
Abstract
Glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1 RAs) are useful tools for treating type 2 diabetes mellitus. In their recent position statement, the American Diabetes Association and European Association for the Study of Diabetes recommend GLP1-RAs as add-on to metformin when therapeutic goals are not achieved with monotherapy, particularly for patients who wish to avoid weight gain or hypoglycemia. GLP1-RAs differ substantially in their duration of action, frequency of administration and clinical profile. Members of this class approved for clinical use include exenatide twice-daily, exenatide once-weekly, liraglutide and lixisenatide once-daily. Recently, two new once-weekly GLP1-RAs have been approved: dulaglutide and albiglutide. This article summarizes properties of short- and long-acting GLP-1 analogs, and provides useful information to help choose the most appropriate compound for individual patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1 RAs) are useful tools for treating type 2 diabetes mellitus (T2DM). In their recent position statement, the American Diabetes Association and European Association for the Study of Diabetes recommend GLP1-RAs as add-on to metformin when therapeutic goals are not achieved with monotherapy, particularly for patients who wish to avoid weight gain or hypoglycemia [1]. This article summarizes properties of short- and long-acting GLP-1 analogs, providing useful information for choosing the most appropriate compound for individual patients.
Methods
The present paper is based on a review of recent publications on GLP-1 RA therapy and data from controlled clinical trials undertaken to investigate properties, functions, efficacy and safety of GLP RAs. Searches of PubMed were conducted for articles published between December 2013 and July 2014 using the terms “GLP-1 receptor agonist therapy”, “GLP-1 and extraglycemic effects”, “lixisenatide”, “exenatide”, “liraglutide”, “dulaglutide”, “albiglutide”, and “long-acting GLP-1 RA”.
For the introduction, we considered articles published between 1996 and 2013 on the biology and physiology of the incretin hormones and their role in the pathophysiology of T2DM. We focused on recent reviews on GLP-1 RA, meta-analyses and controlled clinical trials (January 2005 to October 2014). In particular, we analyzed controlled clinical trials comparing short- and long-acting GLP-1 RA, GLP-1 RA versus insulin, and GLP-1 versus oral agents. We also examined two meta-analyses: one around the efficacy and safety of incretin therapy, and the other comparing exenatide once-weekly or liraglutide once-daily with insulin glargine, exenatide twice-daily or placebo.
Our goal was to analyze the therapies for diabetes in use today and emphasize the mechanism and clinical efficacy of GLP-1 RA therapy. We analyze the molecules which are actually approved by Food Drug Administration (FDA) and European Medicines Agency (EMA).
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
The Physiological Role of Incretins
The incretin notion is based on the observation that the insulin response from ingested glucose is larger and more sustained than that from intravenously administered glucose, suggesting that substances produced in the gastrointestinal tract in response to meals (“incretins”) stimulate insulin release [2, 3]. Two incretins have been identified: gastric inhibitory polypeptide (GIP), which is secreted by enteroendocrine K-cells in the proximal gut, and glucagon-like peptide-1 (GLP-1), which is secreted mainly by L-cells located in the distal ileum. Within minutes of eating, the active forms of GIP and GLP-1 are released into the circulation and act by binding and activating specific G-protein coupled receptors expressed on β-cells and other targets, which rapidly increases exocytosis of insulin granules. Both GIP and GLP-1 are then rapidly inactivated by the ubiquitous serine protease dipeptidyl peptidase-4 (DPP-4). Long-term effects include stimulation of insulin synthesis [4], an increase in β-cell proliferation and a reduction in apoptosis [5]. GLP-1 also improves the glycemic profile by inhibiting glucagon secretion, delaying gastric emptying, and reducing food intake. GLP-1 may also improve glucose disposal in peripheral tissues (Fig. 1) [6–9]. GLP-1 may have an affect on tissues that are not directly involved in glucose metabolism, including protection against myocardial ischemia or reperfusion injury [10, 11]. In blood vessels, it protects against endothelial dysfunction [12], and promotes endothelium-independent artery relaxation [13]. It may have renal protective effects through increases in diuresis and natriuresis [14, 15]. These actions may lower blood pressure and have favorable effects on markers of cardiovascular risk, including brain natriuretic peptide and plasminogen activator inhibitor [16].

Physiology of GLP-1 secretion and action on GLP-1 receptors in different organs and tissues. GLP-1 is produced postprandially by intestinal L-cells. Through activation of insulin receptors on β-cells GLP-1 (like GIP) stimulates insulin biosynthesis and secretion and inhibits glucagon secretion in the pancreas, which in turn reduces hepatic gluconeogenesis. GLP-1 release also exerts protective effects on the heart and brain. Insulin sensitivity in the periphery is increased by improved insulin signaling and reduced gluconeogenesis. GI gastrointestinal, GIP gastric inhibitory polypeptide, GLP-1 glucagon-like peptide-1. Modified with permission from Pratley and Gilbert [106]
Pathophysiological Mechanism
In subjects with normal glucose tolerance, the incretin effect accounts for about two-thirds of the insulin response to an oral load, whereas in patients with T2DM this value is less than 20% [3, 17]. Thus, the incretin response may be particularly important during the postprandial period and impaired response may lead to postprandial hyperglycemia.
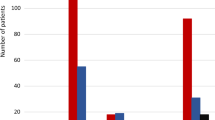
The hypothesis that meal-induced GLP-1 secretion is impaired in patients with T2DM versus control subjects is controversial. A large cross-sectional study by Toft-Nielsen et al. [18] showed that meal-induced GLP-1 responses were significantly reduced in patients with T2DM; however, in other studies they were similar to those in healthy participants (Fig. 2), and were not significantly different in a meta-analysis of 189 patients with T2DM and 217 healthy controls [19].

Responses of "total" GLP to oral glucose or mixed meals in patients with T2DM and control subjects. Integrated responses of "total" GLP to oral glucose or mixed meals based on individual studies reporting integrated incremental "total" GLP-1 responses in patients with T2DM and an appropriate control group (weight-matched, non-diabetic participants) and using non-specific assays that measured intact and DPP-4-degraded forms of GLP-1. The response in patients with T2DM (mean ± SEM) is expressed as percentage of the mean value in the control group. *P < 0.05 versus control. The numbers in bars indicate the number of patients with T2DM (upper row) and control participants (lower row) studied. Light blue oral glucose, dark blue mixed meal. DPP-4 dipeptidyl peptidase-4, GLP glucagon-like peptide, T2DM type 2 diabetes mellitus, SEM standard error of mean. Modified with permission from Nauck et al. [19]
A study performed under hyperinsulinemic–euglycemic clamp conditions, to maintain the same glucose and insulin levels in diabetic patients and matched control subjects, showed that GLP-1 response to oral glucose was reduced in patients with T2DM [20]. Because high glucose levels are known to induce DPP-4 expression [21], it has been hypothesized that chronic hyperglycemia may increase GLP-1 clearance, causing lower levels of circulating active GLP-1 [22]. However, no reduction in elimination rates of GLP-1 has been observed in patients with T2DM and mild-to-moderate hyperglycemia [23]. Thus, there appears to be some variation in GLP-1 secretion and/or inactivation, and in some cohorts the GLP-1 response was somewhat reduced, whereas in other studies such differences were not as apparent (Fig. 2) [19]. Impairment of the GLP-1 axis could be the consequence, rather than the cause, of hyperglycemia, establishing a vicious cycle that contributes to the maintenance of elevated glucose levels in T2DM, rather than to the pathogenesis of the disease.
Incretin-Based Therapies
Twenty-seven years after the first publication by Nauck in Diabetologia [17], our understanding of the role of incretins in the pathophysiology of T2DM has made great advances [22]. We now recognize that, although both GIP and GLP-1 stimulate insulin secretion in response to glycemic excursions, GLP-1 also influences gastric emptying, satiety and glucagon secretion [24].
Native GLP-1 has not advanced as a therapeutic agent because of its rapid degradation by DPP-4 [25]. The therapeutic potential of GLP-1 has been realized using two pharmacologic approaches; first, mimicking and focusing on GLP-1 via GLP-1 receptor agonists; and second, inhibiting the action of DPP-4 via DPP-4 inhibitors [26, 27].
A relevant difference between the DPP-4-resistant GLP-1 RAs and DPP-4 inhibitors is the route of administration: GLP-1 RAs require subcutaneous injection, whereas all DPP-4 inhibitors are oral agents, which may be preferred by patients. However, subcutaneous injection of GLP-1 RAs stimulates insulin secretion more strongly than oral ingestion of DPP-4 inhibitors [28]. This difference is also due to the fact that, although DPP-4 inhibition results in supra-physiological levels of endogenous GLP-1, GLP-1 RAs provide pharmacological levels of stimulation and more glucose-lowering efficacy [6, 24, 28]. Data from animal studies suggest that the effects of systemic versus local intestinal inhibition of DPP-4 activity may be different [29]. DPP-4 inhibition may influence glycemia by activating incretin receptors, preventing the release of bioactive peptides and affecting parasympathetic control of the digestive tract [29]. In addition, unlike DPP-4 inhibitors, GLP-1 RAs slow gastric emptying, increase satiety and promoting weight loss [6, 24, 28]. The difference may be explained by the effect of DPP-4 inhibitors on the degradation of GIP and neuropeptide Y, which have opposing effects on gastric motility and satiety [24].
The extraglycemic effects of incretin-based treatments are also promising. β-cell function is improved during treatment with incretin agents, and pre-clinical models show beneficial effects on β-cell regeneration and function. The positive effects of incretins on β-cells may explain, at least partly, the remission of diabetes documented in obese patients undergoing some types of bariatric surgery. Different bariatric surgery procedures result in distinct anatomical rearrangements of the gut axis with different responses in terms of gut hormone levels and remission of diabetes. The ability of GLP-1 to enhance postprandial insulin secretion in patients who have undergone Roux-en-Y bypass surgery may also result in the hyperinsulinemic hypoglycemia experienced by some patients [30].
The potential cardiovascular benefits of incretins have attracted much attention. Reduction of blood pressure, improvement in lipid profile and endothelial/myocardial function have been documented in several pre-clinical and clinical studies, supporting potential beneficial effects on cardiovascular outcomes [31]. Data from animal models suggest that the cardioprotective and vasodilatory effects of GLP-1 are independent of the cyclic adenosine monophosphate (cAMP)-linked GLP-1 receptor and are likely mediated by the GLP-1 (9–36) metabolite [32].
Lønborg et al. showed that exenatide had a positive effect on myocardial salvage at the time of reperfusion in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention [33]. The mechanism of exenatide-mediated protection against reperfusion injury is yet to be fully clarified. Two key phenomena in reperfusion injury appear to be the loss of mitochondrial integrity [34] and myocyte hypercontracture associated with sarcolemmal rupture [35]. A large body of experimental research suggests that reperfusion injury may be ameliorated by activation of a receptor-mediated survival pathway [36]. This pathway is a target for GLP-1-mediated cardioprotection through activation of phosphoinositide-3-kinase [10]. However, other possible targets for exenatide have been identified, including increased glucose uptake, inhibition of apoptotic factors, and activation of cAMP and cyclic guanosine monophosphate (cGMP). Thus, the cardioprotective action of GLP-1 receptor stimulation may occur through a number of pathways encompassing effects on metabolism, contractility and apoptosis. Other studies have shown that subcutaneous exenatide protects ischemia–reperfusion-induced endothelial dysfunction through the opening of adenosine triphosphate-sensitive potassium channels (KATP channels) [37]. Ischemia–reperfusion impairs endothelium-dependent vasodilatation; however, pre-treatment with exenatide protects the endothelium from this injury [37]. The endothelial protective effect of exenatide is almost completely prevented when a KATP channel blocker is administered before exenatide, suggesting that this effect of GLP-1 RA is mediated by KATP channel opening. Overall, the available results suggest that GLP-1 and its receptor agonists exert ischemic preconditioning through a nitric oxide (NO)-dependent pathway, of which KATP channels are key effectors. These findings represent the first set of evidence in human subjects for the effects of exenatide on pharmacological endothelial preconditioning, and provide a mechanistic explanation for this phenomenon. Additional studies are needed to investigate the mechanisms and their potential clinical implications in greater detail [37].
GLP-1 receptors are widely expressed in the central nervous system (CNS) where they are generally associated with the regulation of appetite and satiety; however, data from pre-clinical models of Alzheimer’s disease suggest that GLP-1 may have neurotrophic and neuroprotective actions, and reduce amyloid-beta accumulation, thus encouraging the successful translation of these data into new treatments for patients with neurodegenerative CNS disorders [38].
Short-Acting GLP-1 RAs
Exenatide
Exenatide was the first incretin agent to be approved for glycemic control in diabetes. The sequence of this 39-amino acid synthetic peptide is based on exendin-4 from the lizard Heloderma suspectum (Gila monster), sharing 53% homology with human GLP-1 [39]. It binds to the pancreatic GLP-1 receptor and has many of the glucoregulatory properties of human GLP-1 [40], with a substantially longer plasma half-life than GLP-1 due to the presence of an N-terminal serine in exendin-4 instead of alanine [41]. The 5–10 μg dose is administered by subcutaneous injection twice-daily within 1 h of eating a main meal.
Exenatide shares some of the glucoregulatory effects of GLP-1, but is resistant to DPP-4 degradation. It has a number of actions, including enhancing glucose-dependent insulin secretion [42], suppressing postprandial glucagon secretion, slowing gastric emptying [43], and reducing caloric intake [44]. Pre-clinical studies have shown that exenatide also increases pancreatic β-cell mass and clinical studies have shown that it improves β-cell function [45, 46]. The efficacy and safety of exenatide administered in patients with T2DM not adequately controlled with oral agents (i.e., metformin, sulfonylurea, or sulfonylurea plus metformin) has been demonstrated in a series of 30-week clinical studies [47–49]. In these studies, up to 46% of exenatide-treated patients achieved target goals for hemoglobin A1c (HbA1c) ≤7% as prescribed by the American Diabetes Association (ADA) guidelines, compared with up to 13% of placebo-treated patients [50]. Mean change from baseline in body weight in these trials was greater in exenatide-treated patients (−1.6 to −2.8 kg) compared to placebo-treated subjects (−0.3 to −0.9 kg) [47–49].
The efficacy of exenatide as adjunctive treatment in patients with T2DM receiving thiazolidinedione was evaluated in a randomized, double-blind, placebo-controlled trial [51]. After 16 weeks of treatment, patients treated with exenatide showed significant improvements in glycated hemoglobin (HbA1c), fasting plasma glucose (FPG), and homeostasis model assessment of β-cell function (HOMA-B), and promoted weight loss compared with placebo [51–55]. Exenatide also improved daily mean postprandial glucose concentrations (PPG) (based on self-monitored blood glucose). The incidence of mild-to-moderate hypoglycemia was similar in both groups with no severe hypoglycemia reported [51].
Long-term data describing the effects of exenatide in the treatment of patients with T2DM have also been reported [52, 56]. Patients enrolled in phase III clinical trials have completed open-label extensions of up to 3.5 years. In addition to exenatide, patients were also receiving metformin, sulfonylurea, or a combination of the two therapies, as well as other agents that reduce cardiovascular (CV) risk. At baseline, 41% were receiving angiotensin-converting enzyme inhibitors, 38% were receiving 3-hydroxy-3 methylglutaryl coenzyme A reductase inhibitors (“statins”), and 39% were receiving aspirin. At the end of either 3 years or 3.5 years of treatment and follow-up, patients showed significant reductions in HbA1c, FPG, and body weight from baseline [52]. In the 3-year completer group, 46% of patients achieved HbA1c of ≤7% and 30% achieved HbA1c of ≤6.5%. At the 82-week interim analysis, 81% of patients had lost weight [56]. In general, after 3 years of exenatide, overweight/obese patients with greater body mass index (BMI) at baseline lost more weight, with 84% of patients losing weight and 50% of patients losing at least 5% of their body weight. Improvements in HbA1c, FPG, and body weight with exenatide were observed regardless of age and were sustained through 3.5 years of treatment [52]. Over the 3 to 3.5 years of follow-up, patients treated with exenatide, which was generally well tolerated, experienced favorable effects on hepatic injury biomarkers and CV risk factors. Exenatide-treated patients with elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) at baseline significantly improved at 3 years (P < 0.001), while patients with normal ALT and AST values at baseline had little or no change. In the 3.5-year completer group, triglycerides, total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, and systolic and diastolic blood pressure all showed significant improvements relative to baseline. The most frequently reported adverse events (AEs) over the course of the study were mild-to-moderate nausea (59%) and hypoglycemia (40%) [52].Generally, the incidence of nausea was highest during the first few weeks of treatment, and a significant reduction was reported after this initial period [52, 56]. A single event of severe hypoglycemia occurred in 1 patient who was taking concomitant metformin and sulfonylurea [52]. A review of current clinical data shows the estimated incidence of acute pancreatitis related to any drug therapy is approximately 0.1% to 2% [57]. Acute pancreatitis has been reported in patients treated with GLP-1 receptor agonist therapy, although no causal relation has been found. A retrospective, cohort study found that patients with T2DM (N = 337,067) may have nearly a threefold greater risk of acute pancreatitis compared with patients without diabetes (N = 337,067) [58]. During the clinical development of exenatide, the incidence of acute pancreatitis in exenatide-treated patients was lower than that observed in patients receiving insulin or placebo [59]. A recent claims-based active drug safety surveillance system revealed that the risk of acute pancreatitis in patients treated with exenatide or sitagliptin (relative risk: 1.0 for either agent) was comparable to that of patients treated with metformin or glyburide [60]. In addition, post hoc analyses of serious adverse events reported in clinical trials have not found an increased risk of pancreatitis with GLP-1 receptor agonists [61]. Although postmarketing surveillance data with exenatide are not sufficient to definitively establish drug-related causality [59, 60], exenatide therapy should be stopped in patients who exhibit symptoms of acute pancreatitis.
Exenatide was compared with the insulin analogs biphasic insulin aspart 70/30 (BIAsp 30) [62] and insulin glargine [63]. Results from these trials suggest that exenatide was not inferior to insulin therapy in terms of HbA1c reduction, and may provide better postprandial glycemic control together with body weight decrease [64]. It has been suggested that the insulin dosages administered in these studies may not have been optimal [65]. The mean daily dose in the study comparing exenatide to BIAsp 30 [62] was 24.4 units, which reduced the mean HbA1c level by 1.0%. Meanwhile in the INITIATE study [66], the daily dose was threefold higher (78.5 units) and this provided a 2.8% reduction in HbA1c. The daily dose in the study comparing exenatide to insulin glargine [63] was 25 units compared to 47 units in the treat-to-target trial [67]. Thus, it remains to be determined how exenatide compares with optimal insulin dosing [68].
From studies mentioned above, exenatide treatment was not inferior to insulin in HbA1c reduction and provided better control of PPG, making it a potential alternative treatment for T2DM.
Lixisenatide
Lixisenatide is a selective GLP-1 receptor agonist with once-daily administration that was approved in Europe in 2013 for the treatment of T2DM [69–72]. Data from phase II/III studies reveal that lixisenatide 20 µg significantly lowers HbA1c and reduces postprandial hyperglycemia. Two-hour postprandial glucose excursions were reduced by approximately 5 mmol/l compared to placebo after a standard meal [73].
Once-daily lixisenatide was significantly better at controlling PPG after a standard solid breakfast compared with liraglutide in a 28-day clinical trial [74], confirming previous findings [69, 72, 75]. Compared with liraglutide, lixisenatide was also significantly better at reducing postprandial levels of insulin, C-peptide and glucagon. Whereas both lixisenatide and liraglutide lowered HbA1c and body weight over the course of the 28-day study, differences in their efficacy over the course of the day were apparent, and there was also a possible difference in their pharmacokinetic profiles [69, 76]. Lixisenatide was better at controlling morning glycemia, while liraglutide provided better fasting and postprandial control.
Long-Acting GLP-1 RA
Liraglutide
Liraglutide is a human GLP-1 analog in which lysine 34 is substituted with arginine, and lysine 26 has a C16 acyl chain attached [77]. These modifications improve the absorption and extend the half-life compared to native GLP-1, allowing once-daily administration. After subcutaneous administration, maximum concentrations are achieved in 9–14 h, and half-life is 13 h [78, 79]. Reductions in HbA1c ranged from 0.6% to 1.6% in clinical trials of liraglutide administered once-daily at 0.6 to 1.8 mg, alone or in combination with other agents [80–84].
The 26-week LEAD-6 trial (effect of liraglutide or exenatide added to an ongoing treatment on blood glucose control in subjects with type 2 diabetes) compared once daily liraglutide 1.8 mg to twice daily exenatide 10 µg in patients with T2DM inadequately controlled with metformin, a sulfonylurea, or both. In this trial liraglutide was associated with significantly greater reductions in mean HbA1c (−1.1% vs. −0.8%) and fasting plasma glucose (−29 vs. −11 mg/dL) (P < 0.0001 for both) [85]. In a 14-week extension of this trial, patients who had switched to liraglutide had additional reductions in mean fasting glucose, as well as HbA1c (from 7.2% at week 26 to 6.9% at week 40), whereas patients who continued to receive liraglutide maintained the HbA1c reductions achieved in the 26-week trial [85]. In other trials, reductions in FPG with liraglutide ranged from 13 to 43 mg/dL [80–84]. In the LEAD-6 trial, liraglutide had a greater effect on fasting glucose, while exenatide was more effective on PPG [85]. As with exenatide, liraglutide is associated with dose-dependent weight loss, ranging from 1.0 to 3.2 kg in clinical trials [80, 82–85], including those examining treatment regimens combining liraglutide with a sulfonylurea, which when given as monotherapy is associated with weight gain [83]. Patients whose BMI exceeded 35 kg/m2 derived the greatest absolute benefit (weight loss up to −4.4 kg). Reductions in systolic blood pressure with liraglutide 0.6–1.8 mg ranged from 0.6 to 6.7 mmHg [80–84]. In the LEAD-6 extension trial, switched patients experienced further reductions in systolic blood pressure (−2.2 ± 0.88 mmHg; P = 0.0128) [86]. Compared with placebo or active comparators, liraglutide significantly improved markers of β-cell function, including HOMA-B, proinsulin–insulin ratio, and proinsulin–C-peptide ratio [81–84]. Compared with placebo, liraglutide significantly increases first-phase insulin secretion and maximum β-cell insulin secretory capacity [87].
Exenatide
Exenatide has been developed also as a once-weekly formulation [exenatide long-acting release (exenatide LAR)] that is approved for treating T2DM [88–91]. In the exenatide LAR formulation, the active peptide is incorporated in poly-(d,l-lactic-co-glycolic acid) matrix that provides controlled delivery [92], allowing steady-state concentrations to be achieved in 6–10 weeks and providing a median half-life of 2 weeks [64, 90]. FPG levels are improved after 2 weeks of treatment [89]. Studies have shown that exenatide once-weekly and liraglutide provide better glycemic control than exenatide twice-daily [85, 90, 91]. Two randomized open-label studies found significantly better glycemic control with the once-weekly formulation compared to the twice-daily formulation [90, 91]. The once-weekly formulation reduced HbA1c by 1.6% after 24 weeks [91] and 1.9% after 30 weeks [90], compared to 0.9% after 24 weeks and 1.5% after 30 weeks with the twice-daily formulation. Weight loss was similar in all groups compared [90, 91].
In the DURATION-6 study, a 26-week, open-label, randomized, parallel-group study conducted at 105 sites in 19 countries, Buse et al. compared the efficacy and safety of liraglutide once-daily (1.8 mg) with exenatide once-weekly (2 mg) in 912 patients with T2DM [93]. They found that exenatide LAR and liraglutide both improved glycemic control and were associated with weight loss. Reductions in HbA1c and weight loss were greater in the liraglutide group than in the exenatide LAR group, while adverse gastrointestinal events and withdrawals due to adverse events were more frequent in the liraglutide group. The incidence of mild hypoglycemia was similar in both groups, and no major hypoglycemic events were reported. Patient-reported outcomes improved in both groups.
Exenatide and liraglutide have provided better glycemic control than other anti-hyperglycemic drugs in comparative studies. Exenatide LAR was more effective than maximum-labeled doses of exenatide twice-daily [90, 91], sitagliptin and pioglitazone [88], and insulin glargine [89] in patients treated with oral anti-hyperglycemic drugs. Exenatide once-weekly reduced HbA1c to a greater extent than sitagliptin in drug-naive patients; it was not inferior to metformin, but did not achieve non-inferiority to pioglitazone [93]. The maximum-labeled dose of liraglutide (1.8 mg) provided better glycemic control than exenatide twice-daily [85], sitagliptin [95], insulin glargine [83], and submaximal doses of glimepiride [80] and rosiglitazone [81]. The reductions in HbA1c noted for these long-acting GLP-1 receptor agonists in comparator-controlled trials were generally greater than those of oral anti-hyperglycemic drugs and basal insulin [81–83, 88–91, 93–95].
It is important to note that, in the studies mentioned above, liraglutide was administered at the maximum dose of 1.8 mg and no studies have compared exenatide LAR with liraglutide twice-daily 1.2 mg or determined the relative efficacies of the available injectable therapies for glycemic control.
Scott et al. [96] performed a network meta-analysis estimating the relative difference in HbA1c for exenatide, exenatide LAR, insulin glargine and liraglutide 1.2 and 1.8 mg compared to placebo based on a combination of direct and indirect clinical evidence. The analysis suggests that exenatide LAR and both doses of liraglutide are associated with clinically important improvements in HbA1c, as shown previously in clinical trials.
While the direct comparison identified a significantly greater HbA1c reduction for liraglutide 1.8 mg compared to exenatide LAR, this network meta-analysis, which also includes indirect data from additional trials, did not identify important differences in HbA1c reduction between the treatments [96].
Albiglutide
One of the newer long-acting GLP-1 RA is albiglutide, which was approved by the FDA in April 2014. It is a dimer of two copies of 30-amino acid fused to human albumin, and a single amino acid substitution (glycine to alanine), and achieves resistance to DPP-4 degradation [97]. The efficacy and safety of albiglutide is demonstrated in the HARMONY clinical trials. Data from these trials have shown that albiglutide, in monotherapy or as add-on to other diabetes therapies, lowered HbA1c levels when compared with sitagliptin, glimepiride, pioglitazone and insulin lispro [98].
Comparison data between albiglutide and lispro insulin in HARMONY-6 trial, in which it met non-inferiority criteria, suggest that this long-acting GLP-1 RA is a valid alternative to lispro insulin in add-on basal insulin. Another finding was the weight loss ability of albiglutide compared to weight gain in patients treated with lispro insulin [99].
In the HARMONY-7 clinical trial, it was demonstrated that liraglutide at a dose of 0.6 mg titrated to 1.8 mg was more effective than albiglutide (at a dose of 30 mg titrated to 50 mg), but gastrointestinal adverse event was more frequent in liraglutide treatment [100].
Dulaglutide
Another once-weekly GLP-1 RA molecule approved for the treatment of T2DM is dulaglutide. It consists of a link between two GLP-1 analog chains and immunoglobulin G fragment. This structure confers a slower absorption and reduced rate of renal clearance [101]. The AWARD (Assessment of weekly Administration of Dulaglutide) clinical trials assessed the efficacy and safety of dulaglutide as monotherapy and as add-on diabetes therapy. This newer molecule is compared to other hypoglycemic medications such as short-acting exenatide, liraglutide, sitagliptin, metformin and insulin lispro. The results have shown a reduction of HbA1c raging from −0.78 to −1.51% [102]. In particular in the AWARD-6 trial, the efficacy of dulaglutide was comparable to its primary competitor, liraglutide. The reduction of HbA1c was −1.42% with dulaglutide and −1.36% with liraglutide. A significant greater reduction of weight loss was obtained in liraglutide group, although both molecules produced significant weight loss from baseline [103]. Other clinical trials are investigating the efficacy of dulaglutide in combination with insulin glargine and the safety in patients with moderate and severe chronic kidney disease.
Conclusion
Although most of the benefits of GLP-1 can be exerted by both long-acting and short-acting GLP-1 analogs, the short-acting preparation of exenatide offers the additional benefit of greater decelerating gastric emptying, which appears to be the key factor driving the reduction of postprandial glycemia [104]. Such additional “flattening” of postprandial glycemia seems to complement the predominant reduction of fasting glycemia achieved with a long-acting insulin. In the study by Buse et al., the short duration of exenatide action is illustrated by the fact that glycemic excursions following lunch—the meal that did not directly follow an injection of exenatide—did not differ from those with placebo [85].
It should be noted that exenatide twice-daily (BID) and liraglutide, which were compared in the LEAD-6 study, have different half-lives. Exenatide has a half-life of 2–4 h, which is similar to insulin aspart (3–5 h) or lispro (2–5 h); whereas the half-life of liraglutide (13 h) is comparable to that of detemir (14 h). As a consequence, exenatide BID appears to be more suitable for the treatment of patients with predominantly postprandial hyperglycemia, whereas liraglutide as well as the other long-acting GLP-1 RAs would be more suitable for patients with predominantly fasting hyperglycemia.
Interestingly, with regard to the effects on gastric motility, glucose profiles and studies with long-acting GLP-1 analogs have suggested that tachyphylaxis—a weakening response over time—may occur with increasing drug exposure and concentrations. Comparatively, with the fluctuating plasma levels of exenatide observed with twice-daily injections, deceleration of gastric emptying is fully maintained.
Thus, although most of the current developments in the field of incretin mimetics aim to increase half-lives and extend injection intervals, these agents in combination with basal insulin preparations may be a promising area for short-acting compounds. Perhaps for this reason, clinical trials of additional short-acting incretin mimetics, such as lixisenatide, are ongoing, with the aim of combining these drugs with basal insulin. On the other hand, the potential advantages of long-acting GLP-1 analogs include a more pronounced reduction of fasting glucose, less frequent injections and lower rates of nausea [85].
Clearly, glycemic control is not the only goal of modern diabetes therapy. Insulin treatment often increases body weight, whereas incretin mimetics promote weight loss, which is recommended for most patients with this condition. Results from a pilot study [105], suggest that the weight-lowering effect of exenatide may predominate over the insulin-induced weight gain. For this reason, adding a GLP-1 analog may help to increase quality of life during insulin therapy by compensating for its tendency to cause weight gain.
It should be emphasized that when added to either sulfonylureas or insulin, GLP-1 RAs are associated with increased risk of hypoglycemia. Therefore, accurate titration of insulin doses by glucose-self-monitoring is recommended when such combinations are prescribed.
References
American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care. 2013;36(Suppl 1):S11–66.
Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab. 1964;24:1076–82.
Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–8.
Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA. 1987;84:3434–8.
Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144:5149–58.
Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–705.
Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7-36) amide in type 2 (noninsulin-dependent) diabetic patients. J Clin Endocrinol Metab. 1996;81:327–32.
Nauck MA, Niedereichholz U, Ettler R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273:E981–8.
Näslund E, Barkeling B, King N, et al. Energy intake and appetite are suppressed by glucagon-like peptide-1 (GLP-1) in obese men. Int J Obes Relat Metab Disord. 1999;23:304–11.
Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes. 2005;54:146–51.
Thrainsdottir I, Malmberg K, Olsson A, Gutniak M, Rydén L. Initial experience with GLP-1 treatment on metabolic control and myocardial function in patients with type 2 diabetes mellitus and heart failure. Diabetes Vasc Dis Res. 2004;1:40–3.
Nyström T, Gutniak MK, Zhang Q, et al. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol Endocrinol Metab. 2004;287:E1209–15.
Nyström T, Gonon AT, Sjöholm A, Pernow J. Glucagon-like peptide-1 relaxes rat conduit arteries via an endothelium-independent mechanism. Regul Pept. 2005;125:173–7.
Yu M, Moreno C, Hoagland KM, et al. Antihypertensive effect of glucagon-like peptide 1 in Dahl salt-sensitive rats. J Hypertens. 2003;21:1125–35.
Gutzwiller JP, Tschopp S, Bock A, et al. Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J Clin Endocrinol Metab. 2004;89:3055–61.
Vilsbøll T, Zdravkovic M, Le-Thi T, et al. Liraglutide, a long-acting human glucagon-like peptide-1 analog, given as monotherapy significantly improves glycemic control and lowers body weight without risk of hypoglycemia in patients with type 2 diabetes. Diabetes Care. 2007;30:1608–10.
Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52.
Toft-Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab. 2001;86:3717–23.
Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia. 2011;54:10–8.
Mannucci E, Ognibene A, Cremasco F, et al. Glucagon-like peptide (GLP)-1 and leptin concentrations in obese patients with Type 2 diabetes mellitus. Diabet Med. 2000;17:713–9.
Pala L, Mannucci E, Pezzatini A, Silvia C, et al. Dipeptidyl peptidase-IV expression and activity in human glomerular endothelial cells. Biochem Biophys Res Commun. 2003;310:28–31.
Mannucci E, Pala L, Ciani S, et al. Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia. 2005;48:1168–72.
Vilsbøll T, Agersø H, Krarup T, Holst JJ. Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. J Clin Endocrinol Metab. 2003;88:220–4.
Nauck MA. Incretin-based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. Am J Med. 2011;124(1 Suppl):S3–18.
Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. 1995;44:1126–31.
Amori RE, Lau J, Pittas AG. Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta-analysis. JAMA. 2007;298:194–206 (Review).
Stonehouse A, Okerson T, Kendall D, Maggs D. Emerging incretin based therapies for type 2 diabetes: incretin mimetics and DPP-4 inhibitors. Curr Diabetes Rev. 2008;4(2):101–9 (Review).
Chia CW, Egan JM. Incretin-based therapies in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2008;93(10):3703–16 (Epub 2008 Jul 15. Review). doi:10.1210/jc.2007-2109.
Waget A, Cabou C, Masseboeuf M, et al. Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology. 2011;152:3018–29.
Salehi M, Prigeon RL, D’Alessio DA. Gastric bypass surgery enhances glucagon-like peptide 1-stimulated postprandial insulin secretion in humans. Diabetes. 2011;60:2308–14.
Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E. Effects of glucagon-like peptide-1 receptor agonists on cardiovascular risk: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2014;16:38–47.
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117:2340–50.
Lønborg J, Vejlstrup N, Kelbæk H, et al. Impact of acute hyperglycemia on myocardial infarct size, area at risk, and salvage in patients with STEMI and the association with exenatide treatment: results from a randomized study. Diabetes. 2014;63:2474–85.
Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol. 2010;105:151–4.
Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M, Inserte J, Agulló L, Cabestrero A. The end-effectors of preconditioning protection against myocardial cell death secondary to ischemia-reperfusion. Cardiovasc Res. 2006;70:274–85.
Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118:1915–9.
Ha SJ, Kim W, Woo JS, et al. Preventive effects of exenatide on endothelial dysfunction induced by ischemia-reperfusion injury via KATP channels. Arterioscler Thromb Vasc Biol. 2012;32:474–80.
Li Y, Duffy KB, Ottinger MA, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1205–19.
Nielsen LL, Young AA, Parkes DG. Pharmacology of exenatide (synthetic exendin-4): a potential therapeutic for improved glycemic control of type 2 diabetes. Regul Pept. 2004;117:77–88.
Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology. 1995;136:3585–96.
Ritzel U, Leonhardt U, Ottleben M, et al. A synthetic glucagon-like peptide-1 analog with improved plasma stability. J Endocrinol. 1998;159:93–102.
Kolterman OG, Buse JB, Fineman MS, et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2003;88:3082–9.
Kolterman OG, Kim DD, Shen L, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm. 2005;62:173–81.
Szayna M, Doyle ME, Betkey JA, et al. Exendin-4 decelerates food intake, weight gain, and fat deposition in Zucker rats. Endocrinology. 2000;141:1936–41.
Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–6.
Bunck MC, Diamant M, Cornér A, et al. One-year treatment with exenatide improves beta-cell function, compared with insulin glargine, in metformin-treated type 2 diabetic patients: a randomized, controlled trial. Diabetes Care. 2009;32:762–8.
Buse JB, Henry RR, Han J, Exenatide-113 Clinical Study Group, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27:2628–35.
DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–100.
Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28:1083–91.
American Diabetes Association. Standards of medical care. Diabetes Care. 2009;32(suppl 1):S13–61.
Zinman B, Hoogwerf BJ, Durán García S, et al. The effect of adding exenatide to a thiazolidinedione in suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med. 2007;146:477–85.
Klonoff DC, Buse JB, Nielsen LL, et al. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr Med Res Opin. 2008;24:275–86.
Aschner P, Kipnes MS, Lunceford JK, Sitagliptin Study 021 Group, et al. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006;29:2632–7.
Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, Sitagliptin Study 020 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care. 2006;29:2638–43.
Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P, Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther. 2006;28:1556–68.
Blonde L, Klein EJ, Han J, et al. Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes Obes Metab. 2006;8:436–47.
Balani AR, Grendell JH. Drug-induced pancreatitis: incidence, management and prevention. Drug Saf. 2008;31:823–37.
Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care. 2009;32:834–8.
Idris I. FDA reviews incidences of acute pancreatitis in patients taking Byetta. Diabetes Obes Metab. 2008;10:96–8.
Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin. 2009;25:1019–27.
Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E. Glucagon-like peptide-1 receptor agonists and pancreatitis: a meta-analysis of randomized clinical trials. Diabetes Res Clin Pract. 2014;103:269–75.
Nauck MA, Duran S, Kim D, et al. A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia. 2007;50:259–67.
Heine RJ, Van Gaal LF, Johns D, GWAA Study Group, et al. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med. 2005;143:559–69.
Kim D, MacConell L, Zhuang D, et al. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care. 2007;30:1487–93.
Home PD. Comment on: Nauck MA, Duran S, Kim D et al (2007) A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia 50:259–267. Diabetologia. 2007;50:1561–2.
Raskin P, Allen E, Hollander P, INITIATE Study Group, et al. Initiating insulin therapy in type 2 Diabetes: a comparison of biphasic and basal insulin analogs. Diabetes Care. 2005;28:260–5.
Riddle MC, Rosenstock J, Gerich J, Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26:3080–6.
Nathan DM, Buse JB, Davidson MB, Heine RJ, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2006;29:1963–72.
Distiller LA, Ruus P, the ACT6011 Study Group. Pharmacokinetics and pharmacodynamics of a new GLP-1 agonist AVE0010 in type 2 diabetes patients (Abstract). Diabetes. 2008;57(Suppl. 1):A154.
Christensen M, Knop FK, Vilsbøll T, Holst JJ. Lixisenatide for type 2 diabetes mellitus. Expert Opin Investig Drugs. 2011;20:549–57.
Werner U, Haschke G, Herling AW, Kramer W. Pharmacological profile of lixisenatide: a new GLP-1 receptor agonist for the treatment of type 2 diabetes. Regul Pept. 2010;164:58–64.
Seino Y, Min KW, Niemoeller E, Takami A, EFC10887 GETGOAL-L Asia Study Investigators. Randomized, double-blind, placebo-controlled trial of the once-daily GLP-1 receptor agonist lixisenatide in Asian patients with type 2 diabetes insufficiently controlled on basal insulin with or without a sulfonylurea (GetGoal-L-Asia). Diabetes Obes Metab. 2012;14:910–7.
Ratner RE, Rosenstock J, Boka G, Silvestre L. Post-meal pharmacodynamic profile of AVE0010, a once-daily GLP-1 receptor agonist, in patients with type 2 diabetes inadequately controlled on metformin (Abstract). Diabetologia. 2009;52(Suppl. 1):S60.
Kapitza C, Forst T, Coester HV, Poitiers F, Ruus P, Hincelin-Méry A. Pharmacodynamic characteristics of lixisenatide once daily versus liraglutide once daily in patients with type 2 diabetes insufficiently controlled on metformin. Diabetes Obes Metab. 2013;15:642–9.
Ratner RE, Rosenstock J, Boka G, DRI6012 Study Investigators. Dose-dependent effects of the once-daily GLP-1 receptor agonist lixisenatide in patients with Type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled trial. Diabet Med. 2010;27:1024–32.
Watson E, Jonker DM, Jacobsen LV, Ingwersen SH. Population pharmacokinetics of liraglutide, a once-daily human glucagon-like peptide-1 analog, in healthy volunteers and subjects with type 2 diabetes, and comparison to twice-daily exenatide. J Clin Pharmacol. 2010;50:886–94.
Vilsbøll T. Liraglutide: a once-daily GLP-1 analogue for the treatment of type 2 diabetes mellitus. Expert Opin Investig Drugs. 2007;16:231–7.
Elbrønd B, Jakobsen G, Larsen S, et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes Care. 2002;25:1398–404.
Agersø H, Jensen LB, Elbrønd B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45:195–202.
Garber A, Henry R, Ratner R, LEAD-3 (Mono) Study Group, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet. 2009;373:473–81.
Marre M, Shaw J, Brändle M, LEAD-1 SU study group, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with Type 2 diabetes (LEAD-1 SU). Diabet Med. 2009;26:268–78.
Nauck M, Frid A, Hermansen K, LEAD-2 Study Group, et al. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care. 2009;32:84–90.
Russell-Jones D, Vaag A, Schmitz O, Liraglutide Effect and Action in Diabetes 5 (LEAD-5) met+SU Study Group, et al. Liraglutide vs insulin glargine and placebo in combination with metformin and sulfonylurea therapy in type 2 diabetes mellitus (LEAD-5 met+SU): a randomised controlled trial. Diabetologia. 2009;52:2046–55.
Zinman B, Gerich J, Buse JB, LEAD-4 Study Investigators, et al. Efficacy and safety of the human glucagon-like peptide-1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD-4 Met+TZD). Diabetes Care. 2009;32:1224–30.
Buse JB, Rosenstock J, Sesti G, LEAD-6 Study Group, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet. 2009;374:39–47.
Buse JB, Sesti G, Schmidt WE, Liraglutide Effect Action in Diabetes-6 Study Group, et al. Switching to once-daily liraglutide from twice-daily exenatide further improves glycemic control in patients with type 2 diabetes using oral agents. Diabetes Care. 2010;33:1300–3.
Vilsbøll T, Brock B, Perrild H, et al. Liraglutide, a once-daily human GLP-1 analogue, improves pancreatic B-cell function and arginine-stimulated insulin secretion during hyperglycaemia in patients with Type 2 diabetes mellitus. Diabet Med. 2008;25:152–6.
Bergenstal RM, Wysham C, Macconell L, DURATION-2 Study Group, et al. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION-2): a randomised trial. Lancet. 2010;376:431–9.
Diamant M, Van Gaal L, Stranks S, et al. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION-3): an open-label randomised trial. Lancet. 2010;375:2234–43.
Drucker DJ, Buse JB, Taylor K, DURATION-1 Study Group, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet. 2008;372:1240–50.
Blevins T, Pullman J, Malloy J, et al. DURATION-5: exenatide once weekly resulted in greater improvements in glycemic control compared with exenatide twice daily in patients with type 2 diabetes. J Clin Endocrinol Metab. 2011;96:1301–10.
DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P. Encapsulation of exenatide in poly-(d, l-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes Technol Ther. 2011;13:1145–54.
Buse JB, Nauck M, Forst T, et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): a randomised, open-label study. Lancet. 2013;381:117–24.
Russell-Jones D, Cuddihy RM, Hanefeld M, DURATION-4 Study Group, et al. Efficacy and safety of exenatide once weekly versus metformin, pioglitazone, and sitagliptin used as monotherapy in drug-naive patients with type 2 diabetes (DURATION-4): a 26-week double-blind study. Diabetes Care. 2012;35:252–8.
Pratley RE, Nauck M, Bailey T, 1860-LIRA-DPP-4 Study Group, et al. Liraglutide versus sitagliptin for patients with type 2 diabetes who did not have adequate glycaemic control with metformin: a 26-week, randomised, parallel-group, open-label trial. Lancet. 2010;375:1447–56.
Scott DA, Boye KS, Timlin L, Clark JF, Best JH. A network meta-analysis to compare glycaemic control in patients with type 2 diabetes treated with exenatide once weekly or liraglutide once daily in comparison with insulin glargine, exenatide twice daily or placebo. Diabetes Obes Metab. 2013;15:213–23.
Trujillo JM, Nuffer W. Albiglutide: a new GLP-1 receptor agonist for the treatment of type 2 diabetes. Ann Pharmacother. 2014;48(11):1494–501.
Lindamood CA, Taylor JR. Emerging new therapies for the treatment of type 2 diabetes mellitus: glucagon-like peptide-1 receptor agonists. Clin Ther. 2015;37:483–93.
Rosenstock J, Fonseca V, Gross JL, et al. Advancing basal insulin replacement in type 2 diabete inadequately controlled with insulin glargine plus oral agents: a comparison of adding albiglutide, a weekly GLP-1 receptor agonist, versus thrice daily prandial insulin lispro. Diabetes Care. 2014;37:2317–25.
Pratley RE, Nauck MA, Barnett AH, et al. Once-weekly albiglutide versus once-daily liraglutide in patients with type 2 diabetes inadequately controlled on oral drugs (HARMONY7):rarandomised, open-label, multicentre, non-inferiority phase 3 study. Lancet Diabetes Endocrinol. 2014;2:289–97.
Blevins T, Wysham C, Arakaki R, et al. Efficacy and safety of dulaglutide added onto pioglitazone and metformin versus exenatide in type 2 diabetes in a randomized controlled trial (AWARD-1). Diabetes Care. 2014;37:2159–67.
UmpierrezG PovedanoS, Manghi FP, et al. Efficacy and safety of dulaglutide monotherapy versus metformin in type 2 diabetes in a randomized controlled trial (AWARD-3). Diabetes Care. 2014;37:2168–76.
Povedano ST, Dungan KM, Forst T, et al. Once-weekly dulaglutide versus once-daily liraglutide in metformin- treated patients with type 2 diabetes (AWARD-6):randomised, open-label, phase 3, non-inferiority trial. Lancet. 2014;384:1349–57.
Linnebjerg H, Park S, Kothare PA, et al. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept. 2008;151:123–9.
Arnolds S, Dellweg S, Clair J, et al. Further improvement in postprandial glucose control with addition of exenatide or sitagliptin to combination therapy with insulin glargine and metformin: a proof-of-concept study. Diabetes Care. 2010;33:1509–15.
Pratley RE, Gilbert M. Targeting incretins in type 2 diabetes: role of GLP-1 receptor agonists and DPP-4 inhibitors. Rev Diabet Stud. 2008;5:73–94.
Acknowledgments
The authors did not receive any funding or sponsorship for the publication of this article. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. Editorial support for this paper was provided by EDRA srl and was funded by Astrazeneca Italy. The authors would like to thank Invernizzi Fondazione for scientific support provided to AnnaChiara Uccellatore at IRCCS MultiMedica. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Annachiara Uccellatore and Ilaria Dicembrini declare no conflict of interest. Stefano Genovese has participated in clinical research, scientific advisory boards, served as a consultant or received honoraria for: Abbott Diabetes Care, Rome, Italy; AstraZeneca, Basiglio (MI), Italy; Boehringer Ingelheim, Milano, Italy; Bristol Myers Squibb, Rome, Italy; Eli Lilly, Sesto Fiorentino (FI), Italy; Janssen, Cologno Monzese (MI), Italy; Lifescan, Milano, Italy; Merck Sharp & Dohme, Rome, Italy; Novartis, Origgio (MI), Italy; Novo Nordisk, Rome, Italy; Takeda, Rome, Italy. Edoardo Mannucci has received consultancy fees, speaking honoraria, and/or research grants from AstraZeneca, Basiglio, Italy; BMS, Rome, Italy; Eli Lilly, Indianapolis, USA, and Sesto Fiorentino, Italy; Janssen, Amsterdam, the Netherlands, and Milan, Italy; Merck, Rome, Italy; Novartis, Origgio, Italy; Novo Nordisk, Rome, Italy; Sanofi, Milan, Italy; and Takeda, Rome, Italy. Antonio Ceriello has advisory board membership at Bayer Healthcare, Basel, Switzerland and Milan, Italy; Bristol Myers Squibb, Rome, Italy; Danone, Amsterdam, The Netherlands; Eli Lilly, Indianapolis, USA, Madrid, Spain and Sesto Fiorentino, Italy; Janssen, Amsterdam, the Netherlands and Milan, Italy; Medtronic, Milan, Italy; Merck Sharp & Dome, Rome, Italy; Novartis, Origgio, Italy; Novo Nordisk, Copenaghen, Denmark; OM Pharma, Basel, Switzerland; Roche Diagnostics, Milan, Italy; Sanofi, Milan, Italy; Takeda, London, UK; and Unilever, Amsterdam, The Netherlands; has received consultancy for Bayer Pharma, Milan, Italy; Lifescan, Milan, Italy; Novartis, Origgio, Italy; and Roche Diagnostics, Milan, Italy; has lectured for Astra Zeneca, Milan, Italy; Bayer Healthcare, Basel, Switzerland and Milan, Italy; Bayer Pharma, Milan, Italy; Boehringer Ingelheim, Milan, Italy; Bristol Myers Squibb, Rome, Italy; Eli Lilly, Indianapolis, USA, Madrid, Spain and Sesto Fiorentino, Italy; Merck Sharp & Dome, Rome, Italy; Mitsubishi, Tokyo, Japan; Novartis, Origgio, Italy; Novo Nordisk, Copenaghen, Denmark; Nutricia, Amsterdam, The Netherlands; Sanofi, Paris, France, Barcelona, Spain and Milan, Italy; Servier, Paris, France; and Takeda, Rome, Italy; and has received research grants from Mitsubishi, Tokyo, Japan; Novartis, Origgio, Italy; and Novo Nordisk, Copenhagen, Denmark.
Compliance with ethics guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Uccellatore, A., Genovese, S., Dicembrini, I. et al. Comparison Review of Short-Acting and Long-Acting Glucagon-like Peptide-1 Receptor Agonists. Diabetes Ther 6, 239–256 (2015). https://doi.org/10.1007/s13300-015-0127-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-015-0127-x