
Abstract
Introduction
Administered basal insulin markedly influences the fasting plasma glucose (FPG) level of individuals with type 1 diabetes. Insulin degludec (IDeg) and insulin glargine U300 (IGlar U300) are now available as ultra-long-acting insulin formulations, but whether or how their glucose-stabilizing effects differ remains unclear. We will compare the effects of these basal insulins on parameters related to blood glucose control, with a focus on day-to-day glycemic variability, in individuals with type 1 diabetes treated with multiple daily injections.
Methods
A multicenter, randomized, open-label, crossover, comparative study (Kobe Best Basal Insulin Study 2) will be performed at 13 participating institutions in Japan. A total of 46 C-peptide-negative adult outpatients with type 1 diabetes will be randomly assigned 1:1 by a centralized allocation process to IGlar U300 (first period)/IDeg (second period) or IDeg (first period)/IGlar U300 (second period) groups, in which subjects will be treated with the corresponding basal insulin for consecutive 4-week periods. The basal insulin will be titrated to achieve an FPG of less than 130 mg/dL initially and then less than 110 mg/dL if feasible. In the last week of each period, plasma glucose will be determined seven times a day by self-monitoring of blood glucose (SMBG) and intraday and day-to-day glucose excursions will be determined by flash glucose monitoring (FGM). The primary end point is comparison of day-to-day glycemic variability as evaluated by the standard deviation (SD) of FPG during the last week of each treatment period. Secondary end points include the coefficient of variance of FPG, the frequency of severe hypoglycemia as evaluated by SMBG, the duration of hypoglycemia as evaluated by FGM, intraday glycemic variability calculated from both SMBG and FGM data, and the administered insulin dose.
Planned Outcomes
The results of the study will be submitted for publication in a peer-reviewed journal to report differences in the effects of two ultra-long-acting basal insulins, IDeg and IGlar U300.
Conclusion
This head-to-head comparison will be the first study to compare the effects of IDeg and IGlar U300 on day-to-day FPG variability in C-peptide-negative individuals with type 1 diabetes.
Trial Registration
Registered in University Hospital Medical Information Network (UMIN) Clinical Trials Registry as 000029630 on 20 June 2017.
Funding
Novo Nordisk Pharma Ltd.
Similar content being viewed by others

Introduction
Background
In patients with type 1 diabetes who undergo basal-bolus insulin therapy with multiple daily injections, basal insulin greatly influences the stability of blood glucose levels in the fasting state. Insulin glargine U100 (IGlar U100; A21Gly, B31Arg, B32Arg human insulin at 100 U/mL) is a long-acting insulin analogue that has been widely and safely administered as a basal insulin [1, 2]. A more highly concentrated formulation of insulin glargine (IGlar U300, 300 U/mL) is now available and has been shown to have a longer duration of action and a more even activity profile compared with IGlar U100 [3, 4]. Insulin degludec (IDeg; Des (B30) LysB29 (γ-Glu Nε-hexadecandioyl) human insulin) is another ultra-long-acting insulin analogue. The duration of action for IDeg, estimated at approximately 42 h, is much longer than that for IGlar U100 [5, 6]. Fasting plasma glucose (FPG) variability, or day-to-day glycemic variability, has recently gained the attention of physicians because it has been associated with micro- and macrovascular complications [7, 8], hypoglycemia [8, 9], Alzheimer’s disease [10], as well as cancer incidence and cancer-related death [11] in individuals with diabetes. The differences in the effects of IGlar U300 and IDeg, in particular their effects on day-to-day glycemic variability, have remained unclear, however. We therefore designed the current study to determine the standard deviation (SD) of FPG levels in individuals with type 1 diabetes treated with basal-bolus insulin therapy including either IGlar U300 or IDeg.
Objectives
The aim of this study is to compare the effects of IDeg and IGlar U300 on day-to-day FPG variability in C-peptide-negative individuals with type 1 diabetes and thereby to reveal any clinically important differences between these two basal insulins. In addition, we will investigate the effects of these two insulins on other parameters related to blood glucose control such as duration of hypoglycemia and nocturnal glucose changes with the use of flash glucose monitoring (FGM).
Methods
Study Design and Ethics Compliance
This randomized, crossover, open-label, parallel-group, multicenter study (Kobe Best Basal Insulin Study 2) will be performed over 2 months with Japanese subjects with type 1 diabetes in accordance with the Declaration of Helsinki and its amendments. The trial was registered with the University Hospital Medical Information Network Clinical Trials Registry (UMIN-CTR) as UMIN 000029630 on 20 June 2017, and it has been approved by the ethics committees of 13 participating institutions listed in “Acknowledgements”. The study is to be conducted according to the Ethical Guidelines for Clinical Research published by the Japanese Ministry of Education, Culture, Sports, Science, and Technology and Ministry of Health, Labor, and Welfare (2014 revision), the ICH Consolidated Guideline for Good Clinical Practice, and applicable local laws and regulations. Written informed consent will be obtained from all subjects prior to their randomization.
Sample Selection
Patients must satisfy all of the following inclusion criteria and not meet any exclusion criteria. Inclusion criteria for the trial include (1) individuals with type 1 diabetes aged at least 20 years whose serum C-peptide immunoreactivity is less than 0.2 ng/mL (as confirmed at least twice); (2) treatment for at least 1 year with basal-bolus insulin injections, with IGar U100, IGlar U300, or IDeg as the basal insulin and a rapid-acting insulin analogue or regular insulin as the bolus insulin; (3) the ability to perform self-monitoring of blood glucose (SMBG); and (4) the ability to perform FGM. Exclusion criteria include (1) a glycated hemoglobin (HbA1c) level of 9.0% or higher; (2) use of medications that affect glucose metabolism (such as beta-blockers, corticosteroids, and monoamine oxidase inhibitors); (3) a history of myocardial infarction, angina, coronary bypass surgery, or heart failure within the previous 6 months; (4) severe hypertension (systolic blood pressure of at least 180 mmHg or diastolic blood pressure of at least 100 mmHg); (5) severe liver dysfunction (serum aspartate aminotransferase or alanine aminotransferase levels of at least 2.5 times the upper limit of normal); (6) severe renal impairment (serum creatinine concentration of at least 2.0 mg/dL); (7) frequently recurring severe hypoglycemia or hospitalization because of serious hypoglycemia or diabetic ketoacidosis within the previous year; (8) proliferative diabetic retinopathy with a high risk of hemorrhage; (9) existing or possible pregnancy or breastfeeding or no use of an adequate contraceptive method (adequate contraceptive measures as recommended by local regulation or practice); (10) a diagnosis of cancer; (11) a complicating psychiatric disorder; (12) alcoholism or other drug addiction; (13) diabetes other than type 1, or type 1 diabetes with remaining insulin secretory capacity; (14) rejection of SMBG or FGM; (15) declaration by an investigator that the individual is otherwise inappropriate for the study. The use of previous FGM or CGM is not included in exclusion criteria. Severe hypoglycemia is defined as an event associated with central nervous system manifestations during which the patient requires the assistance of another person. The judgment of frequent recurrence of hypoglycemia will be made by each attending physician. Participants are required both to be able to understand instructions for SMBG and FGM and to provide informed consent in order to be enrolled.
Rationale for Planned Sample Size
We previously found that the mean value for the SD of FPG was approximately 10 mg/dL smaller with IDeg than with IGlar U100 in individuals with type 1 diabetes [12]. To determine the sample size for the present study, we performed a preliminary analysis of the day-to-day variability in FPG for individuals with type 1 diabetes treated with IDeg as the basal insulin at Kobe University Hospital. The mean and SD values for the SD of FPG in 30 patients over 7 days were determined to be 48.5 and 30.8 mg/dL, respectively. The SD for the intrapatient difference was calculated on the basis of \( \sqrt {30.8^{2} + 30.8^{2} } \) and the noninferiority margin was set as ± 20 mg/dL. At a significance level of 5% and power of 80%, the sample size needed is 19 patients per group. Taking into account subject dropout and withdrawal of informed consent during the study period, we set the sample size at 23 patients per group, for a total of 46 patients.
Intervention
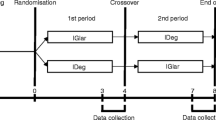
Enrollment and follow-up visits are outlined in Fig. 1. Individuals found to satisfy the study criteria will be randomly assigned to the IGlar U300 (first period)/IDeg (second period) and the IDeg (first period)/IGlar U300 (second period) groups by a centralized allocation process. In the IGlar U300/IDeg group, the basal insulin will be switched after 4 weeks from IGlar U300 (Lantus XR, SoloSTAR; Sanofi, Paris, France) to IDeg (Tresiba, FlexTouch; Novo Nordisk, Bagsvaerd, Denmark). In the IDeg/IGlar U300 group, the basal insulin will be switched after 4 weeks from IDeg to IGlar U300. The participants will be directed to determine their plasma glucose level four times a day (before breakfast, lunch, and dinner as well as at bedtime) by SMBG during the entire trial period. According to the half-life of IDeg (25.4 h) and IGlar U300 (19.0 h), 3 weeks is considered to be enough to washout the effect of previous insulin. The last week of each treatment period constitutes the data collection phase, during which the participants will be directed to determine their plasma glucose level seven times a day (before breakfast, 2 h after breakfast, before lunch, 2 h after lunch, before dinner, 2 h after dinner, and at bedtime). All subjects will be provided with the same device (OneTouch VerioVue; Johnson & Johnson, New Brunswick, NJ, USA) for SMBG and directed on its use. They will also be equipped with an FGM device (Freestyle Libre Pro; Abbott Diabetes Care, Alameda, California, USA) by medical staff on a visit midway through each treatment period, and the device will be removed by the subjects after 2 weeks for collection of data for the final week. Participants will store the detached FGM device until the next visit, when the data will be uploaded at the hospital or clinic. The data of FGM would be blinded to participants until the study end.

Study design. Eligible patients are randomly allocated to the IDeg/IGlar U300 group (upper arm) or the IGlar U300/IDeg group (lower arm). The basal insulin will be switched after 4 weeks as indicated. SMBG and FGM data will be collected and analyzed for the last week of each treatment period, and the insulin dose will be determined
Subjects will be instructed to administer basal insulin at the same time of day in both treatment periods. It should be started at a dose 10% lower than the previous insulin dose at switching in order to prevent unexpected hypoglycemia. If the FPG level is less than 130 mg/dL for more than 3 days, the addition of 1 U of basal insulin will be allowed (self-titration). On the other hand, if FPG is less than 70 mg/dL even for 1 day, the subject will be instructed to reduce the basal insulin dose by 2 U in order to avoid hypoglycemia. Attending physicians will be available to advise patients on basal insulin dose. The type of bolus insulin preparation will not be changed during the trial, and the preprandial bolus insulin dose will be adjusted by each participant. The target plasma glucose level before breakfast, lunch, and dinner as well as at bedtime will initially be less than 130 mg/dL and will subsequently be reduced to less than 110 mg/dL for individuals capable of achieving a reduction on the basis of patients-centered approach. Participants will be directed to avoid hypoglycemia (less than 70 mg/dL) at any time during the day.
Measurements and Planned Outcomes
The primary end point of the study is the day-to-day variability of FPG level as evaluated by the SD of the plasma glucose concentration determined by SMBG before breakfast during the last week of each 4-week treatment period (Fig. 1). Secondary end points include (1) the coefficient of variance for the plasma glucose level determined by SMBG before breakfast during the last week of each 4-week treatment period as an indicator of interday glycemic variability independent of FPG level itself; (2) the intraday glycemic variability calculated from the seven daily measurements of plasma glucose by SMBG or from FGM data during the last week of each treatment period; (3) the frequency of hypoglycemic events; (4) the duration of hypoglycemia determined by FGM; and (5) the administered basal and bolus insulin dose (the mean for the last week of each treatment period).
Safety Reporting
All adverse events that occur during the intervention period must be reported irrespective of their suspected causal relation to the study drug. They must be followed up until they have disappeared or for at least 2 weeks after completion of the study period (after discontinuation) and will be assessed with respect to insulin type, onset, outcome, severity, and causal relation to the study drug.
Data Analysis
Intergroup differences of normally or non-normally distributed data will be tested for significance with the unpaired Student’s t test or Mann–Whitney U test, respectively. A P value of less than 0.05 will be considered statistically significant. All statistical analysis will be performed with SPSS version 22.0 software.
Discussion
Several studies have shown that FPG variability is associated with diabetic complications, severe hypoglycemia, or death [7, 8], suggesting that stabilization of FPG should be a therapeutic target for individuals with diabetes. Moreover, administered basal insulin can greatly influence FPG variability, especially in C-peptide-negative individuals with type 1 diabetes treated with basal-bolus insulin injection. We previously compared IDeg and IGlar U100 with regard to their effects on FPG variability in such patients and found that IDeg yielded a lower FPG level and smaller day-to-day variability in FPG at a lower daily dose compared with IGlar U100 [12]. The longer and more stable action of IDeg is due both to its slower absorption from the injection site into the circulation as a result of its formation of soluble multihexameric chains and to its prolonged retention in the circulation as a result of its binding to albumin in blood [13]. On the other hand, IGlar U300 is a newly developed ultra-long-acting basal insulin that is superior to IGlar U100 in terms of its longer action and more even effect [3, 4]. As a result of its threefold more concentrated formulation, the microprecipitates of subcutaneously injected IGlar U300 have a longer residence time at physiological pH compared with IGlar U100 [3]. Although both IDeg and IGlar U300 are now available in clinical practice, differences in the effects of these ultra-long-acting basal insulins have not been established. The results of euglycemic glucose clamp studies to compare the stability of the glucose-lowering effects of injected IDeg and IGlar U300 have not been consistent [14,15,16]. Recently, the efficacy and safety of IDeg and IGlar U300 were compared in a head-to-head randomized controlled clinical trial in type 2 diabetes (BRIGHT Trial) [17]. The trial revealed that the two basal insulins have similar effects on HbA1c improvement (in primary outcome), and less occurrence of hypoglycemia in IGlar U300 than in IDeg (in secondary outcome) if target FPG level was set as 80–100 mg/dl. The information on the difference of fasting glycemic variability has still not been reported to date.
The current study is the first randomized controlled trial to directly compare the two ultra-long-acting basal insulins with regard to their effects on day-to-day glycemic variability in C-peptide-negative patients with type 1 diabetes. The results of our study should prove informative for selection of the best basal insulin for the treatment of type 1 diabetes in the clinical setting.
Conclusion
This head-to-head study will be the first trial to compare the effects of IDeg and IGlar U300 on day-to-day FPG variability in C-peptide-negative individuals with type 1 diabetes.
References
Goykhman S, Drincic A, Desmangles JC, et al. Insulin glargine: a review 8 years after its introduction. Expert Opin Pharmacother. 2009;10:705–18.
Gerstein HC, Bosch J, Dagenais GR, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. 2012;367:319–28.
Becker RH, Dahmen R, Bergmann K, et al. New insulin glargine 300 Units · mL−1 provides a more even activity profile and prolonged glycemic control at steady state compared with insulin glargine 100 Units · mL−1. Diabetes Care. 2015;38:637–43.
Dailey G, Lavernia F. A review of the safety and efficacy data for insulin glargine 300 units/ml, a new formulation of insulin glargine. Diabetes Obes Metab. 2015;17:1107–14.
Heise T, Nosek L, Bøttcher SG, et al. Ultra-long-acting insulin degludec has a flat and stable glucose-lowering effect in type 2 diabetes. Diabetes Obes Metab. 2012;14:944–50.
Birkeland KI, Home PD, Wendisch U, et al. Insulin degludec in type 1 diabetes: a randomized controlled trial of a new-generation ultra-long-acting insulin compared with insulin glargine. Diabetes Care. 2011;34:661–5.
Hirakawa Y, Arima H, Zoungas S, et al. Impact of visit-to-visit glycemic variability on the risks of macrovascular and microvascular events and all-cause mortality in type 2 diabetes: the ADVANCE trial. Diabetes Care. 2014;37:2359–65.
Zinman B, Marso SP, Poulter NR, et al. Day-to-day fasting glycaemic variability in DEVOTE: associations with severe hypoglycaemia and cardiovascular outcomes (DEVOTE 2). Diabetologia. 2018;61:48–57.
Niskanen L, Virkamäki A, Hansen JB, et al. Fasting plasma glucose variability as a marker of nocturnal hypoglycemia in diabetes: evidence from the PREDICTIVE study. Diabetes Res Clin Pract. 2009;86:e15–8.
Li TC, Yang CP, Tseng ST, et al. Visit-to-visit variations in fasting plasma glucose and HbA1c associated with an increased risk of Alzheimer disease: Taiwan Diabetes Study. Diabetes Care. 2017;40:1210–7.
Lin CC, Li CI, Liu CS, et al. Annual fasting plasma glucose variation increases risk of cancer incidence and mortality in patients with type 2 diabetes: the Taichung Diabetes Study. Endocr Relat Cancer. 2012;19:473–83.
Nakamura T, Sakaguchi K, So A, et al. Effects of insulin degludec and insulin glargine on day-to-day fasting plasma glucose variability in individuals with type 1 diabetes: a multicentre, randomised, crossover study. Diabetologia. 2015;58:2013–9.
Jonassen I, Havelund S, Hoeg-Jensen T, et al. Design of the novel protraction mechanism of insulin degludec, an ultra-long-acting basal insulin. Pharm Res. 2012;29:2104–14.
Heise T, Nørskov M, Nosek L, et al. Insulin degludec: lower day-to-day and within-day variability in pharmacodynamic response compared with insulin glargine 300 U/mL in type 1 diabetes. Diabetes Obes Metab. 2017;19:1032–9.
Bailey TS, Pettus J, Roussel R, et al. Morning administration of 0.4 U/kg/day insulin glargine 300 U/mL provides less fluctuating 24-hour pharmacodynamics and more even pharmacokinetic profile compared with insulin degludec 100 U/mL in type 1 diabetes. Diabetes Metab. 2018;44:15–21.
Heise T, Kaplan K, Haahr HL. Day-to-day and within-day variability in glucose-lowering effect between insulin degludec and insulin glargine (100 U/mL and 300 U/mL): a comparison across studies. J Diabetes Sci Technol. 2018;12:356–63.
Rosestock J, Cheng A, Ritzel R, et al. More similarities than differences testing insulin glargine 300 Units/mL versus insulin degludec 100 Units/mL in insulin-naïve type 2 diabetes: the randomized head-to-head BRIGHT trial. Diabetes Care. 2018;41:2147–54.
Acknowledgements
The authors would like to thank all participants of as well as investigators and staff supporting this study.
Principal Investigators at the Participating Centers
Kazuhiko Sakaguchi (Kobe University Hospital and Obara Hospital), Minoru Kishi (Nishiwaki Municipal Hospital), Akihiko Takeda (Shinko Hospital), Yoichi Tominaga (Takatsuki General Hospital), Tomoaki Nakamura (Kobe Rosai Hospital), Yasuo Kuroki (Kobe Century Memorial Hospital), Tomokazu Matsuda (Kaisei Diabetes Clinic), Keiji Iida (Hyogo Prefectural Kakogawa Medical Center), Michiko Kajikawa (Yodogawa Christian Hospital), Takeshi Ohara (Hyogo Brain and Heart Center), Kazuki Yokota (Yokota Medical Clinic), Kenta Hara (Kita-harima Medical Center), Sanshiro Tateya (Kakogawa Central City Hospital).
Funding
This study, including article processing charges, is funded by Novo Nordisk Pharma Ltd. No drugs will be donated or funded by the sponsor.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Authorship Contributions
Hiroshi Miura led the drafting of the manuscript. Kazuhiko Sakaguchi, Yuko Okada, Yoshikazu Tamori, and Wataru Ogawa reviewed the manuscript and study design and contributed to the final draft. Other authors will recruit participants.
Disclosures
Kazuhiko Sakaguchi has received research support from Novo Nordisk Pharma and lecture fees from Novo Nordisk Pharma and Sanofi. Yuko Okada has received research support from Novo Nordisk Pharma and Sanofi and lecture fees from Novo Nordisk Pharma and Sanofi. Yushi Hirota has received research support from Novo Nordisk Pharma and Sanofi and lecture fees from Novo Nordisk Pharma and Sanofi. Yoshikazu Tamori has received research support from Novo Nordisk Pharma and Sanofi and lecture fees from Novo Nordisk Pharma and Sanofi. Wataru Ogawa has received research support from Novo Nordisk Pharma and Sanofi, and lecture fees from Novo Nordisk Pharma and Sanofi. Akihiko Takeda has received lecture fees from Novo Nordisk Pharma and Sanofi. Yoichi Tominaga has received lecture fees from Novo Nordisk Pharma and Sanofi. Tomoaki Nakamura has received lecture fees from Novo Nordisk Pharma and Sanofi. Yasuo Kuroki has received lecture fees from Novo Nordisk Pharma and Sanofi. Tomokazu Matsuda has received lecture fees from Novo Nordisk Pharma and Sanofi. Keiji Iida has received lecture fees from Novo Nordisk Pharma and Sanofi. Takeshi Ohara has received lecture fees from Novo Nordisk Pharma and Sanofi. Kenta Hara has received lecture fees from Novo Nordisk Pharma and Sanofi. Sanshiro Tateya has received lecture fees from Novo Nordisk Pharma and Sanofi. All other authors, Hiroshi Miura, Natsu Otowa-Suematsu, Tomoko Yamada, Anna So, Hisako Komada, Minoru Kishi, Michiko Kajikawa and Kazuki Yokota declare no potential conflicts of interest.
Compliance with Ethics Guidelines
The investigators will provide an explanation of the study to each eligible participant using the informed consent document. The study is to be conducted according to the Ethical Guidelines for Clinical Research published by the Japanese Ministry of Education, Culture, Sports, Science, and Technology and Ministry of Health, Labor, and Welfare (2014 revision), the ICH Consolidated Guideline for Good Clinical Practice, and applicable local laws and regulations; and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Data Availability
Data sharing is not applicable to this article as it describes a study protocol.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7182485.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Miura, H., Sakaguchi, K., Okada, Y. et al. Effects of Insulin Degludec and Insulin Glargine U300 on Day-to-Day Fasting Plasma Glucose Variability in Individuals with Type 1 Diabetes: A Multicenter, Randomized, Crossover Study (Kobe Best Basal Insulin Study 2). Diabetes Ther 9, 2399–2406 (2018). https://doi.org/10.1007/s13300-018-0523-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-018-0523-0