
Abstract
Diabetes mellitus with autosomal dominant inheritance, i.e., maturity-onset diabetes of the young (MODY), is a genetic form of diabetes mellitus. The MODY phenotype is associated with gene mutations leading to pancreatic β-cell dysfunction. Here, we present the clinical case of a 50-year-old proband with familial diabetes mellitus in five generations (proband, her mother, grandmother, great-grandfather, and son). This disease is most likely associated with the novel Ser6Arg mutation in the HNF1A gene, which was identified in four family members. The mutation was not detected in MODY patients (126 subjects), in patients with type 2 diabetes mellitus (188 subjects), and in a general population sample (564 subjects).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM) with autosomal dominant inheritance, i.e., maturity-onset DM of the young (MODY), is a heterogeneous group of diseases caused by gene mutations that resulting in pancreatic β-cell dysfunction [1,2,3,4]. Verification of MODY allows for successful patient management, ensuring a healthy pregnancy and provision of genetic counseling to families [5–7]. Examination of relatives of MODY probands makes it possible to diagnose hyperglycemia in the preclinical phase.
To date, 14 genes have been identified to be associated with MODY. Mutations in each of these genes lead to the development of different MODY subtypes, differing in population distribution, in clinical features, and in management strategies [1, 8,9,10]. Despite the significant variation in the prevalence of individual forms of the disease in different populations, mutations in the genes coding for hepatocyte nuclear factor 1α (HNF1A) and glucokinase (GCK) are dominant types of mutation associated with MODY, namely, subtypes MODY3 (MODY-HNF1A) and MODY2 (MODY-GCK), respectively [11]. These subtypes account for up to 90% of all MODY cases [12]. In the UK, the Netherlands, and Denmark, the most common form of monogenic diabetes is MODY-HNF1A, whereas in Spain, Italy, France, Germany, and the Czech Republic, MODY-GCK prevails [13]. In Russia, the incidence of MODY-GCK and MODY-HNF1A is approximately equal [14].
In this report, we describe a clinical case in a family with MODY-HNF1A associated with a novel mutation in HNF1A.
Case Description
A 50-year-old woman with DM has been regularly visiting our department and her condition closely monitored. During the initial examination in September 2015, she complained of a burning sensation in the legs during the day, periodic headaches, and fatigue.
At the age of 12 years, symptoms of dry mouth and weakness appeared following the consumption of large amounts of carbohydrate-containing foods. These complaints lasted for approximately 2 months. During a routine medical check-up, the girl received a diagnosis of fasting (7 mmol/L) and postprandial hyperglycemia (11 mmol/L) and glycosuria; she did not have excess body weight, and there was no ketosis or ketoacidosis. Her weight and height corresponded to her age, and there were no signs of insulin resistance, such as acanthosis nigricans. Concomitant pathology and cardiovascular diseases were absent. Autoimmune processes were not detected, and antibodies to B cells were absent. As recommended by the physicians, she went on a diet restricting the intake of easily digestible carbohydrates. The patient was advised to participate in physical activities on a regular basis. Until 1987, she was normoglycemic on this adjusted lifestyle. In 1987, at age 21 years, she became pregnant for the first time, and in week 22 of the pregnancy, a diagnosis of gestational DM was made. Insulin therapy as the basal-bolus regimen was initiated. In 1988, stimulated delivery (36 weeks) resulted in a birth of a boy weighing 4000 g. After the delivery, the insulin therapy was discontinued, and the patient remained on a strict diet. From 1988 to 1994, she was normoglycemic following this regimen of a strict diet and a regular physical activity. In 1994, at the age of 28 years, stress-associated carbohydrate metabolism decompensation developed, with fasting glycemia (10 mmol/L), postprandial glycemia (16 mmol/L), glycosuria, ketonuria, and a weight loss of 7 kg. Insulin therapy as the basal-bolus regimen was resumed and is currently maintained. The patient uses insulin Degludec at the dose of 10 U/day and insulin aspart at 1 U per carbohydrate unit (CU) (averaging 18–20 U/day). From 1994 to 2002, fasting glycemia was in the range of 6–8 mmol/L, and postprandial glycemia ranged from 6 to 10 mmol/L. In 2002, she was diagnosed as non-proliferative diabetic retinopathy with peripheral polyneuropathy. She had no other diabetic complications or comorbidities. Since 2012, the patient has not been examined by an ophthalmologist; therefore, the stage of retinopathy at this time is not known. Testing for pancreas islet cell antibodies (ICAs) and anti-glutamic acid decarboxylase (GAD) antibodies yielded negative results. The C-peptide level was 338 pmol/L (reference range 298–2350), and glycated hemoglobin (HbA1c) was 7.7%. Blood biochemical analysis identified a moderate increase in low-density lipoprotein (LDL) cholesterol: 3.4 mmol/L (reference values are < 2.6 mmol/L). The body–mass index (BMI) is 24.5 kg/m2.
Due to a family history of DM, the son of the proband limited his consumption of easily digestible carbohydrates in childhood and adolescence and was monitored regularly for glycemia; hyperglycemia has not been detected to date. In the same periods, there were no problems with body weight. At age 21, he developed stress-associated symptoms of headaches, fatigue, and sleep disorder. Examination identified fasting (8–10 mmol/L) and postprandial (12.8 mmol/L) hyperglycemia, resulting in a diagnosis of DM. Treatment with sulfonylurea (SU) preparations was recommended, but the patient opted for the basal-bolus regimen of insulin therapy. Currently, he uses insulin degludec at 6 U/day and insulin aspart at 1 U/CU (averaging 15 U/day). Antibodies to β-cells and GAD are absent. The basal C-peptide level is low: 260 pmol/L (reference range 298–2350); HbA1c is 7.1%. Blood biochemical analysis revealed an increase in LDL cholesterol (3.2 mmol/L; reference values < 2.6 mmol/L). No diabetes-related complications have been detected. The respiratory, cardiovascular, and digestive systems of the patient are free of pathologies. Pulse in the feet is adequate, and the sensitivity is normal. Signs of insulin resistance are absent, and the BMI is 22.4 kg/m2.
The mother of the proband is now 72 years old. The diagnosis of DM in her daughter contributed to regular control of glucose. Hyperglycemia was diagnosed at the age of 45 years during a routine examination; no complaints were registered, and she had no problems with weight. Postprandial hyperglycemia was usually observed. The patient attained and maintained normoglycemia via diet therapy for 2 years, then by taking gliclazide for 5 years. At age 52 years (1997), carbohydrate metabolism decompensation presented, mostly as postprandial hyperglycemia; insulin actrapid was prescribed (instead of gliclazide) at 0.5 U/CU, which the patient is currently taking. The mother of the proband was diagnosed with diabetic peripheral polyneuropathy with dyslipidemia. Among comorbidities, there are arterial hypertension since age 40 years (without permanent hypotensive therapy) and esophagus diverticulum.
The proband, her mother, and her son have received higher education, do not smoke, and adhere to the dietary and physical exercise regimen recommended for DM.
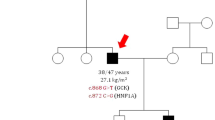
In the family history of the proband, DM was also present in her maternal grandmother and great-grandfather, i.e., in five generations (Fig. 1a). The disease in both relatives manifested at the age of 65–66 years against the background of long-term stressful conditions. The great-grandfather took the medications of SU (carbutamidum) and died at age 74 years after repeated acute cerebrovascular disease. The grandmother also took SU (gliclazide) medications in doses smaller than the average therapeutic ones, with good glycemic control, and died at the age of 91; her medical history included arterial hypertension and a repeated acute cerebrovascular disease.

a Family history and the novel variant of the gene encoding hepatocyte nuclear factor 1α (HNF1a) in a family with inherited diabetes mellitus (DM). Asterisk indicates medically examined HNF1a Ser6Arg mutation carriers. b Conserved amino acid sequence of the HNF1a gene among various species. c Sequence with amino acid substitution
The proband, her son, and her mother presented with dyslipidemia at a young age, and other relatives who had DM developed macrovascular complications at an advanced age. Almost all of these patients achieved and maintained normoglycemia for some time via the recommended diet; this result is indicative of a non-aggressive onset of this type of DM. In the proband, her son, and her mother, all of whom used pharmacotherapy to manage their DM, stress-induced carbohydrate metabolism decompensation eventually developed, leading to dose escalation.
Because of the characteristic features of the disease (familial aggregation of DM, dyslipidemia, and macrovascular complications, a blood glucose increase at normal body mass, the use of small doses of insulin, the stable C-peptide level, and no anti-islet antibodies), MODY-HNF1A was suspected.
The study protocol was approved by the local Ethics Committee of the Institute of Internal and Preventive Medicine (a branch of the Institute of Cytology and Genetics, the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia). Written informed consent to be examined and to participate in the study was obtained from each patient. For individuals younger than 18 years, the informed consent was signed by a parent or legal guardian. Genomic DNA for sequencing was isolated from leukocytes of the venous blood by phenol–chloroform extraction [15].
Forward and reverse primers for exons and exon boundaries were designed in the Primer-Blast software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The oligonucleotides are shown in Electronic Supplementary Material Table 1. PCRs were set up using BioMaster LR HS-PCR (2×) (BiolabMix, Novosibirsk, Russia). The PCR program consisted of an initial denaturation at 94 °C for 3 min, followed by 35 cycles at 94 °C for 30 s, 68 °C for 30 s, and 72 °C for 50 s. The PCR products were evaluated by electrophoresis in a 5% polyacrylamide gel after visualization with an ethidium bromide solution. A 100-bp DNA ladder (SibEnzyme, Novosibirsk, Russia) was run on each gel as molecular size markers. The amplicons were purified using Agencourt AMPure Xp beads (Beckman Coulter, Palo Alto, CA). The sequencing reactions were carried out on an automated ABI 3500 DNA sequencer (Thermo-Fisher Scientific, Waltham, MA) with the BigDye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific). The sequences were analyzed using Vector NTI® Advance software (Thermo Fisher Scientific). The hg19 version of the human genome served as a reference sequence for the alignment.
The method of direct automated Sanger sequencing was applied to the DNA of the proband and to the DNA of the available members of her family, i.e., the proband’s mother, son, and normoglycemic granddaughter aged 2 months, to analyze exons and splice sites of the HNF1A gene. The examination revealed a novel heterozygous Ser6Arg substitution (AGC > AGA) in exon 1 of the HNF1A gene in all family members tested. This substitution is located in a highly conserved region. The affected position is also conserved among species (Fig. 1b) and is located in the domain responsible for protein dimerization [16]. The possible functional and significant effects of HNF1A missense variants were predicted by means of in silico tools PolyPhen-2 v.2.2.5 [17], SIFT, and PROVEAN [18]. The Web services PolyPhen-2 v.2.2.5 and PROVEAN/SIFT predict a possible impact of an amino acid substitution on the structure and function of a human protein. The detected variant was predicted to be ‘potentially pathogenic’ by PolyPhen-2, with a score of 0.815, and ‘damaging’ by SIFT with a score of 0.001.
In the HUNT2 study,, the potentially pathogenic mutation Ser6Arg was detected in codon 6, but at another nucleotide position, AGC > AAC, in a Norwegian patient with MODY [19]. It was not detected in 838 Norwegian non-diabetic HUNT2 control subjects.
We tested the HNF1A gene for Ser6Arg in three groups. The first group consisted of 564 randomly selected patients, the second group comprised 188 patients with DM type 2 (DM2), and the third group comprised 126 patients with the MODY phenotype. All analyses were conducted at the Institute of Internal and Preventive Medicine, Novosibirsk.
The general patient group included in the analysis was selected from a survey of the population interviewed within the framework of the HAPIEE project [20], Novosibirsk, Russia (9360 participants, aged 45–69 years, and 50% men, 97% Caucasians). A total of 564 randomly selected patients (mean age 54.2 ± 0.4) were included in our study. The DM2 group consisted of 68 women and 87 men [mean age 59.0 ± 6.7 years; mean BMI 31 kg/m2 (men) and 32 kg/m2 (women)]. The DM2 sample was arbitrarily selected from the HAPIEE project [20]. We used the criteria of the American Diabetes Association (http://care.diabetesjournals.org/content/40/Supplement_1) for the diagnosis of DM2. A total of 126 patients with the MODY phenotype (mean age 23.8 ± 2.6 years) underwent a full medical examination a some specified time between 2014 and 2017, including a biochemical blood test; determination of HbA1c, C-peptide, GAD, ICA, thyroid status, and microalbuminuria; abdominal and renal ultrasonography; real-time continuous glucose monitoring with Medtronic Paradigm MMT-722 (Meditronic, Dublin, Ireland); and genetic tests.
Ser6Arg was genotyped using the TaqMan SNP assay (BioLabMix, Novosibirsk, Russia) and the StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, USA).
Testing for the Ser6Arg substitution in the HNF1A gene yielded negative results in the population sample of HAPIEE, in the sample of 188 subjects with DM2, and among the 126 patients with a clinical pattern of MODY.
Discussion
Young patients with a carbohydrate metabolism imbalance may receive a diagnosis of type 1 diabetes mellitus (DM1), DM2, or rarer monogenic forms of DM [21]. The onset of our patient’s disease was characterized by a lack of some features of the most common types of DM. The absence of ketonuria and ketoacidosis is atypical; no antibodies to GAD or ICA were detected, which are present in more than 70% of patients with manifestations of DM1 [22]. The C-peptide level was normal, but this result can be observed at the early stages of DM1. The patient was not overweight and did not have clinical manifestations of insulin resistance, such as acanthosis nigricans, which is characteristic of early-onset DM2 [23]. The diagnosis of MODY was supported by the DM diagnosis in five generations, stable C-peptide secretion, and dyslipidemia at a young age without overweight.
This case shows specific features of DM associated with the novel potentially pathogenic mutation Ser6Arg in the HNF1A gene. This gene encodes one of the transcription factors regulating the expression of genes associated with lipid and carbohydrate metabolism and the synthesis of acute-phase proteins [3]. This may account for the presence of dyslipidaemia and, as a consequence, cardiovascular diseases in patients with mutations in the HNF1A gene. Patients with diabetes and HNF1A mutation carriers have a glucose-induced disturbance of insulin secretion. The fasting C-peptide level remains within the reference range for a long time, as seen in this clinical case, and no insulin resistance is detected [6]. Until the age of 10 years, the majority of carriers of other HNF1A mutations have fasting normoglycemia; in adolescents, fasting glycemia is slightly higher than the upper limit of the reference range followed by progressive impaired glucose tolerance and postprandial hyperglycemia most of the time [24], as seen in the clinical case under study. Generally, the disease manifests itself mostly in the post-pubertal period; the average age of hyperglycemia diagnosis is 21–26 years. Localization of the mutation in gene HNF1A is known to affect the age at which the MODY diagnosis is confirmed. Thus, the average age of MODY diagnosis is 7 years less if the mutation is located in exons 1–6 of the HNF1A gene (in contrast to exons 8–10 of the gene) [8].
In the clinical case presented, the substitution was detected in exon 1, the age of hyperglycemia detection in the proband was 12 years, but DM manifested itself at the age of 21 years.
The earliest clinical marker for the disease may be glycosuria, which is related to impaired renal glucose reabsorption [1]. There have been documented cases of micro- and macrovascular complications in MODY-HNF1A patients, which highlights the need to control blood glucose levels in these patients [25]. The relatives of the proband also showed macrovascular complications.
In addition to the development of DM, mutations in the HNF1A gene are related to liver neoplasms, such as hepatocellular adenoma (a rare benign hepatic tumor that generally occurs in young women using oral contraceptives), and, in rare cases, to hepatocellular carcinomas [26]. Generally, clinical manifestations of MODY-HNF1A range from subclinical hyperglycemia to pronounced decompensation of carbohydrate metabolism among different patients and even among relatives in the same family with a pathogenic mutation [10].
SU preparations (SUP) are the main treatment. HNF1A mutation carriers are known to be more sensitive to the hypoglycemic effects of SUP than are DM2 patients [1]. In the case under study, the proband’s mother was able to achieve normoglycemia when taking SUP, also for extended periods. According to the literature, approximately 50% of MODY-HNF1A patients with progressive disease require insulin therapy [27].
Manifestation of DM during pregnancy, accompanied by a substantial increase in glycemia, the lack of ketosis, development of microvascular complications (retinopathy and neuropathy) with a short clinical course, and a combination of hyperglycemia with dyslipidemia are the specific signs of DM in the patient under study.
Conclusion
This case reveals unusual features of DM associated with the novel Ser6Arg substitution in the HNF1A gene (subtype MODY3). This nucleotide change was not detected in a general population sample and was able to cause MODY3 in the proband and her family. This case illustrates the particular importance of a personalized approach to diagnosis and treatment for identifying an atypical course of DM in the young with a family history of hyperglycemia.
Abbreviations
- DM:
-
Diabetes mellitus
- DNA:
-
Deoxyribonucleic acid
- GAD:
-
Anti-glutamic acid decarboxylase
- HNF1A:
-
Gene of hepatocyte nuclear factor 1α
- ICA:
-
Pancreas islet cell antibodies
- LDL:
-
Low-density lipoprotein
- MODY:
-
Maturity onset diabetes of the young
References
Lachanse CH. Practical aspects of monogenic diabetes: a clinical point of view. Can J Diabetes. 2016;40:368–75.
Rubio-Cabezas O, Hattersley AT, Njolstad PR, et al. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2014;15(20):47–64.
Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 2011;34(8):1878–84.
Stanik J, Dusatkova P, Cinek O, et al. De novo mutations of GCK, HNF1a and HNF 4a may be more frequent in MODY than previously assumed. Diabetologia. 2014;57:480–4.
Voevoda MI, Ivanova AA, Shakhtshneider EV, et al. Molecular genetics of maturity-onset diabetes of the young. Ther Arch. 2016;88(4):117–24.
Pihoker C, Gilliam MK, Ellard S, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1a, HNF4a, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab. 2013;98:4055–62.
Agarwal SK, Khatri S, Prakash N, et al. Maturity onset diabetes of young. JIACM. 2002;3(3):271.
Thanabalasingham G, Pal A, Selwood MP, et al. Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care. 2012;35:1206–2012.
McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem. 2013;50(Pt 5):403–15.
Bonnefond A, Philippe J, Durand E, et al. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS One. 2012;7(6):e37423. doi: 10.1371/journal.pone.0037423
Delvecchio M, Ludovico O, Menzaghi C, et al. Low prevalence of HNF1A mutations after molecular screening of multiple MODY genes in 58 Italian families recruited in the pediatric or adult diabetes clinic from a single Italian hospital. Diabetes Care. 2014;37:258–60.
Thanabalasingham G, Owen KR. Diagnosis and management of maturity onset diabetes of the young (MODY). BMJ. 2011;343:d6044. doi: 10.1136/bmj.d6044
Shields BM, Hicks S, Shepherd MH, et al. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504–8.
Kuraeva TL, Sechko EA, Eremina IA, Ivanova ON, Proko’ev SA. MODY3 in the child with type 2 diabetes mellitus phenotype: case report. Diabetes mellitus. 2013;2(59):88–93.
Sambrook J, Russell DW. Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc. 2006;(1). doi: 10.1101/pdb.prot4455
Mendel DB, Crabtree GR. HNF-1, a member of a novel class of dimerizing homeodomain proteins. J Biol Chem. 1991;266(2):677–80.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Eide SA, Raeder H, Johansson S, et al. Prevalence of HNF1A (MODY3) mutations in a Norwegian population (the HUNT2 Study). Diabet Med. 2008;25(7):775–81. https://doi.org/10.1111/j.1464-5491.2008.02459.x.
Pajak A, Szafraniec K, Kubinova R, et al. Binge drinking and blood pressure: cross-sectional results of the HAPIEE Study. PLoS One. 2013;8(6):e65856. https://doi.org/10.1371/journal.pone.0065856.
Ovsyannikova AK, Rymar OD, Shakhtshneider EV, et al. ABCC8-related maturity-onset diabetes of the young (MODY 12). Diabetes Ther. 2016;7:591–600.
Shields BM, Peters JL, Cooper C, et al. Is there an evidence base for the clinical features used to differentiate type 1 from type 2 diabetes? A systematic review of the literature. Diabetologia. 2014;57(11):439–40.
Shields BM, McDonald TJ, Ellard S, et al. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia. 2012;55:1265–72.
Bellanné-Chantelot C, Carette C, Riveline J, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2008;57:503–8.
Steele AM, Shields BM, Shepherd M, et al. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med. 2010;27:157–61.
Zucman-Rossi J, Jeannot E, Nhieu JT, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43(3):515–24.
Wobser H, Bonner C, Nolan JJ, et al. Downregulation of protein kinase B/Akt-1 mediates INS-1 insulinoma cell apoptosis induced by dominant-negative suppression of hepatocyte nuclear factor-1alpha function. Diabetologia. 2006;49:519–26.
Acknowledgements
Funding
This work and article processing charges have been supported by the Russian Science Foundation (No. 14-15-00496-P). The English language was corrected and certified by Shevchuk Editing. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. The authors thank the patients for participation in this study.
Disclosures
Alla K. Ovsyannikova, Oksana D. Rymar, Dinara E. Ivanoshchuk, Svetlana V. Mikhailova, Elena V. Shakhtshneider, Pavel S. Orlov, Ekaterina S. Malakhina and Mikhail I. Voevoda have nothing to disclose.
Compliance with Ethics Guidelines
The study protocol was approved by the local Ethics Committee of the Institute of Internal and Preventive Medicine (a branch of the Institute of Cytology and Genetics, the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia). Written informed consent to be examined and to participate in the study was obtained from each patient. For individuals younger than 18 years, the informed consent was signed by a parent or legal guardian.
Data Availability
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Content
To view enhanced content for this article go to http://www.medengine.com/Redeem/9AFCF06002DED385.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ovsyannikova, A.K., Rymar, O.D., Ivanoshchuk, D.E. et al. A Case of Maturity Onset Diabetes of the Young (MODY3) in a Family with a Novel HNF1A Gene Mutation in Five Generations. Diabetes Ther 9, 413–420 (2018). https://doi.org/10.1007/s13300-017-0350-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-017-0350-8