypoglycemia
Two severe hypoglycemia episodes were reported during the study, which involved two patients in the IGlar arm. Mean annualized rates of total and nocturnal hypoglycemia over 24 weeks were similar between treatments (Table 2), as were the rates for documented symptomatic hypoglycemia (mean [SD]: LY IGlar 10.2 [17.7]; IGlar 13.1 [22.1]; P = 0.35). Additionally, no statistical differences between arms were observed for overall incidence of total hypoglycemia (LY IGlar 69%; IGlar 69%; P = 0.92), nocturnal hypoglycemia (LY IGlar 50%; IGlar 46%; P = 0.47), or documented symptomatic hypoglycemia (LY IGlar 56%; IGlar 57%; P = 0.93).
Adverse Events
Adverse events, including allergic reactions, reported during the 24-week treatment period are summarized in Table 3. Similar incidences of AEs, including potentially treatment-related AEs, and SAEs were observed in the LY IGlar and IGlar arms. The most frequently reported AEs were nasopharyngitis (8.5%) and upper respiratory tract infection (3.9%). The incidence of allergic reactions was similar between treatment groups, and most events were mild or moderate in severity. One death occurred, which was not considered by the investigator to be related to the study drug or study procedures. The patient, a 69-year-old man on IGlar, experienced an SAE of cardiogenic shock with a fatal outcome at week 3. His medical history included T2D with prior IGlar use, hyperlipoproteinemia, hypertension, and arrhythmia.
Insulin Antibodies
The overall proportions of patients with detectable insulin antibodies were similar between treatment groups (Table 2). Additionally, the median insulin antibody percentage binding in the LY IGlar group was similar to that in the IGlar group at baseline and at weeks 4, 12, and 24 (all patients, Fig. 2). For patients with detectable insulin-antibody levels at endpoint, there were no significant treatment-by-endpoint insulin antibody level interactions for HbA1c (P = 0.22), insulin dose (P = 0.92), or total hypoglycemic events (P = 0.87), indicating no significant correlation between these outcomes and antibody levels.

Percentage binding of detectable anti-insulin glargine antibodies over 24 weeks (all patients with detectable antibodies). Data are shown as the median with the 25th, 75th percentiles. P values for treatment comparisons are derived from the Wilcoxon rank-sum test. Data points are slightly offset along the X-axis to avoid overlapping error bars. IGlar insulin glargine (Lantus), LOCF Last observation carried forward, LY IGlar Lilly insulin glargine, n number of patients with detectable insulin antibodies
Laboratory Assessments
No clinically meaningful changes from baseline in any laboratory values were identified within or between groups at 24 weeks (data not shown).
Patient-Reported Outcomes
In the FAS population, no differences in patient-reported health outcomes between treatment groups were identified, as determined from responses to the ITSQ. Specifically, LS mean overall scores at any visit and scores for each domain, including those for glycemic control, lifestyle flexibility, hypoglycemic control, and inconvenience, were similar for the LY IGlar and IGlar treatment arms (Table 2).
Discussion
ELEMENT 5 was the second of two phase III clinical trials comparing the efficacy and safety of LY IGlar to IGlar used in combination with OAMs in patients with T2D. The results from this study are particularly relevant for Asian patients with T2D, who were largely represented in the study population of ELEMENT 5, in contrast to the earlier T2D trial (ELEMENT 2) [26]. Also, patients on basal insulins other than IGlar were included in the present analysis. Outcomes between treatments were generally consistent with the overall findings from the initial T2D study [26] and those from a related study in patients with type 1 diabetes (ELEMENT 1) [24]. Both of the earlier studies were phase III, parallel assignment RCTs. ELEMENT 1 was a 52-week study involving 535 patients with baseline HbA1c of ≤ 11%. ELEMENT 2 was a 24-week study involving 756 insulin-naïve (baseline HbA1c of ≥ 7% and ≤ 11%) or IGlar-treated (baseline HbA1c of ≤ 11%) patients on ≥ 2 oral antihyperglycemic medications with a target FPG the same as that of the current study (≤ 5.6 mmol/L). Both ELEMENT 1 and ELEMENT 5 had open-label designs, which accommodated presentation of the treatment and reference products using the associated prefilled pen devices. ELEMENT 2, on the other hand, employed double-blind masking using syringes and vials concealed by a container closure assembly. All three studies met their primary endpoint, noninferiority of LY IGlar to IGlar, and also demonstrated equivalent significant HbA1c reductions and similar safety profiles between treatments with no apparent clinically meaningful differences. The current findings further support similarity between these biological drug substances, as explained in regulatory guidance [20, 21].
Compared with ELEMENT 2, the current study included a broader Asian population. Asian patients represent a large global proportion of patients with T2D, as evidenced in the recent International Diabetes Federation (IDF) Diabetes Atlas showing that China and India lead the rankings for the number of adults with diabetes (114 million and 73 million, respectively), followed by the USA (30 million). Moreover, India reportedly has the highest projected growth in this age group over the selected interval (2017–2045) (> 60 million vs. 5.4 million people [China and the USA]) [37]. Distinct characteristics of Asian versus non-Asian patients with T2D include a lower BMI with greater adiposity or visceral fat relative to BMI and younger age of onset with increased insulin resistance and/or impairment in insulin secretory function, the relative contributions of which can vary among Asian subpopulations [38,39,40,41]. Moreover, Asians have an increased risk of particular microvascular and macrovascular complications, which can also vary regionally [38,39,40,41]. Because of interethnic differences in the pathophysiology of T2D and its high prevalence in Asian populations, it is important to study the effects of antihyperglycemic therapies in Asian patients. Three recent publications have reviewed and/or analyzed previously reported IGlar treatment results in Asian patients from multiple RCTs or from a combination of RCTs and real-world data [42,43,44]. These studies suggest the need for timely initiation of basal insulin [42, 44] or a combination of basal insulin/prandial therapy (i.e., incretin mimetics or rapid-acting insulin) [43] in Asian patients with T2D.
ELEMENT 5 included Asian patients from India, Korea, and Taiwan (representative of South and East Asian populations), and the results indicate that these demographic differences did not contribute to meaningful differential clinical outcomes between treatments in the overall population. Efficacy results, including HbA1c reduction and proportions of patients reaching prespecified HbA1c targets, were similar between treatments and occurred at similar dose levels.
Efficacy outcomes can be informally compared across the racially distinct overall populations of the different ELEMENT T2D studies. These comparisons reveal somewhat higher HbA1c values at the endpoint with less reduction from baseline and lower percentages of patients achieving HbA1c targets in both ELEMENT 5 treatment groups. These findings parallel corresponding results for Asian patients with T2D from recent insulin-initiation studies, including a meta-analysis of Asian versus non-Asian adults initiating IGlar therapy [43] and a comparison of East Asian, Asian Indian, and non-Asian subgroups from the PARADIGM insulin initiation and intensification study [45]. ELEMENT 5 also included a lower percentage of insulin-naïve patients than ELEMENT 2 (45 vs. 60%, respectively). This may further influence efficacy outcomes, as shown in ELEMENT 2, in which insulin-naïve subgroups had lower HbA1c, a greater reduction from baseline, and more patients, percentage-wise, achieving HbA1c targets at endpoint as compared numerically with prior IGlar-treated subgroups [26]. Thus, differences in race and prior insulin status across studies may have influenced the observed findings for these outcomes.
Daily mean and individual 24-week 7-point SMBG results were similar between treatment arms of the current study, with the exception of the result for the premorning meal (FPG), which was significantly lower for IGlar. No FPG differences were observed at other visits during the study, and no differences in fasting glucose variability or differential treatment effects on FPG across demographic subgroups were observed at 24 weeks. Thus, considering the totality of results, the 24-week FPG difference was not expected to be clinically relevant. No endpoint FPG treatment differences were found in ELEMENT 2, but significantly lower SMBG for LY IGlar at the midday premeal time point (P < 0.05) was reported [26].
Safety profiles at 24 weeks were also generally similar between treatments, as reported in earlier studies [24, 26], including incidence of AEs and incidence and rates of hypoglycemia. Likewise, similar immunogenicity profiles were demonstrated, as shown previously [28], with no association between clinical outcomes and antibody levels. Weight increases, which are common with insulin therapy [46], were also similar between treatments and were comparable to those of the earlier T2D study [26].
There are a number of limitations to this study. The open-label design of the study could have influenced patient- or investigator-initiated actions. This design was necessitated by use of the distinctive pen devices, in which case blinding would have required a double-dummy design, imposing substantial injection burden on participants. Additionally, correlation of the differences in efficacy outcomes across the ELEMENT T2D trials with those for Asian versus non-Asian patients in related studies is limited by the mixed racial constitution of the trials. Moreover, the ELEMENT 5 study population included both East and South Asians, who have shown some distinctions in their respective pathogenesis of diabetes. Clinical studies comparing treatments in fully Asian populations will be important going forward to better characterize racial differences in insulin response and further assess optimal treatment regimens in these genetically distinct populations. Additionally, this report does not include analyses of treatment responses within racial subgroups. These are reported separately [47, 48].
Conclusions
In conclusion, results of the ELEMENT 5 study show that LY IGlar and IGlar, when administered QD with ≥ 2 OAMs, have similar efficacy and safety in a more racially diverse T2D population than previously studied, which included a sizeable Asian component. Inclusion of patients on other basal insulin background therapies further diversified patient representation. These findings provide additional support for the efficacy, safety, and tolerability of QD LY IGlar in patients with T2D. Ongoing assessment of the effectiveness and prolonged safety of newly developed biosimilar/follow-on insulins, such as LY IGlar, in clinical practice will be important as these agents gain more widespread use.
References
International Diabetes Federation. Global guideline for type 2 diabetes. 2012. https://www.iapb.org/wp-content/uploads/Global-Guideline-for-Type-2-Diabetes-IDF-2012.pdf. Accessed 28 Nov 2018.
Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm—2018 executive summary. Endocr Pract. 2018;24:91–120.
American Diabetes Association. Standards of medical care in diabetes—2018. Diabetes Care. 2018;41[Suppl 1]:S1–159.
Hilgenfeld R, Seipke G, Berchtold H, Owens DR. The evolution of insulin glargine and its continuing contribution to diabetes care. Drugs. 2014;74:911–27.
Heinemann L, Linkeschova R, Rave K, Hompesch B, Sedlak M, Heise T. Time-action profile of the long-acting insulin analog insulin glargine (HOE901) in comparison with those of NPH insulin and placebo. Diabetes Care. 2000;23:644–9.
Lepore M, Pampanelli S, Fanelli C, et al. Pharmacokinetics and pharmacodynamics of subcutaneous injection of long-acting human insulin analog glargine, NPH insulin, and ultralente human insulin and continuous subcutaneous infusion of insulin lispro. Diabetes. 2000;49:2142–8.
Ratner RE, Hirsch IB, Neifing JL, Garg SK, Mecca TE, Wilson CA. Less hypoglycemia with insulin glartine in intensive insulin therapy for type1 diabetes. US. Study group of insulin glargine in type 1 diabetes. Diabetes Care. 2000;23:639–43.
Yki-Järvinen H, Dressler A, Ziemen M, HOE 901/3002 Study Group. Less nocturnal hypoglycemia and better post-dinner glucose control with bedtime insulin glargine compared with bedtime NPH insulin during insulin combination therapy in type 2 diabetes. HOE 901/3002 Study Group. Diabetes Care. 2000;23:1130–6.
Riddle MC, Rosenstock J, Gerich J, Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26:3080–6.
Rosenstock J, Dailey G, Massi-Benedetti M, Fritsche A, Lin Z, Salzman A. Reduced hypoglycemia risk with insulin glargine: a meta-analysis comparing insulin glargine with human NPH insulin in type 2 diabetes. Diabetes Care. 2005;28:950–5.
Yki-Järvinen H, Kauppinen-Mäkelin R, Tiikkainen M, et al. Insulin glargine or NPH combined with metformin in type 2 diabetes: the LANMET study. Diabetologia. 2006;49:442–51.
Rosenstock J, Fonseca V, Schinzel S, Dain MP, Mullins P, Riddle M. Reduced risk of hypoglycemia with once-daily glargine versus twice-daily NPH and number needed to harm with NPH to demonstrate the risk of one additional hypoglycemic event in type 2 diabetes: evidence from a long-term controlled trial. J Diabetes Complicat. 2014;28:742–9.
European Medicines Agency. Abasaglar (previously Abasria). 2014. https://www.ema.europa.eu/en/medicines/human/EPAR/abasaglar-previously-abasria. Accessed 28 Nov 2018.
Pharmaceutical and Medical Devices Agency. Review reports: drugs. http://www.pmda.go.jp/english/review-services/reviews/approved-information/drugs/0001.html#select9. Accessed 28 Nov 2018.
US Food and Drug Administration. Basaglar (insulin glargine injection). 2015. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205692Orig1s000TOC.cfm. Accessed 28 Nov 2018.
James J, Pollom RK, Hadjiyianni I, Buchholz G, Reed BL. Biosimilar insulins: what do you need to know? Int Diabetes Nurs. 2017;14:32–5.
Kim AP, Bindler RJ. The Future of biosimilar insulins. Diabetes Spectr. 2016;29:161–6.
World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 2009. http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed 28 Nov 2018.
European Medicines Agency. Guideline on similar biological medicinal products. 2014. https://www.ema.europa.eu/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Accessed 28 Nov 2018.
European Medicines Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues. 2015. https://www.ema.europa.eu/documents/scientific-guideline/guideline-non-clinical-clinical-development-similar-biological-medicinal-products-containing_en-0.pdf. Accessed 28 Nov 2018.
US Food and Drug Administration. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. 2015. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 28 Nov 2018.
US Food and Drug Administration. Guidance for industry: quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. 2015. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291134.pdf. Accessed 28 Nov 2018.
US Food and Drug Administration. Guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product. 2016. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed 28 Nov 2018.
Blevins TC, Dahl D, Rosenstock J, et al. Efficacy and safety of LY2963016 insulin glargine compared with insulin glargine (Lantus®) in patients with type 1 diabetes in a randomized controlled trial: the ELEMENT 1 study. Diabetes Obes Metab. 2015;17:726–33.
Linnebjerg H, Lam EC, Seger ME, et al. Comparison of the pharmacokinetics and pharmacodynamics of LY2963016 insulin glargine and EU- and US-approved versions of Lantus insulin glargine in healthy subjects: three randomized euglycemic clamp studies. Diabetes Care. 2015;38:2226–33.
Rosenstock J, Hollander P, Bhargava A, et al. Similar efficacy and safety of LY2963016 insulin glargine and insulin glargine (Lantus®) in patients with type 2 diabetes who were insulin-naïve or previously treated with insulin glargine: a randomized, double-blind controlled trial (the ELEMENT 2 study). Diabetes Obes Metab. 2015;17:734–41.
Hadjiyianni I, Dahl D, Lacaya LB, Pollom RK, Chang CI, Ilag LL. Efficacy and safety of LY2963016 insulin glargine in patients with type 1 and type 2 diabetes previously treated with insulin glargine. Diabetes Obes Metab. 2016;18:425–9.
Ilag LL, Deeg MA, Costigan T, et al. Evaluation of immunogenicity of LY2963016 insulin glargine compared with Lantus® insulin glargine in patients with type 1 or type 2 diabetes mellitus. Diabetes Obes Metab. 2016;18:159–68.
Byrd RA, Owens RA, Blackbourne JL, et al. Nonclinical pharmacology and toxicology of the first biosimilar insulin glargine drug product (BASAGLAR®/ABASAGLAR®) approved in the European Union. Regul Toxicol Pharmacol. 2017;88:56–65.
Ilag LL, Costigan TM, Deeg MA, et al. Clinical outcomes of patients with diabetes who exhibit upper-quartile insulin antibody responses after treatment with LY2963016 or Lantus® insulin glargine. Diabetes Ther. 2017;8:545–54.
Linnebjerg H, Lam EC, Zhang X, et al. Duration of action of two insulin glargine products, LY2963016 insulin glargine and Lantus insulin glargine, in subjects with type 1 diabetes mellitus. Diabetes Obes Metab. 2017;19:33–9.
Zhang X, Lam ECQ, Seger ME, et al. LY2963016 insulin glargine and insulin glargine (Lantus) produce comparable pharmacokinetics and pharmacodynamics at two dose levels. Clin Pharmacol Drug Dev. 2017;6:556–63.
Pollom RK, Costigan T, Lacaya LB, et al. Similar efficacy and safety of Basaglar® and Lantus® in patients with type 2 diabetes in age groups (< 65 years, ≥ 65 years): a post hoc analysis from the ELEMENT-2 study. Diabetes Ther. 2018;9:827–37.
World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–4.
Gerstein HC, Yale JF, Harris SB, Issa M, Stewart JA, Dempsey E. A randomized trial of adding insulin glargine vs. avoidance of insulin in people with Type 2 diabetes on either no oral glucose-lowering agents or submaximal doses of metformin and/or sulphonylureas. The Canadian INSIGHT (implementing new strategies with insulin glargine for hyperglycaemia treatment) study. Diabet Med. 2006;23:736–42.
Anderson RT, Skovlund SE, Marrero D, et al. Development and validation of the insulin treatment satisfaction questionnaire. Clin Ther. 2004;26:565–78.
International Diabetes Federation. IDF diabetes atlas. 8th ed. Brussels: International Diabetes Federation; 2017.
Chan JC, Malik V, Jia W, et al. Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA. 2009;301:2129–39.
Bakker LEH, Sleddering MA, Schoones JW, Meinders AE, Jazet IM. Pathogenesis of type 2 diabetes in South Asians. Eur J Endocrinol. 2013;169:R99–114.
Ma RCW, Chan JCN. Type 2 diabetes in East Asians: similarities and differences with populations in Europe and the United States. Ann NY Acad Sci. 2013;1281:64–91.
Rhee E-J. Diabetes in Asians. Endocrinol Metab. 2015;30:263–9.
Deerochanawong C, Bajpai S, Dwipayana IMP, et al. Optimizing glycemic control through titration of insulin glargine 100 U/mL: a review of current and future approaches with a focus on Asian populations. Diabetes Ther. 2017;8:1197–214.
Chan JCN, Bunnag P, Chan SP, et al. Glycaemic responses in Asian and non-Asian people with type 2 diabetes initiating insulin glargine 100 units/mL: a patient-level pooled analysis of 16 randomised controlled trials. Diabetes Res Clin Pract. 2018;135:199–205.
Gu T, Hong T, Zhang P, et al. Insulin glargine combined with oral antidiabetic drugs for Asians with type 2 diabetes mellitus: a pooled analysis to identify predictors of dose and treatment response. Diabetes Ther. 2018;9:771–87.
Ji L, Min KW, Oliveira J, Lew T, Duan R. Comparison of efficacy and safety of two starting insulin regimens in non-Asian, Asian Indian, and East Asian patients with type 2 diabetes: a post hoc analysis of the PARADIGM study. Diabetes Metab Syndr Obes. 2016;9:243–9.
Brown A, Guess N, Dornhorst A, Taheri S, Frost G. Insulin-associated weight gain in obese type 2 diabetes mellitus patients: what can be done? Diabetes Obes Metab. 2017;19:1655–68.
Kim SY, Pollom RK, Spaepen E, Huang C-N, Ahn KJ. Similar efficacy and safety of LY2963016 insulin glargine and insulin glargine (Lantus®) in East Asian subpopulation with type 2 diabetes mellitus in the ELEMENT 5 study. Poster session presented at: Korean Diabetes Association International Congress of Diabetes and Metabolism; 2018 Oct 11–13; Seoul, Korea.
Mohan V, Chadha M, Sahay R, et al. Similar efficacy and safety of LY2963016 and Lantus® insulin glargine products in Indian type 2 diabetes mellitus patients (subgroup of ELEMENT 5 study). Poster session presented at: 46th Annual Meeting of Research Society for the Study of Diabetes in India; 2018 Nov 22–25; Ahmedabad, India.
Acknowledgements
The authors acknowledge the investigators and patients who participated in this study.
Funding
This study and the journal’s article processing charges were funded by Eli Lilly and Company (Indianapolis, IN, USA) and Boehringer Ingelheim (Ingelheim am Rheim, Germany). All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing Assistance
The authors acknowledge Karen M. Paulsrud of Eli Lilly and Company for medical writing assistance. This was also funded by Eli Lilly and Company (Indianapolis, IN, USA) and Boehringer Ingelheim (Ingelheim am Rheim, Germany).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
Robyn K. Pollom, Liza L. Ilag, and Lyndon B. Lacaya contributed to the conception and/or design of the study. Ramón Ortiz Carrasquillo contributed to the acquisition of data. Robyn K. Pollom, Liza L. Ilag, Lyndon B. Lacaya, Tina M. Morwick, and Ramón Ortiz Carrasquillo contributed to the analysis and/or interpretation of data for the work and drafting and/or critical revision of the manuscript.
Prior Presentation
This work was previously published as an abstract presented at the American Diabetes Association’s 77th Scientific Sessions, San Diego, CA, USA, June 2017 (A249).
Disclosures
Robyn K. Pollom is an employee and minor stockholder of Eli Lilly and Company. Liza L. Ilag is an employee and minor stockholder of Eli Lilly and Company. Lyndon B. Lacaya is a former employee and minor stockholder of Eli Lilly and Company. Tina M. Morwick is a former employee of Eli Lilly and Company and Boehringer Ingelheim. Ramón Ortiz Carrasquillo has served as a speaker for AstraZeneca, Jansen, Merck (MSD), and Sanofi, and his institution has received funding from Boehringer Ingelheim, Eli Lilly and Company, Merck (MSD), Novartis, Novo Nordisk, and Sanofi for clinical research activities.
Compliance with Ethics Guidelines
All procedures performed were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration, as revised in 2013. The protocol was approved by the appropriate ethics review boards, and informed consent was obtained from all individual participants included in the study.
Data Availability
The data sets analyzed during the current study are not publicly available. Lilly provides access to all individual participant data collected during a trial after anonymization, with the exception of pharmacokinetic or genetic data. Access is provided after a proposal has been submitted and approved by an independent review committee identified for this purpose and after receipt of a signed data-sharing agreement. Data will be provided in a secure data sharing environment for up to 2 years per proposal. To submit a request, go to www.clinicalstudydatarequest.com.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https//doi.org/https://doi.org/10.6084/m9.figshare.7399460.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Pollom, R.K., Ilag, L.L., Lacaya, L.B. et al. Lilly Insulin Glargine Versus Lantus® in Insulin-Naïve and Insulin-Treated Adults with Type 2 Diabetes: A Randomized, Controlled Trial (ELEMENT 5). Diabetes Ther 10, 189–203 (2019). https://doi.org/10.1007/s13300-018-0549-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-018-0549-3