
Abstract
Background
Identification of molecular signatures from omics studies is widely applied in toxicological studies, and the evaluation of potential toxic effects provides novel insights into molecular resolution.
Objective
The prediction of toxic effects and drug tolerance provides important clues regarding the mode of action of target compounds. However, heterogeneity within samples makes toxicology studies challenging because the purity of the target cell in the samples remains unknown until their actual utilization.
Result
Single-cell resolution studies have been suggested in toxicogenomics, and several studies have explained toxic effects and drug tolerance using heterogeneous cells in both in vivo and in vitro conditions. In this review, we presented an understanding of single-cell transcriptomes and their applications in toxicogenomics.
Conclusion
The most toxicological mechanism in organisms occurs through intramolecular combinations, and heterogeneity issues have reached a surmountable level. We hope this review provides insights to successfully conduct future studies on toxicology.
Purpose of the review
Toxicogenomics is an interdisciplinary field between toxicology and genomics that was successfully applied to construct molecular profiles in a broad spectrum of toxicology. However, heterogeneity within samples makes toxicology studies challenging because the purity of target cell in the samples remains unknown until their actual utilisation. In this review, we presented an understanding of single-cell transcriptomes and their applications in toxicogenomics.
Recent findings
A high-throughput techniques have been used to understand cellular heterogeneity and molecular mechanisms at toxicogenomics. Single-cell resolution analysis is required to identify biomarkers of explain toxic effect and in order to understand drug tolerance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toxicology emphasizes alternative methods of risk assessment of chemicals that could be harmful to humans, animals, and microbes. (Liu et al. 2019). It plays an important role in the development of new drugs, evaluation of potential toxic effects, and clinical trials (Liu et al. 2019). These applications promote genomics approaches, including toxicogenomics, to estimate risk assessment in toxicology. Toxicogenomics is an interdisciplinary field between toxicology and genomics that was successfully applied to construct molecular profiles in a broad spectrum of toxicology (Liu et al. 2019). Next-generation sequencing (NGS) technologies have advanced life sciences and enabled a paradigm shift by facilitating personalized diagnosis and precision medicine. Toxicogenomics is also influenced by the high-throughput technology and rapid progress of advanced assay methods of molecular profiling, including DNA, RNA, and protein.
Toxicogenomics has been widely used to improve risk assessment due to the applicability of high-throughput technologies in genomics and in vivo and in vitro models. The advantages of in vivo models commonly used in toxicogenomics are low cost, ease of availability, and resemblance of their biochemistry with that of humans (Moran et al. 2016). However, small size, challenges in analysis, and very different characteristic loads are the reported disadvantages (Moran et al. 2016). The advantages of in vitro models in toxicogenomics are cost-effectiveness and ease of use and downstream processing (Ryan et al. 2016). However, reduced biologically relevant cell-to-cell interactions are a disadvantage (Ryan et al. 2016), In a human-like model, a three-dimensional (3D) system mimics key tissue factors and is much more representative of the in vivo environment (Langhans 2018). An ideal 3D culture model would simulate a tissue-specific physiological or pathophysiological disease-specific microenvironment, in which cells can proliferate, aggregate, and differentiate.
In toxicology, the gene expression measured in models (animal and 3D) with high sensitivity and precision is associated with environmental conditions and cellular heterogeneity. The bulk method, including microarray and RNA sequencing, constructs molecular profiles for assay conditions, but the responses of individual cells indicate heterogeneity, with the fraction of responsive cells being dynamic. In the bulk method, either in 3D or animal models, all cells are mixed in a single pool, and molecular profiles are measured as mean values of all cells. This method is insensitive to the identification of effects caused by masked or rare cell types. Single-cell RNA sequencing (scRNA-seq) measures mRNA expression in responsive cells that are highly sensitive and specific to environmental conditions and cellular heterogeneity.
In this review, we introduced single-cell transcriptomes in toxicology. We further discussed the possibilities of single-cell resolution studies by presenting several such studies. Finally, we provided practical suggestions for the application of single-cell transcriptomes in toxicology.
Platforms for single-cell genomics
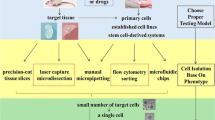
Immunocytochemistry and in situ hybridisation are first-generation single-cell resolution assays. Flow cytometry and image analysis are second-generation assays that provide a powerful approach to assessing cell-to-cell heterogeneity. However, these methods provide information on selected cell behaviors at single-cell resolution and have limitations in molecular profile quantification. In the past few years, improvements in genomics technology have enabled the quantification of the expression of a thousand genes in a single cell via the scRNA-seq technique. Tang et al. provided the first scRNA-seq method (Tang et al. 2009), and several different scRNA-seq protocols have been developed since then, including SMART-seq, CEL-seq, and Drop-seq (Hashimshony et al. 2012; Macosko et al. 2015; Picelli et al. 2013). The three most widely used protocols, based on cell capture strategy, are the plate-based, pooled, and droplet-based methods (Papalexi and Satija 2018) (Fig. 1). The plate-based method uses fluorescence-activated cell sorting (FACS), and selected cells are isolated from specific wells (Papalexi and Satija 2018). This platform has the advantage of using a low number of cells (50–500 single cells) and indicating high sensitivity to gene expression (1000–10,000 genes per single cell), with single-cell tagged reverse transcription sequencing, including SMART-seq and SMART-seq2 (Picelli et al. 2013). However, this method is not fully automated and causes technical noise during the experimental process. The pooled method is a single-cell barcoding strategy that improves throughput (Papalexi and Satija 2018). This strategy was quickly adopted in plate-based approaches and resulted in higher sensitivity and lowered the costs of scRNA-seq (CEL-seq) (Hashimshony et al. 2012). The droplet-based method constructs nano-liter droplets with beads and individual cells, and the beads are loaded with the enzymes required to construct the library (Papalexi and Satija 2018). This method not only allows for unbiased cell capture without the FACS approach, but also increases the number of cells in one run.

Summary of the three most widely used protocols for single cell isolation and library construction. Pooled method uses limiting dilution method in single cell isolation, plate-based method uses FACS isolation, and Droplet method uses microfluidic technology. Libraries are typically generated by specific method
The most suitable platform depends on biological factors. The droplet-based method has the advantage of capturing a large number of cells. This method is applicable to characterize the composition of environmental conditions. The plate-based method is advantageous for characterizing a rare cell population, which can be identified using a known surface marker.
Toxicogenomics recognizes that the molecular signatures of individual cells are accessible through scRNA-seq. Single-cell omics technologies will bring unprecedented resolution and innovation to the examination of biological systems and their perturbations by chemicals.
Cell clustering and cell-type identification
The scRNA-seq technology has made us possible to get a better understanding of cellular heterogeneity. Therefore, cell clustering and cell-type identification can provide novel insights into the biological processes under complex heterogeneity and poorly understood cellular mechanisms. Unsupervised clustering is a useful application of dimension reduction for thousands of cells in toxicological studies. Principle component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE) (van der Maaten and Hinton 2008) are common approaches that capture global and local relationships across cells. Uniform manifold approximation and projection (UMAP) is reported to be the fastest manifold learning implementation (Leland McInnes 2018). The next step is cell typing from the unsupervised clustering results. Traditionally, cell typing of the cluster has two approaches. The first is automatic cell-type identification (Table 1), in which clusters are annotated to specific cell types based on public resources of scRNA-seq data. This approach uses pre-annotated marker genes (or known marker genes) and a reference single-cell map for tissues; MSigDB (Liberzon et al. 2015), DRscDB (Hu et al. 2021), ScType (Ianevski et al. 2022), CellMeSH (Mao et al. 2021), PangladDB (Franzen et al. 2019) and CellMarker (Zhang et al. 2019) provide the relationships between marker genes and cell types. The reference single-cell map uses similar marker gene expression patterns from sources, such as the Gene Expression Omnibus (Barrett et al. 2013), Single Cell Expression Atlas (Papatheodorou et al. 2020), Human Cell Atlas (Regev et al. 2017), human Ensemble Cell Atlas (Chen et al. 2022), and Mouse Cell Atlas (Fei et al. 2022). The advantages of automatic cell-type identification are that it can be used by those without sufficient knowledge of cell markers or biology, applied to individual cells and to cell clusters, and can quickly identify major cell types. The automatic approach recommends starting cell-type annotation. This method is faster than the manual approach and is reproducible for large datasets. The manual approach facilitates challenging cell-type annotations, such as cell subtypes, gradients, highly homogeneous data, or poorly defined clusters.
However, this method cannot be used to estimate novel cell types. In the second approach, manual cell-type identification, clusters are annotated to specific cell types based on canonical markers found in the differentially expressed genes of the cluster. This approach involves annotating each cluster with known marker gene expressions, which are cell type-specific genes. This method can be very time-consuming and requires in-depth knowledge of the marker genes of different cell types. A manual search of the literature and various databases is required. However, rare cells can be identified by manual annotation, and automatic cell-type annotation does not always result in the high-confidence annotation.
Examples of single-cell transcriptomics studies conducted in toxicology
Even the same toxic substance has different effects depending on the type of cell. In toxicology studies, scRNA-seq technology makes us possible to get heterogeneous cellular responses in the tissue microenvironment of chemical exposure. Quinoin is one of the toxins which categorized as ribosome-inactivating proteins (RIPs) found in many edible and flowering plants. Rotondo et al. proposed that that Quinoin reduced the growth of glioblastoma using it as a toxin to kill glioblastoma cells (Rotondo et al. 2021).
2,3,7,8-Tetrachlorodibenzo-P-dioxin (TCDD) has dioxin-like toxicity and affects reproduction, causes birth defects, damages the liver, and suppresses the immune system (Nault et al. 2021). In a test for long-term exposure, the aryl hydrocarbon receptor agonist TCDD was shown to elicit cell-specific responses and alter the relative population sizes. RAS signaling and related pathways were specifically enriched in non-parenchymal cells when exposed to TCDD, and the Kupffer cell subtype highly expressed glycoprotein transmembrane nm (Nault et al. 2021). The testes of adult fish exposed to TCDD during sexual differentiation decreased the number of spermatocytes, spermatids, and spermatozoa. The functional analysis of differentially expressed genes in response to TCDD exposure suggested that this outcome was due to apoptosis of spermatids and spermatozoa (Haimbaugh et al. 2022). Ochratoxin A (OTA) is a health-threatening mycotoxin, and Zhang et al. validated the potential target of OTA and proposed a toxicological mechanism using single-cell resolution (Zhang et al. 2021). Aissa et al. identified a cell population that tolerates EGFR inhibitor, functional heterogeneity of drug-tolerant cells, and effective drug combinations to target persister cell subpopulations (Aissa et al. 2021). Marquina-Sanchez et al. demonstrated that FOXO inhibitor, a small molecule, induces dedifferentiation of both alpha and beta cells (Marquina-Sanchez et al. 2020). These results of toxicology and drug tolerance validate previous studies and provide novel insights into target cell types and molecular mechanisms.
Cell lineage tracing
Cell lineage tracing is an advanced scRNA-seq analysis. Cell lineage tracing has the advantages of being fast to establish the techniques and non-invasive, it is expected to be used in many other studies (Kretzschmar and Watt 2012). By lineage tracing, we can understand the development of tissues, and homeostasis with multiple cell types. It is potent especially when it comes to terms with cell fate and identifies all progeny of individual cells (Kretzschmar and Watt 2012). Cell lineage tracing will help understand possible cell lineages from targeted cells to affected cells and predict marker genes for transition state.
Pseudo-time is the timestamp of the cell along the biological trajectory, following physiological stimulation or perturbation. Most of the large pseudo-time values are differentiated and most of the small values are undifferentiated. In biological samples, cells indicate dynamic behavior upon physiological perturbation, and pseudo-time analysis has the advantage of tracing the biological state transition with pseudotemporal ordering, called trajectory inference. The cellular state transition is divided into three stages: before-transition; pre-transition, at which cell fate commitment occurs; and after-transition stages (Liu et al. 2013). The before-transition stage refers to normal cell types. When sufficiently perturbed in biological systems, some biological pathways are activated, and the cell stage switches to the tipping point, such as toxicity. Monocle is a toolkit for analyzing scRNA-seq data and provides a strategy to order single cells in pseudo-time (Trapnell et al. 2014). Pseudo-time-based trajectory inference of single cells is an unsupervised method that does not require prior knowledge of marker genes.
The cellular heterogeneity of gastric cancer is a leading determinant of therapeutic resistance and treatment failure and the main reason for poor overall survival in patients with gastric cancer. However, the plasticity and high heterogeneity of gastric cancer and the mechanisms involved in tumor differentiation are yet to be elucidated. Kim et al. demonstrated that the intestinal and diffuse types of gastric cancer were classified by different cell lineages, and heterogeneity of gastric cancer was induced by independent cell lineages (Kim et al. 2022). They reported that CCND1 mutations could be responsible for intestinal tumorigenesis and that stemness signatures are promoted by the interaction between diffuse-type tumor cells and cancer-associated fibroblasts (Kim et al. 2022). Moreover, epithelial–myofibroblast transition is a key trans-differentiation process and is associated with poor prognosis in patients with diffuse-type gastric cancer (Kim et al. 2022).
However, a limitation of this method is that gene expression may not reflect the state of an ultimate cell, and feature gene expression may introduce a strong bias in cell ordering (Tritschler et al. 2019). Therefore, adequate application of prior knowledge is essential, and combining pseudo-time and other molecular features provides a deep understanding of cell lineages.
Discussion
Bulk technology-based microarray and NGS revealed the usefulness of molecular profiling in DNA, RNA, and protein resolution, and provided insights into the significance of marker gene selection in vivo and in vitro. However, bulk sequencing has been limited to the heterogeneity of comparisons between biological samples and masked high-risk cell types in samples. The use of heterogeneous samples fundamentally confounds the reliability of data interpretation.
From this perspective, we reviewed single-cell technology, focusing on transcriptome. These high-throughput techniques have been used to understand cellular heterogeneity and molecular mechanisms at a single-cell resolution. Therefore, cell heterogeneity was introduced in toxicology studies as an advanced paradigm for risk assessment.
The most toxicological mechanism in organisms occurs through intramolecular combinations, and heterogeneity issues have reached a surmountable level. Therefore, toxicologists who study biological mechanisms should establish a research strategy for a sample microenvironment prior to the initial study design. We hope this review provides insights into the successful conduction of future studies on toxicology.
References
Aissa AF, Islam A, Ariss MM, Go CC, Rader AE, Conrardy RD, Gajda AM, Rubio-Perez C, Valyi-Nagy K, Pasquinelli M et al (2021) Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat Commun 12:1628. https://doi.org/10.1038/s41467-021-21884-z
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M et al (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41:D991-995. https://doi.org/10.1093/nar/gks1193
Chen S, Luo Y, Gao H, Li F, Chen Y, Li J, You R, Hao M, Bian H, Xi X et al (2022) hECA: the cell-centric assembly of a cell atlas. iScience 25:104318. https://doi.org/10.1016/j.isci.2022.104318
Fei L, Chen H, Ma L, Wang EW, Fang RX, Zhou Z, Sun H, Wang J, Jiang M et al (2022) Systematic identification of cell-fate regulatory programs using a single-cell atlas of mouse development. Nat Genet 54:1051–1061. https://doi.org/10.1038/s41588-022-01118-8
Franzen O, Gan LM, Bjorkegren JLM (2019) PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (oxford). https://doi.org/10.1093/database/baz046
Haimbaugh A, Meyer D, Akemann C, Gurdziel K, Baker TR (2022) Comparative toxicotranscriptomics of single cell RNA-Seq and conventional RNA-Seq in TCDD-exposed testicular tissue. Front Toxicol 4:821116. https://doi.org/10.3389/ftox.2022.821116
Hashimshony T, Wagner F, Sher N, Yanai I (2012) CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2:666–673. https://doi.org/10.1016/j.celrep.2012.08.003
Hu Y, Tattikota SG, Liu Y, Comjean A, Gao Y, Forman C, Kim G, Rodiger J, Papatheodorou I, Dos Santos G et al (2021) DRscDB: a single-cell RNA-seq resource for data mining and data comparison across species. Comput Struct Biotechnol J 19:2018–2026. https://doi.org/10.1016/j.csbj.2021.04.021
Ianevski A, Giri AK, Aittokallio T (2022) Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat Commun 13:1246. https://doi.org/10.1038/s41467-022-28803-w
Kim J, Park C, Kim KH, Kim EH, Kim H, Woo JK, Seong JK, Nam KT, Lee YC, Cho SY (2022) Single-cell analysis of gastric pre-cancerous and cancer lesions reveals cell lineage diversity and intratumoral heterogeneity. NPJ Precis Oncol 6:9. https://doi.org/10.1038/s41698-022-00251-1
Kretzschmar K, Watt FM (2012) Lineage tracing. Cell 148:33–45. https://doi.org/10.1016/j.cell.2012.01.002
Langhans SA (2018) Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front Pharmacol 9:6. https://doi.org/10.3389/fphar.2018.00006
Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P (2015) The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 1:417–425. https://doi.org/10.1016/j.cels.2015.12.004
Liu R, Aihara K, Chen L (2013) Dynamical network biomarkers for identifying critical transitions and their driving networks of biologic processes. Quant Biol 1:105–114. https://doi.org/10.1007/s40484-013-0008-0
Liu Z, Huang R, Roberts R, Tong W (2019) Toxicogenomics: a 2020 vision. Trends Pharmacol Sci 40:92–103. https://doi.org/10.1016/j.tips.2018.12.001
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM et al (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161:1202–1214. https://doi.org/10.1016/j.cell.2015.05.002
Leland M, John H, Nathaniel S, Lukas G (2018) UMAP: Uniform manifold approximation and projection. J Open Sour Softw 3:861. https://doi.org/10.21105/joss.00861
Mao S, Zhang Y, Seelig G, Kannan S (2021) CellMeSH: probabilistic cell-type identification using indexed literature. Bioinformatics. https://doi.org/10.1093/bioinformatics/btab834
Marquina-Sanchez B, Fortelny N, Farlik M, Vieira A, Collombat P, Bock C, Kubicek S (2020) Single-cell RNA-seq with spike-in cells enables accurate quantification of cell-specific drug effects in pancreatic islets. Genome Biol 21:106. https://doi.org/10.1186/s13059-020-02006-2
Moran CJ, Ramesh A, Brama PA, O’Byrne JM, O’Brien FJ, Levingstone TJ (2016) The benefits and limitations of animal models for translational research in cartilage repair. J Exp Orthop 3:1. https://doi.org/10.1186/s40634-015-0037-x
Nault R, Fader KA, Bhattacharya S, Zacharewski TR (2021) Single-nuclei RNA sequencing assessment of the hepatic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cell Mol Gastroenterol Hepatol 11:147–159. https://doi.org/10.1016/j.jcmgh.2020.07.012
Papalexi E, Satija R (2018) Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol 18:35–45. https://doi.org/10.1038/nri.2017.76
Papatheodorou I, Moreno P, Manning J, Fuentes AM, George N, Fexova S, Fonseca NA, Fullgrabe A, Green M, Huang N et al (2020) Expression atlas update: from tissues to single cells. Nucleic Acids Res 48:D77–D83. https://doi.org/10.1093/nar/gkz947
Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R (2013) Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10:1096–1098. https://doi.org/10.1038/nmeth.2639
Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M et al (2017) The human cell atlas. Elife. https://doi.org/10.7554/eLife.27041
Rotondo R, Ragucci S, Castaldo S, Oliva MA, Landi N, Pedone PV, Arcella A, Di Maro A (2021) Cytotoxicity effect of Quinoin, type 1 ribosome-inactivating protein from quinoa seeds, on glioblastoma cells. Toxins (basel). https://doi.org/10.3390/toxins13100684
Ryan SL, Baird AM, Vaz G, Urquhart AJ, Senge M, Richard DJ, O’Byrne KJ, Davies AM (2016) Drug discovery approaches utilizing three-dimensional cell culture. Assay Drug Dev Technol 14:19–28. https://doi.org/10.1089/adt.2015.670
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A et al (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6:377–382. https://doi.org/10.1038/nmeth.1315
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32:381–386. https://doi.org/10.1038/nbt.2859
Tritschler S, Buttner M, Fischer DS, Lange M, Bergen V, Lickert H, Theis FJ (2019) Concepts and limitations for learning developmental trajectories from single cell genomics. Development. https://doi.org/10.1242/dev.170506
van der Maaten L, Hinton G (2008) Visualizing data using t-SNE. J Mach Learn Res 9:2579–2605
Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M et al (2019) Cell marker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 47:D721–D728. https://doi.org/10.1093/nar/gky900
Zhang B, Li H, Zhu L, He X, Luo H, Huang K, Xu W (2021) Single-cell transcriptomics uncovers potential marker genes of ochratoxin A-sensitive renal cells in an acute toxicity rat model. Cell Biol Toxicol 37:7–13. https://doi.org/10.1007/s10565-020-09531-7
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF), a grant funded by the Korean government (MSIT) (NRF-2021R1A2C1009415), and the research fund of Hanyang University (202100000002900).
Author information
Authors and Affiliations
Contributions
SHK: searched the relevant literature and wrote the manuscript. SYC: searched the relevant literature, wrote the manuscript, and supervised the review process.
Corresponding author
Ethics declarations
Conflict of interest
Soo Young Cho declares that he has no conflict of interest. Seon Hwa Kim declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, S.H., Cho, S.Y. Single-cell transcriptomics to understand the cellular heterogeneity in toxicology. Mol. Cell. Toxicol. 19, 223–228 (2023). https://doi.org/10.1007/s13273-022-00304-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13273-022-00304-3