
Abstract
Training set optimization is a crucial factor affecting the probability of success for plant breeding programs using genomic selection. Conventionally, the training set optimization is developed to maximize Pearson’s correlation between true breeding values and genomic estimated breeding values for a testing population, because it is an essential component of genetic gain in plant breeding. However, many practical breeding programs aim to identify the best genotypes for target traits in a breeding population. A modified Bayesian optimization approach is therefore developed in this study to construct training sets for tackling such an interesting problem. The proposed approach is based on Monte Carlo simulation and data cross-validation, which is shown to be competitive with the existing methods developed to achieve the maximal Pearson’s correlation. Four real genome datasets, including two rice, one wheat, and one soybean, are analyzed in this study. An R package is generated to facilitate the application of the proposed approach. Supplementary materials accompanying this paper appear online.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 INTRODUCTION
To ensure a stable food supply for the expanding human population, plant breeding must enhance agricultural productivity through innovative methodologies. Genomic selection (GS) has emerged as a well-established method to accomplish this goal. In the field of plant breeding, GS has demonstrated its utility in improving quantitative traits of crops since it was proposed by Meuwissen et al. (2001). The fundamental concept of GS involves capturing quantitative trait loci using high-density DNA markers spread across the entire genome. Marker effects are estimated through a statistical prediction model using phenotype and genotype data from a training set. Following the model training, genomic estimated breeding values (GEBVs) for any individuals can be predicted solely from their genotype data. Subsequently, GS is implemented based on these computed GEBVs. From a statistical perspective, GS can be conceptualized as constructing a highly efficient predictive model.
Genetic improvement, or breeding progress, has been described as genetic gain, often measured by the difference between a base population and its progeny population (Xu et al. 2017). In the context of GS, the expected genetic gain per year can be defined as \(\Delta G=ir\sigma _\mathrm{{A}}/t\), where \(\Delta G\) is the response to selection, i is the intensity of selection, r is Pearson’s correlation between true breeding values (TBVs) and GEBVs, \(\sigma _\mathrm{{A}}\) is the square root of the additive genetic variance, and t is the breeding cycle time (Heffner et al. 2010). This might partially explain why Pearson’s correlation is usually used as an index to measure the prediction accuracy or ability of a GS scheme. However, many practical plant breeding programs aim to identify the best genotypes for target traits from a breeding population.
It could be sufficient to correctly rank candidates from the most favorable to the least favorable to achieve this goal. The collinearity requirement between GEBVs and TBVs for Pearson’s correlation treating all candidates uniformly can correlate poorly with ranking accuracy (Blondel et al. 2015). Blondel et al. (2015) therefore promoted the use of normalized discounted cumulative gain (NDCG) instead of Pearson’s correlation to measure the prediction ability of a selection strategy. NDCG focuses on high-ranking candidates in ranking and rewards strategies that more strongly assign a high rank to candidates with highly favorable breeding values. NDCG has been commonly employed to measure the ability of search engines to retrieve highly relevant documents from the top search results (Järelin and Kekäläinen 2000).
The GEBV prediction in GS is usually conducted based on two kinds of statistical models such as the whole genome regression (WGR) models (Endelman 2011), and the genomic best linear unbiased prediction (GBLUP) models (VanRaden 2008). Due to the typically larger number of marker effects compared to phenotypic values, the estimation of marker effects becomes a “large-p-and-small-n” problem when employing the WGR model. Consequently, a shrinkage estimation method such as ridge regression, Bayesian estimation methods, and nonparametric approach like random forest are commonly used in model building. Assuming the genotypic value for a specific individual is a linear combination of its marker effects, the GBLUP model treats these genotypic values as random effects within the framework of a linear mixed effects model. For GEBV prediction in the GBLUP model, restricted maximum likelihood estimation (REML) (Covarrubias-Pazaran 2016), and Bayesian estimation (Pérez and de los Campos 2014) are frequently applied. An excellent review regarding the statistical methodology used in GS can be found in Xavier et al. (2016).
The construction of the statistical model plays a pivotal role in GEBV prediction, and its predictive performance is significantly influenced by the composition of the training set. With the considerable reduction in genotyping costs due to advancements in DNA sequencing technology, whereas phenotyping costs have remained relatively high (Bernardo and Yu 2007), optimizing the training set for selective phenotyping emerges as an economical and efficient strategy to enhance the likelihood of success in a GS program (Heslot and Feoktistov 2020). The study on the training set optimization to achieve the maximal Pearson’s correlation between GEBVs and TBVs has drawn much attention. Based on the GBLUP model, Rincent et al. (2012) first promoted the generalized coefficient of determination (CD), presented in Laloë (1993), to identify an optimal training set. Isidro et al. (2015) and Rincent et al. (2017) later extended the CD-based optimization for highly structured populations. The stratified sampling rule proposed by Isidro et al. (2015) for highly structured population was also used in the works of Norman et al. (2018), Sarinelli et al. (2019), de Bem Oliveira et al. (2020), Adeyemo et al. (2020) and Fernández-González et al. (2023). Alternatively, based on the WGR model, Akdemir et al. (2015) presented an optimization criterion by considering the prediction error variance (PEV) for GEBVs, and Ou and Liao (2019) proposed another version derived from the correlation between GEBVs and phenotypic values. An excellent review and other optimization criteria, such as A- and D-optimality, can be found in Akdemir and Isidro-Sánchez (2019). Most recently, Fernández-González et al. (2023) made a comprehensive comparison among these training set optimization methods.
Rather than improving the prediction ability measured by Pearson’s correlation, Tanaka and Iwata (2018) emphasized the identification of the best genotypes from a candidate population. The authors proposed a multistage approach by adopting Bayesian optimization in the GS study. An acquisition function, such as the expected improvement (EI), was constructed to determine new genotypes for phenotyping. The choice of the new genotypes was anticipated to balance the trade-off between exploration and exploitation so that the best genotypes could be identified using as few individuals as possible. Tsai et al. (2021) further proposed several different versions of EI to tackle the same problem and compared the performance of these criteria through detailed simulation studies. The simulation results showed that an augmented version derived from the distribution of predicted genotypic values (PGVs) is advantageous over others regarding the number of individuals required to discover the true best genotype.
The multistage Bayesian optimization approaches required a series of experiments for phenotyping the genotypes selected at each stage. This could be very challenging for most plant breeding programs, mainly due to the time constraint that breeders usually collect phenotype data after a whole growing season of a crop. Therefore, this study aims to develop a practical Bayesian optimization approach for determining an optimal training set for identifying the best genotypes from a candidate population in plant breeding. A simulation-based cross-validation approach was proposed to define optimal training sets that can be phenotyped during a single growing season. Four real genome datasets, including two rice (Oryza sativa L.), one wheat (Triticum aestivum L.), and one soybean (Glycine Max), were analyzed to illustrate the proposed approach.
2 MATERIALS AND METHODS
2.1 The augmented expected improvement
The augmented EI for the distribution of PGVs proposed by Tsai et al. (2021) was introduced. The GBLUP model used in this study is as follows:
where \({\varvec{y}}\) denotes the vector of phenotypic values, \({\upmu }\) the general mean,\( {\varvec{1}}_{n}\) the unit vector of length n, \({\varvec{g}}\) the vector of genotypic values, and \({\varvec{e}}\) the vector of random errors. It is assumed that\(\varvec{ g}\) follows a multivariate normal distribution, denoted by \(\varvec{g}\sim \mathrm{{MVN}}\left( {\varvec{0}},\sigma _\mathrm{{A}}^{2}{\varvec{K}} \right) ,\) where \({\varvec{0}}\) is a zero vector,\( \sigma _\mathrm{{A}}^{2}\) is the additive genetic variance, and \({\varvec{K}}\) stands for a genomic relationship matrix. Moreover, the vector of random errors \(\varvec{e}\sim \mathrm{{MVN}}\left( {\varvec{0}},{\sigma _\mathrm{{e}}^{2}{\varvec{I}}}_{n} \right) \), where \(\sigma _\mathrm{{e}}^{2}\) is the random error variance, and \({\varvec{I}}_{n}\varvec{ }\)is the identity matrix of order n. Here, \({\varvec{g}}\) and \({\varvec{e}}\) are assumed to be mutually independent. In this study, the genomic relationship matrix was obtained as \({\varvec{K}}={\varvec{X}}{\varvec{X}}^{T}\varvec{/}p\), where \({\varvec{X}}\) is the standardized marker score matrix, and p is the number of single nucleotide polymorphism (SNP) markers. That is, \(x_{ij}=\frac{w_{ij}-{\bar{w}}_{j}}{s_{j}}\), where \(x_{ij}\) and \(w_{ij}\) are the respective \(\left( ij \right) \textrm{th}\) elements of \({\varvec{X}}\) and the original marker score matrix, and \({\bar{w}}_{j}\) and \(s_{j}\) are the sample mean and the sample standard deviation for column j in the original marker score matrix. The SNP at each locus is originally coded as −1, 0, or 1 for the homozygote of the minor allele, the heterozygote, and the homozygote of the major allele, respectively. After SNP coding, any missing locus in an individual is imputed by 1 (the coding for the homozygote of the major allele).
Let \({\varvec{y}}_{1} \)and \({\varvec{g}}_{1}\) respectively denote the vectors of phenotypic and genotypic values for the training set, both of which are of order \(n_{1},\) where \(n_{1} \)is the number of individuals in the set. Likewise, \({\varvec{y}}_{2} \)and \({\varvec{g}}_{2}\) denote the corresponding vectors for the remaining \(n_{2}\) individuals not chosen in the training set called as the non-phenotyped or remaining set, where \(n_{1} + n_{2}=n\). Therefore, the GBLUP model of Eq. (1) can be partitioned as follows:
where
The phenotype and genotype data of the training set are used to estimate \(\mu \), \(\sigma _{g}^{2}\) and \(\sigma _\mathrm{{e}}^{2}\), and BLUP for \({\varvec{g}}_{1}\). This is based on the sub-model:
where
These estimated values were denoted as \({\hat{\mu }}\), \(\hat{\sigma }_{g}^{2}\), \({\hat{\sigma }}_\mathrm{{e}}^{2}\) and \({\widehat{\varvec{g}}}_{1}\varvec{. }\)Under the condition that \({\hat{\mu }}\), \({\hat{\sigma }}_{g}^{2}\), \({\hat{\sigma }}_\mathrm{{e}}^{2}\) and \({\widehat{\varvec{g}}}_{1}\) are all assumed to be fixed, and with known values, the distribution of PGVs for the non-phenotyped set is given by:
where \({\widehat{\varvec{\mu }}}_{g}\varvec{= }{\varvec{K}}_{21}{\varvec{(}{\varvec{K}}_{11}\varvec{)}}^{\varvec{-}1}{\widehat{\varvec{g}}}_{1}\) and \({\widehat{\varvec{\varSigma }}}_{g}=\hat{\sigma }_{g}^{2}({\varvec{K}}_{22}-{\varvec{K}}_{21}{\varvec{(}{\varvec{K}}_{11}\varvec{)}}^{-1}{\varvec{K}}_{12}\varvec{)}\).
Tsai et al. (2021) considered the EI criterion based on the distribution of PGVs in (2), mainly because the best genotype for a target trait was defined as the individual with the maximal genotypic value among the candidates. Note that the genotypic values are unobservable but estimable. Let \({\hat{f}}_\mathrm{{Mg}}\) be the maximal value among \({\widehat{\varvec{g}}}_{1}\) and the marginal distribution for each individual of the PGVs is denoted by \({\tilde{{g}}}_{2i}\sim N({\hat{\mu }}_{gi},{\hat{\sigma }}_{gi}^{2})\). The improvement function for \({\tilde{{g}}}_{2i}\) is defined as follows:
From Jones et al. (1998), the expected value of \(\mathrm{{Im}}({\tilde{{g}}}_{2i})\), abbreviated as EI-PGV, has an elegant form given by the following equation:
where \(Z_{gi}=({\hat{\mu }}_{gi}-{\hat{f}}_\mathrm{{{Mg}}})/{\hat{\sigma }}_{gi}\), \(\mathrm {\Phi }\varvec{(\cdot )}\) is the cumulative density function of the standard normal distribution, and \(\phi \mathrm {(\cdot )}\) is the probability density function of the standard normal distribution. Furthermore, Tsai et al. (2021) adopted the augmented EI criterion presented in Huang et al. (2006) to the EI-PGV of Eq. (3) and proposed the modified version as follows:
where
with \(\mathrm {\gamma =1}\), and \(Z_{gi}^{*}=(\hat{\mu }_{gi}-{\hat{f}}_\mathrm{{{Mg}}}^{*})/{\hat{\sigma }}_{gi}\). This criterion was abbreviated as aug-EI-PGV. From Huang et al. (2006), \({\hat{f}}_\mathrm{{{Mg}}}^{*}\) is used to reduce prediction uncertainty, and the augmented term \(\mathrm {1-}\frac{{\hat{\sigma }}_{e}}{\sqrt{\hat{\sigma }_{gi}^{2}+{\hat{\sigma }}_{e}^{2}} }\) accounts for the diminishing return of additional replicates as the prediction becomes more accurate.
2.2 The approach for generating optimal training sets
Because the true genotypic values are unobservable, a Monte Carlo simulation was conducted to generate the values. An m-fold cross-validation process was implemented to determine the priority of candidates selected for the training set. During the cross-validation process, each of the m clusters was progressively and alternately used as the training set. At the same time, the remaining \(m-1 \)clusters were pooled as a non-phenotyped set.
Step 0: For a specific set of parameters in the model (1), generate N sets of \({\varvec{g}}\) and \({\varvec{e}}\) from their corresponding multivariate normal distributions. The simulated values of \({\varvec{g}}\) are treated as true genotypic values for the candidate population. The corresponding phenotypic values are obtained as the sum of the fixed constant \({\upmu }\), the simulated\(\varvec{ g}\), and the simulated \({\varvec{e}}\). Partition the whole candidate population into m exclusive clusters at random. Suppose that each cluster has \(c_{i}\) individuals, so \(\sum _{i=1}^m c_{i} =n\).
Step 1: For each simulated dataset, let the \(c_{i}\) individuals in cluster i serve as the training set. Perform the Bayesian reproducing kernel Hilbert space (RKHS) method in the R package BGLR (Pérez and de los Campos, 2014) using the phenotype and genotype data of the current training set to generate \({\hat{\mu }}\), \({\hat{\sigma }}_{g}^{2}\), \(\hat{\sigma }_\mathrm{{{e}}}^{2}\), and \({\widehat{\varvec{g}}}_{1}\).
Step 2: Treat the union of the remaining \(m-1\) clusters as the non-phenotyped set, and estimate the aug-EI-PGV values by Eq. (4) for the \(n-c_{i}\) individuals in the set using the estimates generated in Step 1.
Step 3: Repeat Steps 1 and 2 for \(i=1, 2, \cdots , m\), resulting in that there are \(m-1\) aug-EI-PGV values for every candidate. The average over the \(m-1\) aug-EI-PGV values is calculated as the priority information for each candidate to be chosen in the optimal training set based on the current simulated dataset.
Step 4: Repeat Steps 1–3 for each of the N simulated datasets generated from Step 0. The mean of the resulting Naug-EI-PGV averages generated in Step 3 is obtained as the selection index for each candidate.
Step 5: For a fixed training set size, candidates are chosen sequentially to compose the optimal training set according to the ranking of the selection index across the entire candidate population without a strong subpopulation structure. However, within each subpopulation of a highly structured candidate population, the stratified sampling rule (Isidro et al. 2015) was implemented to ascertain the quantity of genotypes selected. This rule determines the number of genotypes to be chosen from each subpopulation. The selection of genotypes from each subpopulation is proportional to the number of individuals present in that specific subpopulation of the candidate population, and the genotypes are sequentially selected according to their rankings in the subpopulation.
The parameters for calculating the selection index were given as follows: \({\upmu =100}\), \({\upsigma }_{g}^{2}=25\), and \({\upsigma }_\mathrm{{e}}^{2}={\upsigma }_{g}^{2}(1-h^{2})/h^{2}\). Here, \(h^{2}\) represents the genomic heritability fixed at 0.5. Accordingly, \(N\mathrm {=2500}\) datasets were generated. The number of folds for the cross-validation was fixed at five, and the sizes of the training set under study for each dataset were set to be 25, 50, 100, 150, 200, and 300.
2.3 The metrics for measuring the prediction ability of identifying the best genotypes
Let \({\widehat{\varvec{g}}}_{0}\) denote the resulting BLUPs for the genotypic values by using a training set. From Henderson (1977), the BLUPs of \({\varvec{g}}\) for all of the candidates can be obtained as follows:
where \({\varvec{K}}_\mathrm{{c}}\) is the genomic relationship matrix between the candidate population and the training set, and \({\varvec{K}}_{0}\) is the genomic relationship matrix of the training set. The NDCG index was introduced to measure the prediction ability of the training set to identify the best k genotypes with the most favorable TBVs from a candidate population. From Eq. (1), TBVs of the candidates are equal to \({\mu {\varvec{1}}}_{n}+{\varvec{g}}\). Thus, the ranking according to TBVs is exactly the same as according to genotypic values in \({\varvec{g}}\). Let \(g_{\left( 1 \right) }\ge g_{\left( 2 \right) }\ge ... \ge g_{\left( n \right) }\) be the true genotypic values sorted in decreasing order. Moreover, let \({\widehat{{g}}}_{\left( 1 \right) }, {\widehat{{g}}}_{\left( 2 \right) }\), …\({\widehat{{g}}}_{\left( n \right) }\) be their corresponding BLUPs obtained from Eq. (5). By reordering these BLUPs, it follows that \({\widehat{{g}}}_{\left( \pi _{1} \right) }\ge {\widehat{{g}}}_{\left( \pi _{2} \right) }... \ge {\widehat{{g}}}_{\left( \pi _{n} \right) }\), where \(\pi =\left( \pi _{1},\pi _{2},...,\pi _{n} \right) \) is a permutation of \(\pi _{0}=\left( 1, 2,..., n \right) \) From Blondel et al. (2015), the discounted cumulative gain (DCG) score at position k of the predicted ranking using the training set was defined as follows:
The DCG score at position k of the ideal ranking was defined as:
where f(g) is a monotonically increasing gain function, and \(d\left( i \right) \) is a monotonically decreasing discount function. The linear gain function of \(f\left( g \right) =g\) and the discount function of \(d\left( i \right) =\frac{1}{log_{2}\left( i+1 \right) }\) were used in Eq. (6) and (7). Finally, the NDCG score at position k was defined as follows:
The mean of the \(\mathrm{{NDCG}}@k'\left( g,{\widehat{{g}}} \right) \) for \(k^{'}=1, 2, \cdots , k\) was given as:
Both \(\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) and \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) range from 0 to 1. The higher the value, the higher the prediction ability.
2.4 A simulation study for assessing optimal training sets
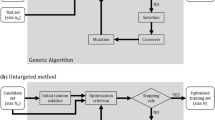
For a given training set, \(\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) in Eq. (8) with \(k=1, 5, 10,\) and \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) in Eq. (9) with \(k =10\) were calculated as the assessment metrics. Based on the GBLUP model in Eq. (1), three scenarios of genomic values were considered for evaluation. The model parameters were fixed at \({\upmu =100,}{\mathrm { \sigma }}_{g}^{2}=25,\) and \({\upsigma }_\mathrm{{{e}}}^{2}={\upsigma }_{g}^{2}(1-h^{2})/h^{2}\) with \(h^{2}=0.2,\) 0.5, and 0.8 for low, intermediate, and high genomic heritability. Accordingly, \(\textrm{1000}\) datasets were generated for each combination of parameters. To address the potential vulnerability to stochastic processes in the proposed simulation-based cross-validation approach, we evaluated 20 different optimal training sets at each of the fixed sizes ranging from 25 to 300 for every simulation scenario. The mean NDCG values obtained from these 20 optimal training sets for the metrics were calculated to assess the method’s performance. Additionally, training sets randomly selected from simulated datasets for each setting were evaluated to compare their performance with the mean performance of the 20 optimal training sets. Consequently, there were 20 different genotype datasets for the optimal training sets, each comprising 1000 distinct phenotype datasets at a fixed training set size. Furthermore, the random training sets consisted of 1000 different genotype and phenotype datasets for the same setting. A flowchart illustrating the simulation study is presented in Fig. 1.
2.5 Analysis of real traits
For each trait–dataset combination, the BLUPs of genotypic values for the candidates using all available phenotypic values were treated as the “true” genotypic values. \(\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) with \(k=1, 5, 10,\) and \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) with \(k =10\) were calculated for an optimal training set at a fixed size. Here, the \(\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) and \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) represent the relative prediction ability of the optimal training set to the entire candidate set. To avoid confusion, they were, respectively, renamed as \(\mathrm{{RE}}\_\mathrm{{NDCG}}@k\) and \(\mathrm{{RE}}\_\mathrm{{mean}}\_\mathrm{{NDCG}}@k\). The relative NDCG values will attain the maximum of 1, if the entire candidate set is used as the training set (Wu et al. 2023).
2.6 A comparison between training set optimization methods
Some training set optimization methods developed for achieving maximal Pearson’s correlation were employed for comparison, including a ranking approach for the CD method (Ou and Liao 2019), PEV (Akdemir et al. 2015), and r-score (Ou and Liao 2019). Moreover, a naive selection method according to the ranking of the average true genomic values, which were obtained from the above simulation-based cross-validation approach, was also considered. The optimal training sets, determined using the PEV and r-score criteria, were generated from the R package SSDFGP (Ou 2022), developed around an exchanging genetic algorithm. Given the potential vulnerability to stochastic processes in training set generation, we also evaluated 20 different training sets at a fixed size for the naïve, PEV, and r-score methods in the simulation study.

The flowchart illustrating the procedure in the simulation study
For those utilizing the CD criterion, we computed the priority information for each candidate to be chosen in the training set. A general form of the GBLUP model, as described in Eq. (1), is given by:
where \({\varvec{X}}\) is the design matrix for the fixed effects \(\varvec{\beta }\), and \({\varvec{Z}}\) is the incidence matrix for the random effects \({\varvec{u}}\). Rincent et al. (2012) presented CD for a contrast \({\varvec{c}}_{i}\) with \({{\varvec{1}}_{n}^{T}{\varvec{c}}}_{i}\varvec{=} 0\) as follows:
where \(\lambda =\frac{\sigma _{e}^{2}}{\sigma _{g}^{2}}\) and \(\varvec{M=}{\varvec{I}}_{n}\varvec{-X}{\varvec{(}{\varvec{X}}^{T}\varvec{X)}}^{-}{\varvec{X}}^{T}\) is the projection matrix orthogonal to the vector space spanned by the columns of \({\varvec{X}}\). Here, \({\varvec{(}{\varvec{X}}^{T}\varvec{X)}}^{-}\) is a generalized inverse of \({\varvec{X}}^{T}{\varvec{X}}\). Accordingly, let \(\varvec{X=}{\varvec{1}}_{n}\) and \(\varvec{Z=}{\varvec{I}}_{n}\), then CD corresponding to the specific GBLUP model in Eq. (1) is reduced to:
where \({\varvec{\bar{J}}}_{n}\) is the square matrix of order n with all elements equal to \(\frac{1}{n}\). Subsequently, let \({\varvec{c}}_{i}=\textrm{ }{\varvec{e}}_{i}-\frac{1}{n}{\varvec{1}}_{n}\), where \({\varvec{e}}_{i}\) is the vector of length n with all elements equal to 0 except theith element equal to 1, for \(i\mathrm {=1, 2, ...,} n\), and \(\lambda \) was fixed at 1. The candidates of the optimal training set using the CD in Eq. (11) were selected sequentially according to the ranking of their CD information values. Hence, the ranking CD method consistently yields identical results, unaffected by stochastic processes as observed in the aug-EI-PGV, naive, PEV, and r-score methods.
The simulation study with \(h^{2}=0.5\) and the analysis of real traits, as described above, were undertaken to compute \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) and \(\mathrm{{RE}}\_\mathrm{{mean}}\_\mathrm{{NDCG}}@k\) with \(k=10\), respectively. The calculations were performed across training set sizes of 25, 50, 100, 150, and 200 for the purpose of comparison.
2.7 Behavior of different estimation methods in actual data analysis
Blondel et al. (2015) formulated genomic selection as the problem of ranking individuals according to their breeding values, as mentioned earlier. In the article, the authors evaluated 10 regression methods and three ranking methods on six datasets by calculating their NDCG@\(k=\)1, 5, 10, and mean_NDCG@\(k=\)10 for 13 traits of the datasets. Their results showed that ordinal McRank (Li et al. 2008), random forest (RF) (Breiman 2001), and RKHS regression (Endelman 2011) had the best performance on at least one of these four NDCG indexes. To explore the behavior of these estimation methods in actual data analysis, the three traits in the soybean dataset were reanalyzed using the optimal training sets with a size ranging from 25 to 200.
2.8 The genome datasets
The phenotype and genotype data analyzed in this study were described as follows.
Tropical rice dataset
This dataset, presented by Spindel et al. (2015), contained 73,147 SNP markers and 363 elite breeding lines belonging to an indica or an indica-admixed group. Phenotypic observations were conducted eight times in 2009–2012, once in the dry season and once in the wet season each year, on grain yield (YLD), flowering time (FT), and plant height (PH), although PH data were not available for the wet season of 2009. Phenotypic values for 35 of the 363 individuals were missing. The adjusted least squares means (ls_means) presented in Spindel et al. (2015) of the 328 individuals were used in this example. One SNP marker was randomly chosen per 0.1-cM interval over each chromosome. This resulted in 10,772 of the 73,147 SNP markers used for this example. From Spindel et al. (2015), employing the subset of markers has been shown to maintain prediction accuracy, as opposed to using the entire marker set. In the present study, utilizing a subset of markers can also expedite the optimization process of r-score and PEV, both of which require computationally intensive algorithms such as a genetic algorithm.
Wheat dataset
This dataset, presented by Kristensen et al. (2019), contained 13,006 SNP markers and 635 F\(_{\textrm{6}}\) winter wheat lines from two breeding cycles. The first breeding cycle had 321 individuals harvested in 2014, while the second had 314 individuals harvested in 2015. The ls_means were collected on the four alveograph quality traits: flour yield (FYD), dough tenacity (DT), dough extensibility (DE), and dough strength (DS). Only 313 wheat lines from the second breeding cycle, with no missing data on the four quality traits, were used in this study. The SNPs were filtered at a missing rate > 0.1 and a minor allele frequency (MAF) < 0.05, leaving 11,214 remaining for further analyses.
44K rice dataset
The rice genome dataset presented by Zhao et al. (2011), originally collected for a genome-wide association study, was reanalyzed here. It contained 44,100 SNP markers and 36 traits of 413 accessions, and this dataset featured a strong subpopulation structure. All SNP markers with a missing rate > 0.05 and a MAF < 0.05 were first removed from the dataset, leaving 34,233 SNP markers. About one-third of these SNP markers (11,043 of 34,233) evenly distributed over each chromosome were selected. Only 300 of the 413 accessions with no missing phenotype data from all three locations—Arkansas (FT-Ark), Faridpur (FT-Far), and Aberdeen (FT-Abe)—were used here to illustrate the proposed approach.
Soybean
This dataset, presented by Stewart-Brown et al. (2019), contained 2,647 SNP markers and 483 recombinant inbred lines with the BLUP values of yield (YLD), protein content (PRC), and oil content (OC). The BLUP values for each genotype were calculated to account for environmental factors and maturity variation. Individuals were classified into four subpopulations and one admixed group, where the admixed group was composed of individuals in sets 9–11 and 12–14 (see Table 1 in Stewart-Brown et al. 2019). SNPs with a missing rate > 0.1 and a MAF < 0.05 were filtered out, leaving 2425 SNPs for 483 individuals retained for further analyses.
3 RESULTS
3.1 Simulation results
The bar plot according to the resulting selection index values of candidates for each dataset based on a single set of selection indexes through 2500 simulation runs, as described in Section 2.2, is displayed in Fig. 2. The figure shows that the selection index patterns seemed similar for the tropical rice and wheat datasets with no strong subpopulation structure. The variation of selection index values was found to be small within a subpopulation, but large between subpopulations in the highly structured datasets of 44K rice and soybean.
The average and standard deviation over the 1000 resulting mean performances of \(\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) with \(k=1,\)5, 10, and \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) with \(k =10\) for the tropical rice dataset in the simulation study are displayed in Fig. 3. The corresponding results for the remaining three datasets are displayed in Figures S1–S3 of the Supplementary Materials. Some general results are summarized as follows: (i) The optimal training sets outperformed their counterpart random training sets in all simulation scenarios. The outperformance was more significant in the two datasets with no strong subpopulation structure (tropical rice and wheat) than those with a strong subpopulation structure (44K rice and soybean). (ii) The performance improved with increasing genomic heritability for a particular training set at a fixed training set size. (iii) The NDCG or mean_NDCG value increased as the size became larger for a particular training set, and the margin gradually decreased as it was close to the size of the entire candidate set. The three curves of the NDCG with \(k=1\), 5, 10 and that of the mean_NDCG with \(k=10\) had very similar trends across the various sizes, and optimal or random sampling training sets in each dataset. (iv) An optimal training set with intermediate heritability \(\left( h^{2}=0.5 \right) \) can perform better than its random counter training set with high heritability \(\left( h^{2}=0.8 \right) \) at some smaller sizes in the tropical rice and wheat datasets. Similar results can be found for the optimal training set with low heritability \(\left( h^{2}=0.2 \right) \) compared with its random counterpart training set with intermediate heritability \(\left( h^{2}=0.5 \right) \) in these two datasets.
3.2 Real trait analysis
The relative NDCG and mean_NDCG values for the traits in the tropical rice dataset are displayed in Table 1. The corresponding results for the remaining three datasets are displayed in Tables S1–S3 of the Supplementary Materials. From the tables, the relative NDCG and mean_NDCG values generally increased as the training set size increased, except that the size changed from 25 to 50 for YLD in the soybean dataset (Table S3). The RE_mean_NDCG@k\( = \)10 fell between RE_NDCG@k\( = \)1 and RE_NDCG@k\( = \)10 for most of the cases. The RE_NDCG@k\( = \)1 \(\ge \) RE_NDCG@k\( = \)5 \(\ge \) RE_NDCG@k\( = \)10 at a fixed training set size for most of the cases in the wheat, 44K rice, and soybean datasets (Tables S1–S3). For the tropical rice dataset, RE_NDCG@k\( = \)1 \(\ge \) RE_NDCG@k\( = \)5 \(\ge \) RE_NDCG@k\( = \)10 at only the size equal to 25 or 50, and RE_NDCG@k\( = \)5 \(\ge \) RE_NDCG@k\( = \)1 \(\ge \) RE_NDCG@k\( = \)10 at the remaining cases (Table 1).

Bar plots of the average aug-EI-PGV for the four datasets with \(h^{2}=0.5\). In the panel of 44K rice, IND denotes Indica, TEJ denotes temperate japonica, and TRJ denotes tropical japonica

The \({\overline{\text {NDCG}}}\) (average) \(\mathrm {\pm }\) SD (standard deviation) over the 1000 resulting mean performances with the training set size equal to 25, 50, 100, 150, 200, and 300 for the four NDCG metrics under three different degrees of genomic heritability (low: \(h^{2}=0.2;\) intermediate: \(h^{2}=0.5;\) high: \(h^{2}=0.8)\) using the aug-EI-PGV and random sampling methods in the tropical rice dataset
It is also observed that the size of the optimal training set needed to achieve at least a specified value in RE_NDCG@k may increase with k. In essence, a more extensive training set is required to achieve a comparable performance in identifying the top genotypes from the candidate population when dealing with larger values of k. To illustrate this observation, the smallest sizes required to achieve RE_NDCG@k with \(k=\)1, 5, and 10 exceeding 0.95 in each trait-dataset combination are highlighted in the tables. Furthermore, the optimal training set exhibits comparable ability to the entire candidate population when its size reaches a fixed number of 300, 200, 200, and 300 for the tropical rice, wheat, 44K rice, and soybean datasets, respectively. Overall, the trait with lower genomic heritability could lead to smaller relative NDCG values, e.g., FT-Far with \(h^{2}=0.4525\) (Table S7 of the Supplementary Materials) in the 44K rice dataset (Table S2), and YLD with \(h^{2}=0.2675\) (Table S7) in the soybean dataset (Table S3).
3.3 Comparison results between the optimization methods
The average performances across 1000 iterations for the metric of \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k(\varvec{g},\widehat{\varvec{g}})\) with k =10 using various methods are presented in Table 2. In the case of the tropical rice and wheat datasets, aug-EI-PGV and CD exhibited similar high-performance levels, surpassing the other methods. Notably, the naive method relying on the true genomic value s showed comparatively inferior performance, underperforming even the random sampling method in the wheat dataset Conversely, for datasets such as the 44K rice and soybean, characterized by a strong subpopulation structure, the four optimization methods of aug-EI-PGV, CD, PEV, and r-score demonstrated competitive capabilities in identifying superior genotypes. Although the naive and random sampling methods still lagged behind the optimization methods, their performance disadvantage was much less pronounced in these datasets compared to the tropical rice and wheat datasets. Overall, performance convergence was observed across all criteria with increasing training set sizes, except for the naive and random methods in the tropical rice dataset.
The resulting RE_mean_NDCG@k with \(k=\)10 for the actual traits in the tropical rice dataset is presented in Table 3. Corresponding outcomes in the remaining three datasets are displayed in Tables S4–S6 of the Supplementary Materials. These results largely aligned with those obtained in the preceding simulation study. The four optimization methods, namely aug-EI-PGV, CD, PEV, and r-score, consistently outperformed the random sampling method in the majority of cases. However, there were a few notable exceptions, particularly at small training set sizes. For instance, both aug-EI-PGV and CD exhibited significantly poorer performance than random sampling at a size of 25 for YLD in the tropical dataset. A similar situation occurred with CD at a size of 50 (Table 3). As for the naive method, its performance proved competitive with the optimization methods in most instances. Nevertheless, the naive method can also yield markedly inferior results compared to the optimization methods in certain cases, such as YLD in the tropical rice dataset (Table 3), FYD in the wheat dataset (Table S4), and OC in the soybean dataset (Table S6). In summary, aug-EI-PGV can be recommended for use in the tropical rice and wheat datasets, while PEV can be advised for the 44K rice and soybean datasets.
3.4 Behavior of the estimation methods in actual data analysis
The resulting\( \mathrm{{RE}}\_\mathrm{{NDCG}}@k\) with \(k=1, 5, 10\) and \(\mathrm{{RE}}\_\mathrm{{mean}}\_\mathrm{{NDCG}}@k\) with \(k=10\) for the trait of YLD in the soybean dataset is displayed in Fig. 4 The corresponding results for the remaining two traits are displayed in Figures S4–S5 of the Supplementary Materials. From Figs. 3 and S1–S3, the average NDCG and mean_NDCG values were shown to increase with the training set size for all four datasets based on the GBLUP model using the Bayesian RKSH method in the simulation study. However, this consistent behavior cannot hold for all of the four estimation methods in the actual data analysis. The four estimation methods under consideration can be classified into two types: (i) statistical methods, including the GBLUP model (Bayesian RKSH method) and RKHS regression; and (ii) machine learning methods, including RF and McRank. From Figures 3 and S4–S5, the two methods in each type had similar behavior, and the fluctuations occurred most often in RE_NDCG@k with \(k=1\) compared with the other NDCGs. More interestingly, drastic fluctuations occurred in the trait of YLD using the two statistical methods of the GBLUP model and RKSH regression. The YLD had much lower genomic heritability (\(h^{2}=0.2675)\) than both PRC (\(h^{2}=0.8331)\) and OC (\(h^{2}=0.8105)\). The genomic heritability can be found in Table S7 of the Supplementary Materials. In consequence, the two machine learning methods can have more robust behavior in estimating various phenotype data than the two statistical methods.
4 DISCUSSION
The main idea of Bayesian optimization is to identify the best individuals among a set of candidates starting from a model trained by a small number of randomly selected individuals with known phenotype and genotype data. Subsequently, selecting individuals with the maximal EI scores augmented to the current training set to improve the ability of identifying the best genotypes in multiple stages. Following this idea, our proposed approach started with a random training set and then calculated the EI scores among the remaining individuals. The average EI score for each candidate over the cross-validation replicates based on the simulated datasets was used as the index to determine individuals for selective phenotyping. In other words, the proposed simulation-based cross-validation approach performed only one time of model training, and then selected potential genotypes based on the resulting EI score. The simulated trait values were specifically generated with the genomic heritability \(h^{2}=0.5\) in the approach. This specification might affect the determination of optimal training sets. It is thus necessary to verify whether the optimal training sets generated with \(h^{2}=0.5\) can be applicable to traits with a wide range of \(h^{2}\).
To investigate whether the performance of the optimal training set can be robust against the parameter specification, we first calculated the selection index with \(h^{2}=0.2\) and 0.8 to compare with the case of \(h^{2}=0.5\). The bar plots according to the resulting selection index values for each dataset are displayed in Figures S6–S7 of the Supplementary Materials. Compared with Fig. 2, the two bar plots with \(h^{2}=0.2\) and 0.8 were similar to that with \(h^{2}=0.5\) for each dataset.

RE_NDCG@k with \(k=\)1, 5, and 10, and RE_mean_NDCG@k with \(k=\)10 values using different estimation methods for YLD (yield) in the soybean dataset
Furthermore, we redid the analysis of the phenotype data of each dataset by using the optimal designs generated from each specification and calculated Pearson’s correlation and mean of absolute differences (MADs) over the paired resulting relative NDCG and mean_NDCG values between the two specifications of \(h^{2}=0.5\) and \(h^{2}=0.2\), and of \(h^{2}=0.5\) and \(h^{2}=0.8\) to evaluate how close their performances were. The MAD was defined as follows:
where (\(a_{i}\),\(b_{i})\) is the paired values in the real data analysis, and M is the total number of pairs. The resulting Pearson’s correlation coefficients and MADs are displayed in Table 4. From which, the relative NDCG or mean_NDCG values obtained from different specifications appeared to be highly correlated. However, the different specifications for generating optimal training sets may still result in slightly different performances, particularly in the case of NDCG@\(k=\)1. The Pearson’s correlations and MADs were also recalculated by excluding the points with a training set size equal to 25 and 50 for FT-Far in the 44K rice dataset, and for YLD in the soybean dataset, due to their relatively small training set sizes and small genomic heritability (FT-Far: 0.4525; YLD: 0.2675, (Table S7)). The results are also displayed in Table 4. From the table, both Pearson’s correlation and MAD improved, as these small-size points are excluded. Overall, the above discussion supports the idea that the optimal training set determined with \(h^{2}=0.5\) can apply to practical genomic selection programs.
As indicated in Sections 3.2 and 3.3, all the optimization methods, except the \(\mathrm{{CD}}\left( {\varvec{c}}_{i} \right) \) in Eq. (11), exhibited vulnerability to stochastic processes in constructing training sets. The ranking approach used in the CD method was also described in Atanda et al. (2021). One primary advantage of the ranking approach is its ability to significantly reduce computational costs. Conceptually, the prediction model is trained using the entire candidate set (\({\varvec{Z}}\) in Eq. (10) which was set to be \({\varvec{I}}_{n})\) by the ranking CD method, potentially leading to insufficient diversity in training set optimization due to the lack of a heuristic algorithm. The CD method, originally outlined in Rincent et al. (2012), was initially paired with a heuristic algorithm. This involved selecting a training set from a candidate population to maximize the mean of squared correlations between the true and estimated genotypic values. Fernández-González et al. (2023) showed that a heuristic-based CD method, known as CD\(_{\textrm{mean}}\), excelled in GEBV prediction, albeit with high computational intensity. A detailed illustration exemplifying CD mean can be found in Alemu et al. (2024). The adoption of heuristic-based CD methods for identifying the best genotypes from a candidate population could be advantageous.
To further explore the robustness of the proposed aug-EI-PGV method against vulnerability to stochastic processes when constructing training sets, Table 5 displays the averages of the standard deviations (calculated from the 20 training sets at a fixed size) over the 1000 iterations for the NDCG metrics in the simulation study with \(h^{2}=0.5\) for the datasets. The results indicate that the aug-EI-PGV method demonstrated greater robustness for datasets lacking a strong subpopulation structure, such as the tropical rice and wheat datasets. Across all datasets, at a fixed training set size, the average standard deviations for estimating the four metrics follow the order: NDCG@\(k=\)1 > NDCG@k\(=\)5 > NDCG@\(k=\)10 > mean_NDCG@\(k=\)10. Overall, the vulnerability to stochastic processes of aug-EI-PGV appears acceptable for constructing training sets in datasets without a strong subpopulation structure. However, for datasets with a strong subpopulation structure, particularly in scenarios with small training set sizes, there remains room for improvement.
The variation of selection index values within a subpopulation in the 44K rice and soybean datasets was reasonably small as shown in Fig. 2. The sampling rule of selecting a proportion of candidates within every subpopulation for these two datasets led to the result that the optimal training sets had a minimal advantage over the random sampling ones. We calculated the average mean performances over the 1000 iterations for the metric of \(\mathrm{{mean}}\_\mathrm{{NDCG}}@k\left( {\varvec{g}},{\widehat{\varvec{g}}} \right) \) with \(k=10\) for the optimal training sets without employing the sampling rule in the two datasets to explore how the inclusion of subpopulation structure information impacts the ability to identify the best genotypes The simulation results and those for the optimal training sets with the sampling rule at sizes equal to 25, 50, 100, 150, and 200 are displayed in Table 6 From the table, the optimal training sets had a slightly better performance at the size of 25, 50, and 100 when the sampling rule was used in the 44K rice dataset, but the margin of benefit diminished and even worsened at the sizes of 150 and 200. For the soybean dataset, the employment of the sampling rule had an advantage only at a small size of 25. The results suggest that considering the subpopulation structure in the training set optimization does not notably enhance performance in identifying the best genotypes. One possible explanation for the limited benefit of incorporating subpopulation structure could be the unpredictable distribution of genotypes within a candidate population. Consequently, implementing the stratified sampling rule might restrict the search space for the best genotypes.
Concerns may arise regarding the computational cost associated with the proposed simulation-based cross-validation approach. As such, we have provided details on the runtime required to complete Steps 1–4 of the approach outlined in Section 2.2 for the four datasets. These results are summarized in Table 7, which demonstrates that the runtime ranged between 22 and 25 min for datasets containing 301, 313, 328, and 401 individuals. This runtime appears manageable for datasets of these sizes. However, it is important to note that the runtime may escalate with an increase in the number of individuals within the candidate population. To illustrate this, we executed the program using a larger wheat genome dataset, as described in Ou and Liao (2019). This wheat dataset comprised 1896 individuals and 5028 SNPs, resulting in a required runtime of 3 h and 15 min (as shown in Table 7). Generally, the runtime tends to increase as the sample size of the candidate population grows. Nonetheless, this time requirement can be mitigated by employing upgraded hardware. Overall, the runtime incurred by the proposed approach seems acceptable for general datasets. In practice, the ranking CD method can be recommended for extremely large datasets.
From Figs. 3 and S1–S3, the NDCG or mean_NDCG value gradually reached the plateau as the training population size exceeds a fixed size for each dataset. This indicates that an appropriate utility function can be applied to connect the NDCG or mean_NDCG value and the size of the optimal training set. A practical training set size can then be accurately interpolated at a fixed acceptable NDCG or mean_NDCG value through the utility function. Most recently, Wu et al. (2023) and Fernández-González et al. (2023), respectively, applied logistic and non-logistic growth curves to determine the sample size for the training set optimization. We will investigate this issue for the optimal training set derived from the proposed Bayesian optimization approach in a future study. An R package, called TSOFIBG, is available from GitHub (https://github.com/huining0312/TSOFIBG) for conducting the proposed approach for the training set optimization and the methods for the rank estimation.
Data Availability
All phenotype and genotype datasets that were analyzed in this study are freely accessible and can be downloaded from Figshare (https://doi.org/10.6084/m9.figshare.22640041.v1).
Change history
17 July 2024
A Correction to this paper has been published: https://doi.org/10.1007/s13253-024-00641-x
References
Adeyemo E, Bajgain P, Conley E, Sallam AH, Anderson JA (2020) Optimizing training population size and content to improve prediction accuracy of FHB-related traits in wheat. Agronomy 10:543
Akdemir D, Isidro-Sánchez J (2019) Design of training populations for selective phenotyping in genomic prediction. Sci Rep 9:1–15
Akdemir D, Sanchez JI, Jannink JL (2015) Optimization of genomic selection training populations with a genetic algorithm. Genet Sel Evol 47:1–10
Atanda SA, Olsen M, Burgueno J, Crossa J, Burgueño J et al (2021) Scalable sparse testing genomic selection strategy for early yield testing stage. Front Plant Sci 12:658978
Alemu A, Åstrand J, Montesinos-López OA, Isidro y Sánchez J, Fernández-Gónzalez J et al (2024) Genomic selection in plant breeding: key factors shaping two decades of progress. Mol Plant 17:552–578
Bernardo R, Yu J (2007) Prospects for genome-wide selection for quantitative traits in maize. Crop Sci 47:1082–1090
Breiman L (2001) Random forest. Mach Learn 45:5–32
Blondel M, Onogi A, Iwata H, Ueda N (2015) A ranking approach to genomic selection. PLoS ONE 10:e0128570
Covarrubias-Pazaran G (2016) Genome-assisted prediction of quantitative traits using the R package sommer. PLOS One 11:e0156744
de Bem Oliveira I, Amadeu RR, Ferrão LFV, Muñoz PR (2020) Optimizing whole-genomic prediction for autotetraploid blueberry breeding. Heredity 125:437–448
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4:250–255
Fernández-González J, Akdemir D, Isidro y Sánchez J (2023) A comparison of methods for training population optimization in genomic selection. Theor Appl Genet 136:30
Heffner EL, Lorenz AJ, Jannink JL, Sorrells ME (2010) Plant breeding with genomic selection: gain per unit time and cost. Crop Sci 50:1681–1690
Henderson CR (1977) Best linear unbiased prediction of breeding values not in the model for records. J Dairy Sci 60:783–787
Heslot N, Feoktistov V (2020) Optimization of selective phenotyping and population design for genomic selection. J Agric Biol Environ Stat 25:601–616
Huang D, Allen TT, Notz WI, Zeng N (2006) Global optimization of stochastic black-box systems via sequential kriging meta-models. J Global Optim 34:441–446
Isidro J, Jannink JL, Akdemir D, Poland J, Heslot N, Sorrells ME (2015) Training set optimization under population structure in genomic selection. Theor Appl Genet 128:145–158
Järelin K, Kekäläinen J (2000) IR evaluation methods for retrieving highly relevant documents, In: Proceedings of the international ACM SIGIR conference on research and development in information retrieval, pp. 41–48
Jones DR, Schonlau M, Welch WJ (1998) Efficient global optimization of expensive black-box functions. J Global Optim 13:455–492
Kristensen PS, Jensen J, Andersen JR, Guzmán C, Orabi J, Jahoor A (2019) Genomic prediction and genome-wide association studies of flour yield and alveograph quality traits using advanced winter wheat breeding material. Genes 210(9):669
Li P, Wu Q, Burges CJ (2008) Mcrank: Learning to rank using multiple classification and gradient boosting, In: Proceedings of the 20\(^{\rm th}\) International Conference on Neural Information Processing Systems, pp. 897–904
Laloë D (1993) Precision and information in linear models of genetic evaluation. Genet Sel Evol 25:1–20
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Norman A, Taylor J, Edwards J, Kuchel H (2018) Optimising genomic selection in wheat: effect of marker density, population size and population structure on prediction accuracy. G3 Genes Genomes Genet 8:2889–2899
Ou JH (2022) TSDFGS: Training set determination for genomic selection, R package version 2.0. Available online at https://cran.r-project.org/package=TSDFGS
Ou JH, Liao CT (2019) Training set determination for genomic selection. Theor Appl Genet 132:2781–2792
Perez P, de los Campos G (2014) Genome-wide regression and prediction with the BGLR statistical package. Genetics 198:483–495
Rincent R, Laloë D, Nicolas S, Altmann T, Brunel D et al (2012) Maximizing the reliability of genomic selection by optimizing the calibration set of reference individuals: comparison of methods in two diverse groups of maize inbreds (Zea mays L.). Genetics 192:715–728
Rincent R, Charcosset A, Moreau L (2017) Predicting genomic selection efficiency to optimize calibration set and to assess prediction accuracy in highly structured populations. Theor Appl Genet 130:2231–2247
Sarinelli JM, Murphy JP, Tyagi P, Holland JB, Johnson JW et al (2019) Training population selection and use of fixed effects to optimize genomic predictions in a historical USA winter wheat panel. Theor Appl Genet 132:1247–1261
Spindel J, Begum H, Akdemir D, Virk P, Collard B et al (2015) Genomic selection and association mapping in rice (Oryza sativa): effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet 11:e1004982
Stewart-Brown BB, Song Q, Vaughn JN, Li Z (2019) Genomic selection for yield and seed composition traits within an applied soybean breeding program. G3 Genes Genomes Genet 9:2253–2265
Tanaka R, Iwata H (2018) Bayesian optimization for genomic selection: a method for discovering the best genotype among a large number of candidates. Theor Appl Genet 131:93–105
Tsai SF, Shen CC, Liao CT (2021) Bayesian approaches for identifying the best genotype from a candidate population. J Agric Biol Environ Stat 26:519–537
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Wu PY, Ou JH, Liao CT (2023) Sample size determination for training set optimization in genomic prediction. Theor Appl Genet 136:57
Xavier A, Muir WM, Craig B, Rainey KM (2016) Walking through the statistical black boxes of plant breeding. Theor Appl Genet 129:1933–1949
Xu Y, Li P, Zou C, Lu Y, Xie C et al (2017) Enhancing genetic gain in the era of molecular breeding. J Exp Bot 68:2641–2666
Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML et al (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2:467
Funding
This research was supported by the Ministry of Science and Technology, Taiwan (grant number MOST 112-2118-M-002-003-MY2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no Conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The funding information section was missing from this article and should have read “This research was supported by the Ministry of Science and Technology, Taiwan (grant number MOST 112-2118-M-002-003-MY2)”.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tu, HN., Liao, CT. A Modified Bayesian Optimization Approach for Determining a Training Set to Identify the Best Genotypes from a Candidate Population in Genomic Selection. JABES (2024). https://doi.org/10.1007/s13253-024-00632-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13253-024-00632-y