
Abstract
The cystine/glutamate antiporter SLC7A11 (also commonly known as xCT) functions to import cystine for glutathione biosynthesis and antioxidant defense and is overexpressed in multiple human cancers. Recent studies revealed that SLC7A11 overexpression promotes tumor growth partly through suppressing ferroptosis, a form of regulated cell death induced by excessive lipid peroxidation. However, cancer cells with high expression of SLC7A11 (SLC7A11high) also have to endure the significant cost associated with SLC7A11-mediated metabolic reprogramming, leading to glucose- and glutamine-dependency in SLC7A11high cancer cells, which presents potential metabolic vulnerabilities for therapeutic targeting in SLC7A11high cancer. In this review, we summarize diverse regulatory mechanisms of SLC7A11 in cancer, discuss ferroptosis-dependent and -independent functions of SLC7A11 in promoting tumor development, explore the mechanistic basis of SLC7A11-induced nutrient dependency in cancer cells, and conceptualize therapeutic strategies to target SLC7A11 in cancer treatment. This review will provide the foundation for further understanding SLC7A11 in ferroptosis, nutrient dependency, and tumor biology and for developing novel effective cancer therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
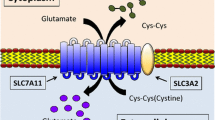
Cysteine is a proteinogenic amino acid that has a versatile role in protein synthesis, posttranslational modification, and redox maintenance (Stipanuk et al., 2006; Combs and DeNicola, 2019). Cysteine serves as the rate-limiting precursor for glutathione, a tripeptide comprised of three amino acids—cysteine, glutamate, and glycine—and the most abundant cellular antioxidant. Cysteine can also act as an antioxidant itself as well as the precursor for other biomolecules with antioxidant properties, such as taurine and hydrogen sulfide (Stipanuk et al., 2006; Combs and DeNicola, 2019). Intracellular cysteine can be synthesized through de novo biosynthesis (via the transsulfuration pathway) or recycled through protein degradation. However, because cancer cells often experience high levels of oxidative stress (Trachootham et al., 2009; Chio and Tuveson, 2017), cysteine supply via de novo biosynthesis or protein catabolism generally cannot meet the high demand for antioxidant defense in cancer cells; therefore, most cancer cells mainly rely on obtaining cysteine from the extracellular environment through nutrient transporters. The intra- and extra-cellular redox environments are very different: while cells generally maintain a reducing environment in the cytosol, the extracellular environment is highly oxidizing. Consequently, extracellular cysteine is highly unstable with only around 30-minute half-life in culture media (Ishii and Bannai, 1985), and is quickly oxidized to cystine (the oxidized dimer form of cysteine). The concentration of extracellular cystine is higher than that of extracellular cysteine by an order of magnitude. Some cancer cells can still effectively take up extracellular cysteine through cysteine transporters under certain conditions; for example, due to the high availability of extracellular cysteine secreted from surrounding bone marrow stromal cells, chronic lymphocytic leukemia cells can actively import cysteine from the extracellular environment (Zhang et al., 2012). However, most cancer cells largely depend on the cystine transporter system x −c to import cystine, which is then converted to cysteine in the cytosol through an NADPH-consuming reduction reaction; cysteine is subsequently used to synthesize glutathione (as well as other biomolecules) (Fig. 1) (Stipanuk et al., 2006; Combs and DeNicola, 2019).

Structure and function of SLC7A11. System x −c functions as a cystine/glutamate antiporter that imports one molecule of cystine in exchange for one molecule of intracellular glutamate. System x −c is a heterodimer consisting of the light chain subunit SLC7A11 and the heavy chain subunit SLC3A2. SLC7A11 mediates the antiporter activity of system x −c , whereas SLC3A2 anchors SLC7A11 to the plasma membrane and maintains SLC7A11 protein stability. Extracellular cystine is imported into the cell through SLC7A11, and then is converted to cysteine through a NADPH-consuming reduction reaction. Subsequently, cysteine is utilized to synthesize GSH through a two-step process. Cysteine forms γ-glutamylcysteine in conjugation with glutamate in the first step catalyzed by γ-GCS. The second step involves GS-mediated enzymatic addition of a glycine molecule to produce GSH. Ferroptosis is induced by excessive accumulation of lipid hydroperoxides in cellular membrane. GPX4 uses GSH to reduce lipid hydroperoxides to lipid alcohols, thereby suppressing ferroptosis, through which GSH is oxidized to GSSG; GSSG is then converted back to GSH via GR-mediated reduction reaction, which consumes NADPH. γ-GCS: γ-glutamylcysteine synthetase, GS: glutathione synthetase, GPX4: glutathione peroxidase 4, GR: glutathione reductase, GSH: reduced glutathione, GSSG: oxidized glutathione, LOOH: lipid hydroperoxide, LOH: lipid alcohol
The system x −c is a sodium-independent antiporter that exports intracellular glutamate and import extracellular cystine at a 1:1 ratio (Fig. 1) (Bannai, 1986; Conrad and Sato, 2012). It consists of two subunits linked via a disulfide bond, including the heavy chain subunit solute carrier family 3 member 2 (SLC3A2; also called CD98 or 4F2hc) and the light chain subunit solute carrier family 7 member 11 (SLC7A11; also commonly known as xCT). SLC7A11, a multi-pass transmembrane protein, mediates the cystine/glutamate antiporter activity in the system x −c (Sato et al., 1999; Koppula et al., 2018), whereas SLC3A2, a single transmembrane protein, is a chaperone to maintain SLC7A11 protein stability and appropriate membrane localization (Fig. 1) (Nakamura et al., 1999). Since SLC3A2 also serves as the chaperone for several other amino acid transporters, its function is not limited to cystine transport (Kandasamy et al., 2018). We will focus on SLC7A11 and refer to this cystine transporter as SLC7A11 throughout this review.
The role of cystine uptake in maintaining cell survival was implicated many years ago by the observation that cystine deprivation in cell culture media can lead to significant cell death in many cancer cell lines (Eagle, 1955a, b); however, the nature of this cell death had remained unclear. It was later shown that cystine starvation results in glutathione depletion in cells and that cystine starvation-induced cell death can be rescued by antioxidant vitamin E, therefore indicating the involvement of an oxidative stress-induced cell death in this context (Bannai et al., 1977). In 2012, it was discovered that pharmacologic blockade of SLC7A11-mediated cystine uptake by compounds such as erastin induces a new form of cell death termed ferroptosis (Dixon et al., 2012). Ferroptosis is an iron-dependent form of regulated cell death induced by lethal accumulation of lipid peroxides in the cellular membrane, and is mechanistically and morphologically distinct from other forms of regulated cell death such as apoptosis and necroptosis (Stockwell et al., 2017; Stockwell and Jiang, 2020). Cells have evolved various defense mechanisms to detoxify these toxic lipid peroxides, prominent among which is glutathione peroxidase 4 (GPX4), a GPX family member that uses reduced glutathione (GSH) as a co-factor to detoxify lipid peroxides to lipid alcohols, thereby suppressing ferroptosis (Fig. 1) (Friedmann Angeli et al., 2014; Yang et al., 2014). As noted above, cysteine is the rate-limiting precursor for GSH synthesis, and intracellular cysteine is mainly supplied by SLC7A11-mediated cystine uptake. Consistent with this, it was shown that removing cystine from cell culture media or SLC7A11 inactivation by genetic ablation or pharmacologic inhibition induces potent ferroptosis in many cancer cells; conversely, SLC7A11 overexpression in cancer cells promotes GSH biosynthesis and ferroptosis resistance (Dixon et al., 2012; Jiang et al., 2015; Zhang et al., 2018b). These studies together establish a critical role of SLC7A11-mediated cystine uptake in suppressing ferroptosis and maintaining cell survival under oxidative stress conditions.
In recent years, ferroptosis as a new cell death mechanism has sparked great interest in the scientific community. Accumulating evidence suggests that ferroptosis, similar to apoptosis, is a key tumor suppression mechanism (Jiang et al., 2015; Jennis et al., 2016; Stockwell et al., 2017; Zhang et al., 2018b; Chu et al., 2019; Liu et al., 2019). Importantly, recent studies also revealed that common cancer therapies, such as immunotherapy and radiotherapy, can induce ferroptosis partly through modulating SLC7A11 expression (Lang et al., 2019; Wang et al., 2019a; Lei et al., 2020; Ye et al., 2020). Correspondingly, considerable interest has been directed toward understanding the role and regulatory mechanisms of SLC7A11 in ferroptosis and tumor biology as well as therapeutically targeting SLC7A11 in cancer therapy. Another fascinating topic in SLC7A11 research in recent years has been to uncover the role of SLC7A11 in inducing nutrient dependency in cancer cells (Goji et al., 2017; Koppula et al., 2017; Muir et al., 2017; Romero et al., 2017; Sayin et al., 2017; Shin et al., 2017; Koppula et al., 2018; Joly et al., 2020; Liu et al., 2020). (Here nutrient dependency refers to the phenomenon wherein metabolic reprogramming renders cancer cells to be highly dependent on certain nutrients for survival and growth, such that removal of such nutrients will selectively kill or halt the growth of cancer cells that are dependent on these nutrients while having less harmful effects on other cancer cells or normal cells.) In this review, we will summarize how diverse regulation of SLC7A11 expression and its transporter activity governs ferroptosis. We will then discuss how SLC7A11 promotes tumor development through its function in inhibiting ferroptosis as well as other ferroptosis-independent functions. We will also explore the metabolic underpinnings of SLC7A11-induced nutrient dependency in cancer cells. Finally, we will discuss how our understanding of SLC7A11 function in ferroptosis and nutrient dependency informs strategies to target SLC7A11 in cancer and highlight important unknown questions for future investigation.
REGULATORY MECHANISMS OF SLC7A11
To ensure appropriate functioning of SLC7A11 in maintaining redox homeostasis, the expression and activity of SLC7A11 are subjected to tight regulation through a variety of mechanisms, including transcriptional regulation by transcription factors and epigenetic regulators, and post-transcriptional regulatory mechanisms to control its mRNA levels, protein stability, subcellular localization, and transporter activity. An emerging theme from recent studies is that various SLC7A11 regulators govern ferroptosis sensitivity through modulating SLC7A11 expression or activity. In this section, we will discuss how different regulatory mechanisms modulate SLC7A11 expression or activity and downstream biological effects mediated by SLC7A11, with a focus on ferroptosis. As further discussed in later sections, some of these SLC7A11 regulators also play important roles in cancer biology, and their dysregulation in cancer can lead to ferroptosis resistance and tumor formation.
Transcriptional regulation of SLC7A11 by transcriptional factors
It is well established that SLC7A11 expression can be induced under various stress conditions, including oxidative stress, amino acid starvation, metabolic stress, and genotoxic stress, likely as an adaptive response to enable cells to restore redox homeostasis and maintain survival under stress conditions (Koppula et al., 2018). Activating transcription factor 4 (ATF4) and/or nuclear factor erythroid 2-related factor 2 (NRF2) represent two major transcription factors that mediate stress-induced SLC7A11 transcription. ATF4, a member of the ATF/CREB family of transcription factors, is induced, primarily by mRNA translation, under various stress conditions such as amino acid starvation, endoplasmic reticulum stress, hypoxia, and viral infection; ATF4 then translocates into the nucleus and regulates the transcription of genes that cope with these stress conditions (Pakos-Zebrucka et al., 2016). For example, amino acid deprivation increases ATF4 mRNA translation through the general control non-derepressible-2 (GCN2)-eukaryotic initiation factor 2α (eIF2α) signaling axis. Consequently, ATF4 binds on the amino acid response elements (AAREs) in gene promoters and promotes the transcription of genes involved in amino acid metabolism and stress response, including SLC7A11 (Fig. 2), thereby enabling cells to cope with amino acid-limiting conditions (Kilberg et al., 2009). Indeed, SLC7A11 expression can be potently induced upon deprivation of various amino acids, including cystine, and amino acid deprivation-induced SLC7A11 expression is largely mediated by ATF4 (Sato et al., 2004). Since cystine starvation induces ferroptosis, whereas SLC7A11 protects cells from ferroptosis (Stockwell and Jiang, 2020), cystine starvation-induced SLC7A11 expression ostensibly acts as an adaptive response to help cells survive under cystine-limiting conditions. In support of this, it was shown that ATF4 promotes ferroptosis resistance by upregulating SLC7A11 (Chen et al., 2017).

SLC7A11 regulation by transcriptional, epigenetic, and post-translational mechanisms. SLC7A11 expression and function can be regulated at multiple levels under basal and stress conditions. Transcriptional activation: ATF4 activates SLC7A11 transcription in response to amino acid deprivation through the GCN2-eIF2α signaling axis. The KEAP1-NRF2 signaling axis regulates oxidative stress-induced SLC7A11 transcription. In response to oxidative stress, KEAP1-mediated NRF2 proteasomal degradation is suppressed, allowing stabilized NRF2 protein to translocate into the nucleus and activate the transcription of SLC7A11 and other genes involved in antioxidant response. SWI/SNF chromatin remodeling complexes facilitate NRF2-mediated transcriptional activation of SLC7A11. H3K9 demethylase KDM3B decreases H3K9 methylation on the SLC7A11 promoter and promotes SLC7A11 transcription. Transcriptional repression: Under basal conditions, p53 and ATF3 repress SLC7A11 transcription. BAP1 deubiquitinates H2Aub on the SLC7A11 promoter and subsequently represses SLC7A11, whereas H2A ubiquitination by PRC1 on the SLC7A11 promoter also represses SLC7A11. p53-mediated nuclear translocation of USP7 results in decreased H2Bub occupancy on the SLC7A11 promoter via deubiquitination, resulting in transcriptional repression. Post-translational regulation: OTUB1 and CD44 form a trimeric complex with SLC7A11 to deubiquitinate SLC7A11 and inhibit SLC7A11 degradation in proteasome, thereby stabilizing SLC7A11 protein, whereas mTORC1 promotes SLC7A11 protein stability by inhibiting its lysosomal degradation. High cell density inhibits mTORC1 and promotes SLC7A11 degradation in lysosomes. Both mTORC2 and AKT inhibit SLC7A11 transporter activity by phosphorylating SLC7A11 at serine 26. ATF4: activating transcription factor 4, GCN2: general control non-derepressible-2, eif2α: eukaryotic initiation factor 2α, KEAP1: Kelch-like ECH associated protein-1, NRF2: nuclear factor erythroid 2-related factor 2, p53: tumor protein p53, ATF3: activating transcription factor 3, BAP1: BRCA1 associated protein-1, PRC1: polycomb repressive complex 1, USP7: ubiquitin-specific-processing protease 7, SWI/SNF: SWItch/sucrose non-fermentable modeling complex, OTUB1: OTU deubiquitinase, ubiquitin aldehyde binding 1, mTORC1: mechanistic target of rapamycin complex 1, mTORC2: mechanistic target of rapamaycin complex 2
Another transcription factor that promotes SLC7A11 transcription is NRF2, which primarily mediates transcriptional programs in response to oxidative stress (Rojo de la Vega et al., 2018). NRF2 protein is unstable under basal conditions, being ubiquitinated by an E3 ubiquitin ligase kelch-like ECH-associated protein-1 (KEAP1) and subsequently undergoing rapid degradation through the proteasome pathway. Oxidative stress impairs NRF2 degradation by KEAP1 and stabilizes NRF2 protein, which then binds on the antioxidant response elements (AREs) in gene promoters and governs the transcription of genes involved in antioxidant defense and redox maintenance, including SLC7A11 (Fig. 2). KEAP1 inactivation leads to NRF2 stabilization under basal conditions, resulting in upregulation of NRF2 target genes (Rojo de la Vega et al., 2018). Recent studies using clustered regularly interspaced short palindromic repeats (CRISPR) screens uncovered KEAP1 as a key regulator of ferroptosis, and it was shown that KEAP1 deficiency promotes ferroptosis resistance (Cao et al., 2019). Considering the critical role of SLC7A11 in inhibiting ferroptosis, it is likely that the ferroptosis resistance caused by KEAP1 deficiency is at least partly mediated by SLC7A11. It should be noted that NRF2 regulates other genes involved in GSH biosynthesis, iron metabolism, and antioxidant responses, which might also play a role in mediating ferroptosis resistance in KEAP1 deficient cells. An extensive discussion of this topic will be beyond the scope of this review, and we refer readers to an excellent recent review focusing on NRF2 signaling in ferroptosis (Anandhan et al., 2020).
ATF4 and NRF2 often act concertedly to regulate stress-induced gene expression. For example, ATF4 and NRF2 interact with each other on the SLC7A11 promoter, and cooperatively regulate SLC7A11 transcription under stress conditions (Ye et al., 2014). It is important to note that the definitive evidence for the requirement of ATF4 or NRF2 to mediate various stress-induced SLC7A11 expression is often lacking (i.e., to show that ATF4 or NRF2 deletion would abolish SLC7A11 induction under a given stress condition). A recent study showed that ATF4 or NRF2 deficiency abolished glucose starvation-induced SLC7A11 expression; surprisingly, erastin-induced SLC7A11 remains intact in ATF4 or NRF2 knockout (KO) cells (Zhang et al., 2019a), suggesting that while glucose starvation-induced SLC7A11 expression depends on ATF4 and NRF2, erastin-induced SLC7A11 expression likely operates through ATF4- or NRF2-independent mechanisms.
SLC7A11 expression can also be repressed by transcriptional regulation. p53 is a transcription factor that plays a key role in tumor suppression (Vousden and Prives, 2009). SLC7A11 was identified as a p53 transcriptional target that is repressed by p53 (Fig. 2) (Jiang et al., 2015). It was further shown that p53 promotes ferroptosis under various ferroptosis-inducing conditions partly by repressing SLC7A11 expression, whereas SLC7A11 upregulation by p53 deficiency promotes ferroptosis resistance (Jiang et al., 2015). As further elaborated in the following section, p53 regulation of ferroptosis via suppressing SLC7A11 expression plays a key role in tumor suppression.
ATF3, another member of the ATF/CREB family of transcription factors, was previously identified to be upregulated upon erastin treatment (Dixon et al., 2014). A subsequent study confirmed erastin-induced ATF3 expression and further showed that ATF3 can bind on the SLC7A11 promoter and repress its expression independent of p53 (Fig. 2), and ATF3 promotes erastin-induced ferroptosis via downregulating SLC7A11 levels (Wang et al., 2020a). Since erastin treatment (or cystine starvation) potently induces SLC7A11 expression (Zhang et al., 2018b) yet ATF3 suppresses SLC7A11 expression (Wang et al., 2020a), it seems unlikely that erastin-induced ATF3 would contribute to erastin-induced SLC7A11 expression; it was further demonstrated that erastin-induced ATF3 does not appear to regulate SLC7A11 levels induced by erastin treatment (Wang et al., 2020a). These data suggest that ATF3 primarily regulates SLC7A11 transcription at basal conditions and does not affect stress-induced SLC7A11 expression even though ATF3 is often induced upon stress. In brief summary, various stress conditions promote SLC7A11 transcription partly through ATF4 and/or NRF2, whereas p53 and ATF3 mainly repress SLC7A11 expression under basal conditions (Fig. 2). Consequently, regulation of SLC7A11 expression by these transcription factors leads to altered cellular sensitivity to ferroptosis.
Epigenetic regulation of SLC7A11 transcription
Epigenetic regulation of gene transcription is primarily achieved by chemical modifications on DNA (such as DNA methylation) and/or DNA-associated histones (such as histone acetylation, methylation, ubiquitination, and phosphorylation) (Jaenisch and Bird, 2003). These modifications are added, removed, and recognized by various epigenetic regulators collectively called writers, erasers, and readers, respectively, resulting in transcriptional activation or repression (Badeaux and Shi, 2013). Specific DNA or histone modifications are often associated with specific types of gene transcription. For example, mono- and tri-methylation at lysine 4 of histone H3 (H3K4me1 and H3K4me3) are associated with transcriptional activation, whereas tri-methylation at lysine 9 and lysine 27 of histone H3 (H3K9me3 and H3K27me3) correlate with transcriptional repression. Epigenetic regulation of gene transcription is critical for controlling cellular homeostasis and development, and its dysregulation results in diseases such as cancer. Recent studies have revealed a critical role of epigenetic regulation of SLC7A11 transcription in governing ferroptosis.
BAP1 is a nuclear deubiquitinase (DUB) that removes histone 2A mono-ubiquitination (H2Aub) at lysine 119, a histone modification that is generally associated with transcriptional repression (Wang et al., 2004; Scheuermann et al., 2010). BAP1 and its interacting proteins form the polycomb repressive deubiquitinase (PR-DUB) complex (Carbone et al., 2013). In a recent study, genome-wide analyses identified SLC7A11 as a key transcriptional target of BAP1 (Zhang et al., 2018b). Mechanistically, the PR-DUB complex binds on the SLC7A11 promoter, allowing BAP1 to deubiquitinate H2Aub on the SLC7A11 promoter and subsequently repress SLC7A11 expression independent of p53 (Fig. 2), resulting in decreased cystine uptake and increased ferroptosis sensitivity; conversely, BAP1 deficiency in cancer cells leads to SLC7A11 upregulation and ferroptosis resistance (Zhang et al., 2018b; Zhang et al., 2019c).
Based on the current model that H2Aub is associated with gene repression (Scheuermann et al., 2010), it would be expected that BAP1, by removing H2Aub, should mainly activate gene transcription. Therefore, the finding that BAP1 represses SLC7A11 expression seems counterintuitive. However, genome-wide analyses showed that BAP1-mediated deubiquitination of H2Aub is associated with both transcriptional activation and repression of target genes (Zhang et al., 2018b; Artegiani et al., 2019). These data raised the question of how BAP1-mediated deubiquitination of H2Aub regulates transcriptional repression. Polycomb repressive complex 1 (PRC1) is a major ubiquitin ligase to mediate H2Aub (Wang et al., 2004). It was shown that, while PRC1 and BAP1 (the writer and eraser of H2Aub, respectively) exert opposing effects on H2Aub levels on the SLC7A11 promoter (i.e., PRC1 inactivation decreases whereas BAP1 inactivation increases H2Aub levels on the SLC7A11 promoter), both PRC1 and BAP1 repress SLC7A11 expression (Fig. 2) (Zhang et al., 2019a). These data are in line with previous observations showing that inactivation of Drosophila PRC1 or BAP1 leads to upregulation of HOX genes (Scheuermann et al., 2010), and suggest a model that a dynamic balance between H2A ubiquitination and deubiquitination by PRC1 and BAP1, rather than H2Aub per se, might be important for repressing the expression of certain target genes such as SLC7A11 and HOX genes.
While H2Aub is associated with transcriptional repression, mono-ubiquitination of histone 2B (H2Bub) at lysine 120 generally correlates with transcriptional activation (Henry et al., 2003). A recent study linked H2Bub-mediated epigenetic mechanisms to transcriptional activation of SLC7A11 and ferroptosis suppression (Wang et al., 2019b). It was shown that erastin treatment decreases global H2Bub levels as well as H2Bub occupancy on the SLC7A11 promoter, resulting in SLC7A11 transcriptional repression (Fig. 2) (Wang et al., 2019b). [As noted above, erastin treatment generally increases SLC7A11 expression. These data suggest that erastin treatment can induce two opposing effects on SLC7A11 transcription: transcriptional activation (through a still unknown mechanism) and transcriptional repression (through decreasing H2Bub occupancy on the SLC7A11 promoter (Wang et al., 2019b) and potentially activating ATF3 (Wang et al., 2020a)]. It seems that erastin-induced SLC7A11 transcriptional activation overrides its transcriptional repression, resulting in increased SLC7A11 expression as a net effect.] It was further demonstrated that a reduction of H2Bub levels upon erastin treatment promotes ferroptosis sensitivity, possibly through repressing SLC7A11 expression. Mechanistically, it was proposed that p53 promotes the nuclear translocation of USP7, a DUB to remove ubiquitin from H2Bub, resulting in decreased H2Bub occupancy on the SLC7A11 promoter and subsequent SLC7A11 transcriptional repression (Fig. 2) (Wang et al., 2019b).
H3K9 methylation has been shown to correlate with transcriptional repression (Stewart et al., 2005). It was recently reported that overexpression of KDM3B, a H3K9 demethylase, decreases H3K9 methylation and upregulates SLC7A11 expression (Fig. 2), presumably by removing this repressive histone mark on the SLC7A11 promoter, resulting in enhanced resistance to erastin-induced ferroptosis (Wang et al., 2020b). As another example, BRD4, a member of the bromodomain and extraterminal domain (BET) family of proteins, is an important reader protein capable of recognizing acetylated histones and recruiting transcription factors to regulate gene transcription. Pharmacological inhibition of BRD4 by its inhibitors such as JQ1 represents a promising therapeutic strategy in cancer treatment (Shi and Vakoc, 2014). A recent study showed that JQ1 can induce ferroptosis both in vitro and in xenograft tumors; furthermore, JQ1 treatment or BRD4 knockdown results in downregulation of SLC7A11 as well as other ferroptosis regulators (Sui et al., 2019), suggesting that BRD4 might activate SLC7A11 transcription via epigenetic mechanisms, whereas JQ1-mediated BRD4 inhibition represses SLC7A11 expression and promotes ferroptosis.
Another critical epigenetic mechanism to control gene transcription involves chromatin remodeling mediated by SWI/SNF complexes, which govern histone insertion, ejection, and nucleosome sliding and therefore control gene transcription (Kadoch and Crabtree, 2015). SWI/SNF complex-mediated chromatin remodeling was recently implicated to regulate SLC7A11 transcription (Ogiwara et al., 2019). It was shown that ARID1A, an integral component of SWI/SNF complexes, binds on the SLC7A11 promoter and facilitates NRF2-mediated transcriptional activation of SLC7A11 through chromatin remodeling by SWI/SNF complexes (Fig. 2); ARID1A deficiency results in SLC7A11 transcriptional repression, impaired cystine uptake and GSH biosynthesis, and subsequently ROS induction (Ogiwara et al., 2019). Together, these recent studies highlight critical roles of histone modifications and chromatin remodeling in governing SLC7A11 expression and ferroptosis.
Posttranslational regulation of SLC7A11
Apart from transcriptional regulation as discussed above, SLC7A11 expression can also be regulated at mRNA levels by nonsense-mediated mRNA decay and microRNAs (Liu et al., 2011; Drayton et al., 2014; Martin and Gardner, 2015; Wu et al., 2017), the detailed mechanisms of which have been discussed in a previous review (Koppula et al., 2018). Below we focus on various aspects of posttranslational regulation of SLC7A11 as revealed by more recent studies.
Earlier studies identified adhesion molecule CD44 variant (CD44v) as a SLC7A11-binding partner that regulates SLC7A11 protein stability, and showed that CD44v inactivation leads to decreased stability and defective cell surface localization of SLC7A11, resulting in compromised SLC7A11 function in regulating GSH synthesis, redox maintenance, and tumor growth and metastasis (Ishimoto et al., 2011; Yae et al., 2012). Since CD44v is neither a ubiquitin ligase nor a DUB, how VD44v regulates SLC7A11 protein stability had remained elusive. A recent study sheds light on this question. OTUB1, a DUB of the ovarian tumor (OTU) family, was identified as a SLC7A11-interacting protein which stabilizes SLC7A11 by preventing SLC7A11 from undergoing ubiquitination and proteasomal degradation (Fig. 2) (Gan, 2019; Liu et al., 2019). It was further shown that OTUB1, CD44v, and SLC7A11 form a trimeric complex and that CD44v promotes SLC7A11 protein stability by maintaining the interaction between OTUB1 and SLC7A11; consequently, inactivation of CD44v or OTUB1 destabilizes SLC7A11 and increases ferroptosis sensitivity (Liu et al., 2019). The potential ubiquitin ligase which mediates SLC7A11 ubiquitination and proteasomal degradation remains to be identified.
Recent evidence also indicated that, as a transmembrane protein localized on the plasma membrane, SLC7A11 is also subjected to regulation by lysosomal degradation. As detailed in a later section, SLC7A11 promotes glucose dependency, leading to the exquisite vulnerability of cancer cells with high SLC7A11 expression (SLC7A11high) to glucose starvation (Goji et al., 2017; Koppula et al., 2017; Shin et al., 2017; Joly et al., 2020; Liu et al., 2020). It is known that cells cultured at high density survive much better than those cultured at low density under glucose starvation. A recent study showed that the better survival of cancer cells under higher density is at least partly achieved by downregulation of SLC7A11 levels (Yamaguchi et al., 2020). Mechanistically, it was proposed that higher cell density inactivates mechanistic target of rapamycin complex 1 (mTORC1), resulting in SLC7A11 degradation in lysosomes (Fig. 2). Although how cell density modulates mTORC1 activity was not revealed in this study, another recent study showed that high cell density activates LATS1/2 kinases, a key component in the Hippo pathway, which then phosphorylates Raptor, an integral component of mTORC1, resulting in mTORC1 inactivation (Gan et al., 2020). How mTORC1 inactivation subsequently promotes SLC7A11 lysosomal degradation remains unknown currently.
mTOR exists in two distinct kinase complexes, mTORC1 and mTOR complex 2 (mTORC2). While mTORC1 coordinates nutrient and energy availability with protein synthesis and cell growth, mTORC2 integrates growth factor signaling to promote cell proliferation and survival mainly through phosphorylating AKT (Kim and Guan, 2019). Notably, SLC7A11 can be regulated not only by mTORC1 (Gan et al., 2020), but also by mTORC2 (Gu et al., 2017). It was shown that mTORC2 interacts with SLC7A11 and phosphorylates serine 26 located at the cytoplasmic region of SLC7A11 in response to growth factor stimulation, resulting in inhibition of its transporter activity (Fig. 2) (Gu et al., 2017). Interestingly, another study revealed that AKT, a major substrate of mTORC2, is capable of directly phosphorylating SLC7A11 on the same site, and AKT-mediated SLC7A11 phosphorylation also inhibits its cystine transport activity (Fig. 2) (Lien et al., 2017).
Other studies revealed that cell surface localization of SLC7A11 is also subjected to regulation. EGF receptor (EGFR) was shown to interact with SLC7A11 and maintain its appropriate localization on the plasma membrane (Tsuchihashi et al., 2016). Consequently, EGFR-expressing glioma cells exhibit increased cystine uptake and GSH biosynthesis as well as enhanced glutamate export, which together promote tumor growth and invasion (Tsuchihashi et al., 2016). Together, these emerging studies established that SLC7A11 can be regulated by diverse posttranslational mechanisms, including regulation of protein stability, localization, and transporter activity.
SLC7A11 PROMOTES TUMOR DEVELOPMENT PARTLY VIA INHIBITING FERROPTOSIS
It is well established that cell death, most notably apoptosis, plays important roles in tumor suppression (Green and Evan, 2002; Igney and Krammer, 2002), and cell death resistance is a hallmark of cancer (Hanahan and Weinberg, 2011). Recent studies have unraveled that ferroptosis, similar to apoptosis, represents another powerful tumor suppression mechanism, which presumably has evolved to eliminate precancerous cells that have been exposed to metabolic stress or nutrient deprivation. Importantly, SLC7A11 has emerged as a central hub linking ferroptosis to its proposed tumor suppression function. In this section, we will discuss how SLC7A11-mediated ferroptosis suppression contributes to tumor development in the context of several tumor suppressors and oncogene pathways.
p53 is the most mutated tumor suppressor in human cancers (Muller and Vousden, 2013). Our mechanistic understanding of p53 function in tumor biology has provided great insights into the role of ferroptosis in tumor suppression. Traditionally, the tumor-suppressive function of p53 has been largely attributed to its roles in inducing cell cycle arrest, senescence, and/or apoptosis. Curiously, a p53 mutant that cannot be acetylated on certain lysine residues (3KR mutant) loses its ability to induce cell cycle arrest, senescence, or apoptosis, yet is still capable of suppressing tumor formation in vivo (Li et al., 2012). This p53 mutant was later discovered to retain its tumor-suppressive function, at least partly by repressing SLC7A11 expression and inducing ferroptosis (Jiang et al., 2015). Mutation of an additional acetylation site at the backdrop of p53 3KR mutant markedly attenuated p53’s capability to suppress tumor formation, repress SLC7A11 expression, and induce ferroptosis in cancer cells (Wang et al., 2016). An African-specific polymorphic mutant of p53 (S47 mutant) provides another evidence to link p53 regulation of SLC7A11 and ferroptosis to tumor suppression. Opposite to p53 3KR mutant, p53 S47 mutant retains its capability to induce cell cycle arrest and apoptosis, yet is unable to suppress tumor development (Jennis et al., 2016). It was further shown that this mutant exhibits impaired ability to repress SLC7A11 expression or induce ferroptosis, suggesting that SLC7A11 downregulation and ferroptosis induction play a role in p53-mediated tumor suppression. Collectively, these studies using different p53 mutants suggest that ferroptosis induction caused by SLC7A11 repression at least partly underlies p53 function in tumor suppression (Fig. 3).

SLC7A11 promotes tumor development through both ferroptosis-dependent and -independent mechanisms. SLC7A11 promotes tumor development partly through detoxifying lipid peroxides and suppressing ferroptosis, which involves both ASCL4-dependent (via GPX4) and -independent (via ALOX12) pathways. Tumor suppressor proteins such as p53 and BAP1 inhibit tumorigenesis partly by suppressing SLC7A11 expression and promoting ferroptosis, while oncogenic KRAS and OTUB1 promote tumor development partly by promoting SLC7A11 levels and inhibiting ferroptosis. SLC7A11 can also promote tumorigenesis via pathways that are not dependent on its function to suppress ferroptosis. KRAS: Kirsten rat sarcoma viral oncogene homolog, OTUB1: OTU deubiquitinase, ubiquitin aldehyde binding 1, p53: tumor protein p53, BAP1: BRCA1 associated protein-1, ACSL4: Acyl-CoA synthetase long chain family member 4, GPX4: glutathione peroxidase 4, ALOX12: arachidonate 12-lipoxygenase
It is well established that SLC7A11 inhibits ferroptosis through importing cystine, promoting GSH biosynthesis, and subsequently facilitating GPX4-mediated detoxification of lipid peroxides (Fig. 1) (Stockwell et al., 2017). Ferroptosis induced by SLC7A11 or GPX4 inactivation can be largely abolished by inactivation of ACSL4, a lipid metabolism enzyme (Doll et al., 2017; Kagan et al., 2017; Stockwell and Jiang, 2020). Interestingly, a recent study proposed that SLC7A11 suppression downstream of p53 promotes ferroptosis through GSH-GPX4- and ACSL4-independent mechanisms (Chu et al., 2019). Mechanistically, it was shown that SLC7A11 interacts with arachidonate 12-lipoxygenase (ALOX12), and this interaction suppresses ALOX12’s lipoxygenase activity to mediate polyunsaturated fatty acid (PUFA) peroxidation, thereby inhibiting ferroptosis; consequently, inactivation of ALOX12, but not ACSL4, abrogates p53-mediated ferroptosis (Chu et al., 2019). Importantly, ALOX12 gene is frequently deleted in human cancers (Chu et al., 2019). It was further demonstrated that deletion of one Alox12 allele in mouse promotes lymphomagenesis in Eμ-Myc models, and that ALOX12 missense mutations derived from human cancers lose their capabilities to oxygenate PUFA and to promote p53-mediated ferroptosis (Chu et al., 2019). This study suggests that, in the context of p53 deficiency, SLC7A11 can promote tumor development through a non-canonical mechanism by directly interacting with ALOX12 and inhibiting ALOX12-mediated ferroptosis, which seems to be independent of its cystine transporting activity (Fig. 3).
BAP1 is another tumor suppressor that is frequently mutated or deleted in several human cancers (Carbone et al., 2013). As discussed above, recent studies identified SLC7A11 as a key downstream target of BAP1 (Zhang et al., 2018b). Functional analyses revealed that, by suppressing SLC7A11 transcription, BAP1 inhibits cystine uptake and GSH biosynthesis and promotes ferroptosis under various ferroptosis-inducing conditions; furthermore, tumor growth suppression caused by BAP1 restoration in BAP1-deficient tumors can be partly abolished by SLC7A11 overexpression or treatment with ferroptosis inhibitor, and BAP1 mutations derived from human cancers abrogate their abilities to suppress SLC7A11 expression and to induce ferroptosis (Zhang et al., 2018b). Therefore, similar to p53, BAP1 suppresses tumor development at least partly by inducing ferroptosis upon SLC7A11 repression (Fig. 3) (Zhang et al., 2019c). However, unlike p53, it seems that BAP1 promotes ferroptosis through the canonical function of SLC7A11 (i.e., through SLC7A11-mediated cystine uptake to promote GSH biosynthesis). Since both p53 and BAP1 regulate SLC7A11 at the transcriptional level, it remains unclear how SLC7A11 shifts between its canonical and non-canonical functions in ferroptosis regulation in these two different contexts.
In cancer cells, not only de-repression of SLC7A11 expression, but also stabilization of SLC7A11 protein can lead to enhanced tumor formation through inhibiting ferroptosis. As discussed in the previous section, OTUB1 deubiquitinates and stabilizes SLC7A11 protein (Liu et al., 2019). Consequently, OTUB1 promotes tumor development partly via stabilizing SLC7A11 and inhibiting ferroptosis (Fig. 3) (Liu et al., 2019). Since OTUB1 is frequently overexpressed in human cancers, high SLC7A11 levels in some cancers might result from post-translational regulation by OTUB1.
Emerging data suggest that SLC7A11-mediated ferroptosis inhibition not only plays a role in tumor development caused by loss of tumor suppressors (such as p53 and BAP1), but also contributes to oncogene-driven tumorigenesis. KRAS is one of the most mutated proto-oncogene in human cancers, and is highly mutated in a variety of cancers, including pancreatic ductal adenocarcinomas (PDAC), lung cancer, and colorectal cancer (Prior et al., 2012). Recent studies showed that oncogenic KRAS promotes SLC7A11 transcription, resulting in increased SLC7A11-mediated cystine uptake and GSH biosynthesis (Fig. 3) (Lim et al., 2019; Hu et al., 2020). Mechanistically, one study suggested that ETS-1 transcription factor mediates oncogenic KRAS-induced SLC7A11 expression via cooperation with ATF4 (Lim et al., 2019), while the other study indicated that oncogenic KRAS promotes SLC7A11 transcription through NRF2 (Hu et al., 2020). It was further shown that genetic ablation or pharmacological inhibition of SLC7A11 markedly attenuates oncogenic KRAS-induced tumor growth in xenograft models (Lim et al., 2019; Hu et al., 2020). More than 90% of PDAC harbor KRAS mutations (Prior et al., 2012). SLC7A11 inactivation, genetically or pharmacologically, was shown to potently induce lipid peroxidation and ferroptotic cell death in pancreatic cancer cells (Daher et al., 2019; Badgley et al., 2020); further studies using genetically engineered mouse models (GEMMs) demonstrated that deletion of Slc7a11 significantly suppresses oncogenic Kras-driven PDAC development with ferroptosis induction in Slc7a11-deficient pancreatic tumors with oncogenic Kras activation (Badgley et al., 2020), suggesting that SLC7A11-mediated ferroptosis suppression likely contributes to KRAS-driven PDAC development.
It should be noted that not all SLC7A11 expression alterations in cancers can be linked to its presumed function to mitigate ferroptosis and promote tumor development. Tumor suppressor ARID1A is frequently mutated in multiple forms of cancers (Jones et al., 2010; Mao and Shih Ie, 2013). As discussed in the previous section, ARID1A-containing SWI/SNF complex promotes SLC7A11 transcription. In contrast to p53 or BAP1 deficiency (wherein tumor suppressor deficiency increases SLC7A11 expression), ARID1A deficiency suppresses SLC7A11 expression in cancer cells (Ogiwara et al., 2019). It was further shown that the suppressed SLC7A11 expression results in decreased cystine uptake and GSH biosynthesis in ARID1A-deficient cancer cells, which induces a vulnerability rendering ARID1A-deficient cancers susceptible for pharmacological inhibition of GSH biosynthesis (Ogiwara et al., 2019). Tumors with p53 gain-of-function mutations present another example wherein tumors actually exhibit reduced SLC7A11 levels. While many tumors harbor p53 loss-of-function mutations or deletions, others contain oncogenic gain-of-function mutations of p53, resulting in the expression of mutant p53 protein in tumors, and such tumors often exhibit poor prognosis and aggressive tumor phenotypes (Muller and Vousden, 2013). Based on the findings that wild-type (WT) p53 represses SLC7A11 expression and p53 deficiency promotes SLC7A11 expression in cancer cells (Jiang et al., 2015), one would expect that p53 mutants with oncogenic functions, similar to p53 deficiency, should promote SLC7A11 expression. In contrast, it was demonstrated that p53 mutants somehow gain additional functional capabilities to inhibit NRF2-mediated transcriptional activation of SLC7A11, resulting in decreased levels of SLC7A11 and GSH and increased ROS and lipid peroxidation in p53 mutant cancer cells, and rendering such tumors more sensitive to SLC7A11 inhibitors such as sulfasalazine (Liu et al., 2017). Whether NRF2 is involved in WT p53-mediated SLC7A11 repression remains to be examined.
To summarize, consistent with the known role of SLC7A11 in suppressing ferroptosis and the emerging concept that ferroptosis is a tumor suppression mechanism, various studies have revealed that loss of tumor suppressors (such as p53 and BAP1) (Jiang et al., 2015; Zhang et al., 2018b), mutation of proto-oncogenes (such as KRAS) (Badgley et al., 2020), or overexpression of proteins with pro-tumorigenic functions (such as OTUB1) (Liu et al., 2019) increases SLC7A11 levels in cancer (by either upregulating its transcription or stabilizing its protein), resulting in ferroptosis suppression and enhanced tumor development (Fig. 3). However, not all tumors show increased SLC7A11 levels, and some tumors (such as ARID1A-deficient tumors or tumors with p53 gain-of-function mutations) even exhibit decreased SLC7A11 levels (Liu et al., 2017; Ogiwara et al., 2019). In this latter scenario, the decreased SLC7A11 levels apparently cannot explain tumor phenotypes in the corresponding cancers; instead, it is proposed that this decreased SLC7A11 expression creates an Achilles heel for therapeutic targeting in tumors with low expression of SLC7A11, as such tumors presumably should be more vulnerable to oxidative stress.
FERROPTOSIS-INDEPENDENT FUNCTIONS OF SLC7A11 IN PROMOTING TUMOR DEVELOPMENT
Although this review so far has somewhat focused on the anti-ferroptosis function of SLC7A11 in tumor biology (partly because ferroptosis is a relatively recently discovered cell death mechanism), we want to emphasize that SLC7A11’s roles in tumor biology clearly go beyond regulating ferroptosis. In this section, we also discuss several other functions of SLC7A11 which likely operate independent of ferroptosis (Fig. 3).
While SLC7A11’s function in suppressing ferroptosis is well established, other studies have shown that SLC7A11 inactivation can also induce apoptosis. It was initially shown that Slc7a11 deficiency in mouse melanocytes induces substantial cell death accompanied with features of apoptosis such as caspase-3 cleavage (Qiao et al., 2008). Subsequent studies revealed that SLC7A11 inhibition can induce apoptosis in cancer cells under different contexts (Liu et al., 2011; Yoshikawa et al., 2013; Dai et al., 2014). While erastin potently induces ferroptosis by inhibiting SLC7A11-mediated cystine uptake (Stockwell et al., 2017), HG106, another recently identified SLC7A11 inhibitor, does not induce ferroptosis but mainly induces apoptosis (Hu et al., 2020). Since these studies suggested that SLC7A11 inactivation-induced apoptosis is likely driven by GSH depletion and ROS induction, it remains to be established how GSH depletion resulting from SLC7A11 inactivation can induce different modes of cell death (ferroptosis vs apoptosis) under different conditions or contexts. The differential effects of erastin and HG106 may relate to their different potencies or off-target effects as well as different cell lines used in these studies (such that SLC7A11 inactivation results in ferroptosis in one cell line but induces apoptosis in another cell line). Future studies using respective cell death inhibitors or cell death deficient cells (such as BAX/BAK or ACSL4 KO cells) are required to address these questions. Furthermore, a side-by-side comparison between erastin and HG106 in the same cell lines will be critical to clarify the differential cell death-inducing effects of these SLC7A11 inhibitors.
In addition, inhibition of SLC7A11, genetically or pharmacologically, has been shown to result in cellular defects typically associated with oncogenic pathway inactivation, including suppression of cell proliferation/growth and invasion, defective clonogenic survival, and decreased anchorage-independent growth, whereas SLC7A11 overexpression can exert the opposite cellular effects (Ishimoto et al., 2011; Yae et al., 2012; Yang and Yee, 2014; Ji et al., 2018; Shin et al., 2018; Hu et al., 2020). Although the underlying mechanisms are often not definitively established, SLC7A11’s ability to promote GSH biosynthesis has been attributed to regulate these cellular functions. In another study, SLC7A11-mediated glutamate export has been linked to the increased invasiveness of breast cancer cells (Dornier et al., 2017).
As noted above, various stress conditions induce SLC7A11 expression to promote cellular adaption to stress conditions. Correspondingly, it is well established that SLC7A11 promotes drug-, chemo-, and radio-resistance in cancer cells. SLC7A11 overexpression has been shown to correlate with or functionally promote resistance to various therapeutic drugs, such as cisplatin, gemcitabine, and MAPK pathway inhibitors (Okuno et al., 2003; Lo et al., 2008; Wang et al., 2018). SLC7A11 also confers resistance to geldanamycin, thereby promoting chemoresistance (Huang et al., 2005). Finally, it was shown that radiation can induce SLC7A11 expression (Xie et al., 2011; Lei et al., 2020), and SLC7A11 overexpression promotes radioresistance, whereas SLC7A11 inhibition enhances radiosensitivity in cancer cells or tumors (Cobler et al., 2018; Nagane et al., 2018; Lei et al., 2020); importantly, the radioresistance caused by SLC7A11 overexpression was largely abolished under ferroptosis inhibitor treatment (Lei et al., 2020), suggesting that SLC7A11 promotes radioresistance mainly through inhibiting ferroptosis. It remains to be determined whether ferroptosis inhibition also plays a role in SLC7A11-mediated drug- or chemo-resistance, although it is generally believed that SLC7A11 promotes these resistance phenotypes through enhancing GSH synthesis.
Most of the cellular functions we have discussed so far relate to cell autonomous effects regulated by SLC7A11. Emerging evidence indicated that SLC7A11 can also exert cell non-autonomous effects on tumor microenvironment through glutamate exporting. A recent study revealed that treatment with VEGF blockade agents in a glioma mouse model leads to increased expression of SLC7A11, and it was proposed that glutamate exported by SLC7A11 from tumor cells then promotes the proliferation and immunosuppressive function of regulatory T-cells (Tregs), which contributes to adaptive resistance to anti-VEGF therapy in glioblastoma (Long et al., 2020). It was further shown that blocking Tregs using anti-CD25 antibody could sensitize tumors to VEGF blockade agents (Long et al., 2020). While most studies have focused on SLC7A11-mediated cystine import, this study suggests that SLC7A11-mediated glutamate export can cross-talk with and modulate tumor immune systems, which in turn influence tumor growth and response to cancer therapies.
In summary, ferroptosis-independent functions of SLC7A11 in tumor biology include its regulation of other non-ferroptotic cell death, cell proliferation, cell invasion, chemo-/drug-/radio-resistance, and tumor immunity, which can be achieved through SLC7A11-mediated cystine import and/or glutamate export.
SLC7A11 INDUCES NUTRIENT DEPENDENCY IN CANCER CELLS
As extensively discussed in previous sections, SLC7A11-mediated cystine uptake is critical for cancer cells to suppress ferroptosis and to maintain redox homeostasis and biomass incorporation. Many cancer cells upregulate SLC7A11 levels to enhance their capability to obtain extracellular cystine. Emerging evidence indicates that, however, this comes at a significant cost for SLC7A11high cancer cells. In our view, these “costs” are mainly derived from two metabolic features associated with cystine uptake mediated by SLC7A11. First, as an antiporter, SLC7A11-mediated cystine uptake is coupled with glutamate export (Fig. 4A). Consequently, a significant amount of intracellular glutamate is exported out of cells in exchange for cystine uptake in SLC7A11high cancer cells. Indeed, it was estimated that 30%–50% of intracellular glutamate is exported by SLC7A11 in exchange for extracellular cystine (Bannai and Ishii, 1988). In addition, due to the oxidizing extracellular environment, extracellular cystine is much more stable than extracellular cysteine, resulting in a much higher concentration of extracellular cystine than that of extracellular cysteine. In order to obtain cysteine, cancer cells primarily rely on taking up extracellular cystine (rather than extracellular cysteine) and then need to convert intracellular cystine to cysteine (Fig. 4A) (this feature is unique in nutrient uptake. For most other nutrients, such as glucose and glutamine, cells can directly take up the nutrients they require through corresponding nutrient transporters). Because the reduction of cystine to cysteine consumes NADPH, cells need to first make redox “investment” (in the form of NADPH) in order to obtain cysteine for maintaining redox homeostasis [which is somewhat similar to “ATP investment” made in glycolysis, wherein two molecules of ATP are “invested” (i.e., consumed) in the first several steps of glycolysis in order to activate glucose for its subsequent generation of four molecules of ATPs in the later steps of glycolysis]. Consequently, these two “costs” associated with SLC7A11-mediated cystine uptake, namely glutamate export and NADPH consumption for cystine reduction, drive SLC7A11high cancer cells to be highly dependent on glutamine and glucose. In this section, we will discuss recent studies leading to our current understanding of SLC7A11-induced glutamine and glucose dependency.

SLC7A11 regulates glucose and glutamine dependencies in cancer cells. (A) SLC7A11 functions to import extracellular cystine in exchange for intracellular glutamate. Extracellular glutamine is imported into the cell and converted to glutamate by Glutaminase. Subsequently, glutamate serves to fuel the TCA cycle via its conversion to α-ketoglutarate and acts as a precursor for GSH synthesis. The other arm of SLC7A11 transporter function imports extracellular cystine. Once imported into the cell, cystine is reduced to cysteine by the consumption of NADPH. Cysteine then serves as the rate limiting precursor for GSH synthesis, and GSH suppresses ROS. NADPH is mainly supplied from glucose via the PPP. Consequently, an imbalance in this metabolic network leads to specific metabolic dependencies in SLC7A11high cancer cells. (B and C) Glutamine dependency: In SLC7A11high cancer cells, increased glutamate export results in a partial depletion of intracellular glutamate that can feed into the TCA cycle. This pushes SLC7A11high cancer cells to uptake more glutamine and activates glutaminase to promote glutamine conversion to glutamate, resulting in glutamine dependency (B). When SLC7A11high cancer cells (with increased glutamate export) are challenged with low glutamine availability, intracellular glutamate is inadequate to fuel the TCA cycle, resulting in anaplesrosis defect and cell growth arrest (C). (D and E) Glucose dependency: In SLC7A11high cancer cells, large amount of extracellular cystine is imported into the cell. Due to its low solubility, buildup of intracellular cystine is likely toxic, forcing cells to quickly reducing cystine to much more soluble cysteine in the cytosol. Because this reaction requires NADPH and cytosolic NADPH is mainly supplied from glucose via the PPP, SLC7A11high cancer cells exhibit glucose-PPP dependency. (D) Limiting glucose availability in SLC7A11high cancer cells leads to NADPH depletion, accumulation of intracellular cystine and other disulfide molecules resulting in disulfide stress and rapid cell death (E). GSH: reduced glutathione, PPP: pentose phosphate pathway, TCA cycle: tricarboxylic acid cycle, αKG: α-ketoglutarate
SLC7A11-induced glutamine dependency in cancer cells
Glutamine is a critical amino acid that supports bioenergetic and biosynthetic processes and maintains redox balance in cancer cells (Hensley et al., 2013). It is also the most abundant amino acid in plasma as well as in cell culture media. Once imported into cells via SLC1A5 and other transporters, glutamine is converted to glutamate via glutaminase. Glutamate has several important functions in cellular metabolism, prominent among which are its roles in anaplerosis [replenishing the tricarboxylic acid (TCA) cycle through its conversion to TCA cycle intermediate α-ketoglutarate (αKG)] and as a precursor for GSH synthesis (Fig. 4A) (Hensley et al., 2013). As noted above, glutamate is also exported out of cells via SLC7A11 in exchange for extracellular cystine. In SLC7A11high cancer cells, high glutamate export results in a partial depletion of intracellular glutamate, which then drives cells through the relief of feedback inhibition to take up more glutamine as well as to activate glutaminase for glutamate replenishment, leading to glutamine dependency in SLC7A11high cancer cells (Fig. 4B). Consequently, SLC7A11high cancer cells are more sensitive to glutamine starvation (Fig. 4C).
This model has been supported by several studies in recent years. An analysis of glutamine uptake and dependency across a wide range of breast cancer cells revealed that, compared to normal mammary epithelial cells and basal breast cancer cells, most basal and claudin-low triple-negative breast cancer (TNBC) cells consume more glutamine and exhibit more glutamine dependency (Timmerman et al., 2013). Differential glutamine dependency among these breast cancer cells does not correlate with the expression levels of known regulators in glutamine metabolism, such as glutaminase; instead, it was shown that the sensitivities of these cells to glutamine deprivation correlate with SLC7A11 expression levels and cystine consumption rates (i.e., compared to other breast cancer cell lines, basal and claudin-low TNBC cells express higher levels of SLC7A11, exhibit more cystine consumption, and are more sensitive to glutamine deprivation) (Timmerman et al., 2013). These data suggest that the glutamine dependency phenotype observed in these TNBC cells is likely driven by the high expression of SLC7A11 and the high rate of SLC7A11-mediated cystine uptake in these cells.
In another study aimed to identify the environmental factors that underlie the less glutamine dependence of tumor cells grown in vivo than those grown in vitro, cystine was identified as the sole factor that causes the higher glutamine dependency for cancer cells cultured in standard cell culture media than the same cells cultured in media that better mimic in vivo conditions; therefore, compared to in vivo conditions, higher levels of cystine in standard cell culture media drive more SLC7A11-mediated cystine uptake and glutamate export, resulting in more glutamine dependency and more cellular sensitivity to glutaminase inhibition (Muir et al., 2017). As discussed in a previous section, SLC7A11 transcription is controlled by the KEAP1-NRF2 signaling axis. In line with this, it was shown that cancer cells with KEAP1 mutation and/or NRF2 hyperactivation exhibit increased glutamine dependency and enhanced sensitivity to glutamine deprivation or glutaminase inhibition, partly via high SLC7A11-mediated glutamate export in these cancer cells (Romero et al., 2017; Sayin et al., 2017).
SLC7A11-induced glucose dependency in cancer cells
One hallmark of metabolic reprogramming in cancer cells is the deregulated uptake of glucose (Pavlova and Thompson, 2016). Upon its transport into cells through glucose transporters (GLUTs), glucose is subsequently shunted into two major metabolic pathways, glycolysis and the pentose phosphate pathway (PPP) (Vander Heiden et al., 2009). Via glycolysis, glucose is metabolized to pyruvate, which can be oxidized by the TCA cycle to generate ATP. By contrast, the PPP uses glucose to generate both ribose-5-phosphate, for nucleic acid synthesis, and NADPH, which provides the reducing power to support reductive biosynthetic reactions and to maintain cellular redox homeostasis.
Glucose is one of the principal nutrients supporting cancer cell survival (Vander Heiden et al., 2009; Boroughs and DeBerardinis, 2015). Several studies independently revealed that SLC7A11high cancer cells are highly dependent on glucose for survival and are exquisitely sensitive to glucose starvation-induced cell death (Goji et al., 2017; Koppula et al., 2017; Shin et al., 2017; Liu et al., 2020). In one study, loss-of-function screens uncovered both SLC7A11 and SLC3A2 as the top genes whose inactivation promotes cell survival under glucose starvation (Shin et al., 2017). In another study, SLC7A11 expression was identified to be highly induced upon glucose starvation (Koppula et al., 2017). As discussed in previous sections, stress-induced SLC7A11 expression generally serves as an adaptive response to protect cells from stress-induced cell death (such as ferroptosis); therefore, it was initially hypothesized that glucose starvation-induced SLC7A11 expression might protect cells from cell death triggered by glucose starvation. Surprisingly, it was subsequently shown that SLC7A11 overexpression drastically promotes glucose starvation-induced cell death, whereas SLC7A11 inactivation significantly suppresses cell death under glucose starvation (Koppula et al., 2017; Shin et al., 2017). Together, these studies revealed that SLC7A11 promotes glucose dependency in cancer cells.
While it is easy to understand the metabolic underpinning of SLC7A11-induced glutamine dependency (given that SLC7A11-mediated glutamate export is directly linked to glutamine metabolism), how SLC7A11 promotes glucose dependency has remained less clear (partly because SLC7A11-associated metabolism is not directly connected to glucose metabolism). Since both glucose and glutamate are important for TCA cycle anaplerosis and mitochondrial respiration, it was initially proposed that, because SLC7A11-mediated glutamate export decreases intracellular glutamate reserves, SLC7A11high cancer cells are forced to have more reliance on glucose to replenish the TCA cycle (presumably through glycolysis) and to maintain mitochondrial respiration, resulting in glucose dependency (Koppula et al., 2017; Shin et al., 2017). In support of this, it was shown that supplementation of αKG, a TCA cycle intermediate derived from glutamate, rescues glucose starvation-induced cell death in SLC7A11high cancer cells (Koppula et al., 2017; Shin et al., 2017). However, other recent data argued against this model. For example, this model would predict that SLC7A11 overexpression should promote glycolytic flux and that SLC7A11high cancer cells should be sensitive to glycolysis inhibitors. However, a recent study showed that SLC7A11 overexpression does not promote glycolytic flux and that 2-deoxyglucose (2DG), a glycolysis inhibitor, even rescues glucose starvation-induced cell death in SLC7A11high cancer cells (the underlying mechanism will be described below) (Koppula et al., 2017; Shin et al., 2017). Furthermore, it was shown that cystine starvation rescues the cell death in SLC7A11high cancer cells under glucose starvation, suggesting that it is cystine import that underlies glucose starvation-induced cell death in SLC7A11high cancer cells (Goji et al., 2017; Joly et al., 2020; Liu et al., 2020).
Mechanistically, it was proposed that, because cystine is highly insoluble, significant buildup of intracellular cystine potentially is toxic to cells, forcing SLC7A11high cancer cells to quickly reduce cystine to much more soluble cysteine once cystine is taken up into cells. Since cystine reduction to cysteine consumes NADPH, this renders SLC7A11high cancer cells to be highly dependent on the glucose-PPP route (which is the major provider of cellular NADPH) to supply NADPH for cystine reduction (Fig. 4D). Consequently, glucose starvation limits NADPH supply, resulting in NADPH depletion, marked accumulation of cystine and other disulfide molecules, ROS accumulation, and rapid cell death (Fig. 4E). It should be noted that the fold changes of these redox defects are much more pronounced than that of glutamate level decrease in SLC7A11high cancer cells under glucose starvation, and thereby correlate better with the rapid and dramatic cell death observed in these cells upon glucose starvation (Liu et al., 2020). In further support of this model, glucose starvation-induced cell death and redox defects in SLC7A11high cancer cells can be substantially blocked by treatments that restore NADPH levels and prevent cystine accumulation, such as treatment with 2DG (which inhibits glycolysis but can still be shunted into the PPP to generate NADPH) (Liu et al., 2020), suggesting that it is cystine accumulation and/or NADPH depletion resulting from cystine uptake that causes the cell death in SLC7A11high cancer cells under glucose starvation.
There are several peculiar features associated with SLC7A11-induced glucose dependency that are worth further discussion. Because SLC7A11 has a well-established role in suppressing ROS under oxidative stress conditions, the observation that SLC7A11 overexpression dramatically increases ROS levels (Goji et al., 2017; Liu et al., 2020) under glucose starvation is rather counterintuitive. Earlier, we introduced the concept that cells need to first make redox “investment” (by supplying NADPH to reduce cystine to cysteine) in order to obtain cysteine for its subsequent utilization in redox maintenance. These data suggest that the failure in appropriately maintaining this redox “investment” (such as under glucose starvation) can have devastating cellular consequences by “bankrupting” the redox system in SLC7A11high cancer cells—somewhat analogous to company bankruptcy resulting from finance investment mismanagement. In addition, as discussed in previous sections, cystine in culture media is generally required for cell survival and cystine starvation is well known to induce ferroptosis; therefore, the observation that cystine starvation also rescues glucose starvation-induced cell death in SLC7A11high cancer cells seems confusing. It should be noted that glucose starvation-induced cell death in SLC7A11high cancer cells is not ferroptosis (Liu et al., 2020) and that SLC7A11high cancer cells have high intracellular cysteine reserves and therefore are generally resistant to cystine starvation (Zhang et al., 2018b; Liu et al., 2020). Consequently, at the time points when cystine starvation has not induced obvious ferroptosis in SLC7A11high cancer cells, it actually prevents glucose starvation-induced cell death in these cells. In other words, in SLC7A11high cancer cells, glucose starvation induces far more acute cellular toxicity than cystine starvation.
Taken together, these studies suggest that SLC7A11 overexpression induces both glutamine and glucose dependency in cancer cells with different underlying mechanisms. While SLC7A11-induced glutamine dependency mainly results from glutamate-derived anaplerosis (Fig. 4B), it seems that SLC7A11-induced glucose dependency largely relates to cystine-associated redox maintenance (although glutamate-derived anaplerosis might still play a role in it) (Fig. 4D). Correspondingly, the cellular phenotypes in SLC7A11high cancer cells in response to glutamine or glucose deprivation seem somewhat different: while glutamine deprivation (or glutaminase inhibition) mainly suppresses the cell growth in SLC7A11high cancer cells (which is in line with anaplerosis defects in these cells) (Romero et al., 2017) (Fig. 4C), glucose starvation induces rapid cell death in SLC7A11high cancer cells (consistent with drastic redox system collapse in this context) (Goji et al., 2017; Koppula et al., 2017; Shin et al., 2017; Liu et al., 2020) (Fig. 4E). In further support of this, N-acetyl cysteine (NAC) largely rescues cell death in SLC7A11high cancer cells under glucose starvation (Liu et al., 2020), but does not rescue the growth defect of these cells upon glutamine deprivation or glutaminase inhibition (Romero et al., 2017). In the next section, we will further discuss how our mechanistic understanding of SLC7A11-induced nutrient dependency can help inform therapeutic strategies to target SLC7A11 in cancer.
THERAPEUTIC STRATEGIES TARGETING SLC7A11 IN CANCER
In recent years, SLC7A11 has emerged as a promising therapeutic target in cancer therapy. To achieve appropriate therapeutic index in cancer therapy, the ideal therapeutic target being inhibited by cancer drugs presumably should have a selective role in tumor growth with a dispensable function in normal physiology, so that the corresponding drugs can produce the desired toxic effects in cancer cells (or tumors) without unwanted side effects in normal cells (or normal tissues). This seems to be the case for SLC7A11. Slc7a11 KO mice are viable, with no obvious phenotype in the major organs (Sato et al., 2005). Subsequent analyses revealed defects in spatial working memory and skin pigmentation in Slc7a11 KO or mutant mice (Chintala et al., 2005; De Bundel et al., 2011); nonetheless, these phenotypes are relatively minor. Considering the essential role of SLC7A11 in defending ferroptosis, the mild phenotype from Slc7a11 KO mice may seem somewhat surprising (as a comparison, germline or postnatal deletion of Gpx4, another essential anti-ferroptosis regulator, results in embryonic or adult lethality, respectively (Yant et al., 2003; Friedmann Angeli et al., 2014)). It is possible that normal cells or tissues can compensate for the loss of SLC7A11 by obtaining intracellular cysteine through de novo cysteine synthesis or cystine (or cysteine) uptake via additional transporters (such as SLC7A9-mediated cystine uptake in the kidney) (Stipanuk, 2004; Kandasamy et al., 2018). Because cancer cells often experience high levels of oxidative stress (Chio and Tuveson, 2017), they have significantly increased needs in antioxidant defense and therefore are much more dependent on SLC7A11-mediated cystine uptake to obtain cysteine and to maintain redox homeostasis than are normal tissues—much analogous to oncogene addiction in cancer development. Consistent with this, SLC7A11 is overexpressed in multiple cancer types, including lung cancer, TNBC, PDAC, renal cell carcinoma, liver cancer, and glioma, and its high expression often correlates with poor prognosis (Timmerman et al., 2013; Ji et al., 2018; Koppula et al., 2018; Zhang et al., 2018a; Zhang et al., 2018b; Badgley et al., 2020). In further support of this, deletion of Slc7a11 in adult mice does not affect normal pancreas development but significantly impairs Kras-driven PDAC development (Badgley et al., 2020).
These two characteristics of SLC7A11, namely its dispensability in normal physiology and its high expression in cancers, suggest that targeting SLC7A11 likely can selectively kill tumor cells and impair tumor growth while sparing normal cells or tissues, therefore nominating SLC7A11 as a promising therapeutic target in cancer treatment. Below, we further discuss recent preclinical studies aimed to target SLC7A11 in cancer. To help readers better understand these works, we have conceptualized these studies into two broad strategies (Fig. 5). The first involves directly inhibiting SLC7A11 transporter activity, whereas the other relates to targeting SLC7A11-associated metabolic vulnerabilities (glucose or glutamine dependency) in cancer. (To make an analogy here, these strategies are similar to those used to target KRAS-driven cancers: while some investigators have focused on directly inhibiting oncogenic KRAS, with the recently developed Kras (G12C) inhibitor AMG510 as a notable example (Canon et al., 2019), others have aimed instead to target KRAS-associated vulnerabilities, for example, the efforts to identify synthetically lethal interactions with KRAS (Cox et al., 2014)).

Therapeutic strategies to target SLC7A11 in cancer. The antiporter function of SLC7A11 offers several strategies for therapeutic targeting. (1) Direct blocking of SLC7A11 cystine transporter activity using its inhibitors such as erastin, IKE, sulfasalazine, sorafenib, and HG106. These drugs inhibit cystine uptake by SLC7A11, thereby inducing lipid peroxidation and ferroptotic cell death. (2) Targeting glucose dependency in SLC7A11high cancer cells by inhibiting glucose uptake using GLUT inhibitors. Decreased glucose availability in in SLC7A11high cancer cells induce disulfide stress, leading to rapid cell death. (3) Targeting glutamine dependency in SLC7A11high cancer cells by using glutaminase inhibitors such as CB-839. Glutaminase inhibition decreases glutamate-derived anaplerosis and induces cell growth arrest in SLC7A11high cancer cells. IKE: imidazole ketone erastin, GLUT: glucose transporter
Several compounds have been identified or characterized as SLC7A11 inhibitors, including erastin, imidazole ketone erastin (IKE), sulfasalazine, sorafenib, and HG106 (Feng and Stockwell, 2018; Hu et al., 2020). Given their capabilities of inducing ferroptosis through blocking SLC7A11-mediated cystine uptake, erastin, IKE, sulfasalazine, and sorafenib are also collectively called class 1 ferroptosis inducers (FINs) in ferroptosis studies. From the therapeutic point of view, each of these compounds has its pros and cons. Erastin probably is the most widely used SLC7A11 inhibitor or class 1 FIN in cell culture studies; however, due to its poor metabolic stability and low solubility in vivo, erastin cannot be used for animal studies (Feng and Stockwell, 2018), preventing its further testing in preclinical or clinical studies. IKE is an erastin analog with nanomolar potency and high metabolic stability that is suitable for animal treatment (Zhang et al., 2019b). Recent studies using GEMMs also demonstrated its efficacy in suppressing Kras-driven PDAC development (Badgley et al., 2020). However, because IKE was developed relatively recently, it has not moved to the clinical trial stage yet. Sorafenib and sulfasalazine, in contrast, are drugs that were already approved by the U.S. Food and Drug Administration (FDA). Previous studies showed that both drugs inhibit SLC7A11 transporter activity, and can induce ferroptosis and suppress tumor growth in vivo (Feng and Stockwell, 2018; Lei et al., 2020; Ye et al., 2020). However, both drugs have mechanisms of action besides inhibiting SLC7A11 and inducing ferroptosis: sorafenib is commonly used in cancer therapy as a multi-kinase inhibitor, whereas sulfasalazine is capable of blocking prostaglandin production and is commonly used for treating patients with arthritis. Therefore, it is critical to determine whether their anti-cancer effect under any given context is indeed caused by SLC7A11 inhibition and ferroptosis induction. Finally, although HG106 was recently identified as a SLC7A11 inhibitor (Hu et al., 2020), whether it can act as a FIN remains to be further examined (so far, all other SLC7A11 inhibitors can induce ferroptosis). Together, although preclinical studies have established the proof of concept to inhibit SLC7A11 in cancer therapy, there is still a significant need to further identify potent and specific SLC7A11 inhibitors, study their mechanisms of action, test them in rigorous preclinical models, and eventually apply them in clinical care.
As another approach, rather than directly inhibiting SLC7A11, various studies have tested targeting metabolic vulnerabilities associated with SLC7A11high cancers. As discussed in the preceding section, SLC7A11high cancer cells are dependent on glutamine for glutamate-derived anaplerosis, which prompted the hypothesis that SLC7A11high cancer cells or tumors may be particularly sensitive to glutaminase inhibition. KEAP1 is frequently mutated in lung cancer, and KEAP1-mutant lung cancer exhibits NRF2 hyperactivation and aberrant expression of SLC7A11 (Cancer Genome Atlas Research, 2012, 2014). Consistent with this, it was shown that glutaminase inhibitor CB-839 suppresses tumor growth of KEAP1-mutant cell line- or patient-derived xenografts (PDXs) much more potently than that of KEAP1-WT counterparts (Romero et al., 2017; Galan-Cobo et al., 2019). In the previous section, we also discussed glucose dependency of SLC7A11high cancer cells; consequently, SLC7A11high cancer cells are exquisitely sensitive to glucose starvation-induced cell death. Likewise, compared with SLC7A11low cancer cells, SLC7A11high cancer cells exhibit much more vulnerability to GLUT inhibitor-induced cell death (Liu et al., 2020). It was further demonstrated that KL-11743, a potent GLUT1/3 dual inhibitor, selectively suppresses SLC7A11high tumor growth in both cell line xenografts and PDXs (Liu et al., 2020). These studies together suggest the possibility that SLC7A11 expression can be used as a biomarker to select cancer patients for glutaminase or GLUT inhibitor treatment.
CONCLUSIONS AND FUTURE PERSPECTIVES
Cancer cells upregulate SLC7A11 expression through diverse mechanisms to enhance their antioxidant defense and to suppress ferroptosis (a tumor suppression mechanism), which is beneficial for tumor growth. However, emerging evidence suggests that building up cysteine-derived antioxidant defense systems also comes at an enormous cost for SLC7A11high cancer, as such efforts require a significant “investment” of cellular resources, including glutamate export for exchange of cystine import and NADPH supply to reduce cystine to cysteine in the cytosol, resulting in glucose- and glutamine-dependency in SLC7A11high cancer cells. Because glucose and glutamine are among the most abundant nutrients in the extracellular environment, under normal conditions, SLC7A11high cancer cells can take up sufficient amounts of glucose and glutamine to meet these demands for their antioxidant defense. On the basis of this, we propose that under most conditions, SLC7A11 primarily exerts a tumor promoting effect, but its overexpression does expose an Achilles’ heel in cancer cells (by rendering them to be more sensitive to glucose or glutamine starvation). We further reason that under glucose or glutamine limiting conditions, SLC7A11 may even exert a tumor-suppressive effect, a hypothesis remaining to be tested. Below we highlight a few other key questions for further understanding and targeting SLC7A11 in tumor biology in future studies.
Until relatively recently, only NRF2 and ATF4 were known to regulate SLC7A11 transcription. In the past two years, we have witnessed a flurry of findings revealing transcriptional regulation of SLC7A11 by diverse transcription factors and epigenetic regulators (Fig. 2). As such, we now have a much deeper understanding of SLC7A11 regulation at the transcriptional level. In contrast, our knowledge of SLC7A11 regulation at the posttranslational level remains relatively rudimentary. In addition, how such posttranslational regulations in turn modulate SLC7A11-mediated downstream biological effects, such as ferroptosis and nutrient dependency, still remains largely unknown. For example, considering important roles of mTORC2 and AKT in promoting cell survival (Manning and Toker, 2017), the findings that mTORC2- or AKT-mediated phosphorylation of SLC7A11 inhibits SLC7A11’s transporter activity may seem somewhat counterintuitive (Gu et al., 2017; Lien et al., 2017). Since SLC7A11 promotes cell death under glucose starvation, it will be interesting to test whether this inhibitory phosphorylation serves to suppress SLC7A11 function in response to glucose starvation, thereby promoting cell survival under glucose limiting conditions. Further studies are needed to further understand these intriguing questions.
Further, considering SLC7A11’s function to import cystine from and export glutamate into extracellular environment, SLC7A11 might play a role in mediating the cross-talk between tumor cells and tumor microenvironment. However, most current studies have focused on the cell autonomous function of SLC7A11 in tumor cells. We envision that future studies will elucidate potential cell non-autonomous functions of SLC7A11 in tumor biology. Likewise, whether and how SLC7A11 function in immune cells or stromal cells in turn modulates tumor cell behavior is also a potentially fascinating topic for future investigation.
The identity of the cystine reductase(s) that consume NADPH to reduce cystine to cysteine still remains elusive. Previous studies implicated that thioredoxin-related protein of 14 kDa (TRP14) and thioredoxin reductase 1 (Txnrd1) are potential cystine reductases (Mandal et al., 2010; Pader et al., 2014). Both TRP14 and Txnrd1 are indeed coupled to NADPH. However, further genetic studies are required to establish them as bona fide cystine reductases (for example, to show that inactivation of the presumed cystine reductase can dramatically increase intracellular cystine levels).
In this review, we summarized two broad strategies to target SLC7A11; one is to directly inhibit SLC7A11-mediated cystine uptake, the other is to target SLC7A11-induced glucose or glutamine dependency (Fig. 5). To move these findings to clinic application, we propose that two questions should be further addressed in future preclinical studies. The first is to identify the specific tumor or genotype context for therapeutic targeting. Based on the recent preclinical studies (Romero et al., 2017; Liu et al., 2020), it is clear that SLC7A11high tumors should be selected for GLUT or glutaminase inhibitors (to target glucose or glutamine dependency, respectively). Since KEAP1 or BAP1 mutant cancers exhibit high SLC7A11 expression (Cancer Genome Atlas Research, 2012, 2014; Zhang et al., 2018b), KEAP1 or BAP1 mutation can be used as potential biomarkers to select SLC7A11high tumors for GLUT or glutaminase inhibition. On the other hand, it remains unclear whether SLC7A11high or SLC7A11low tumors should be selected for SLC7A11 inhibitor treatment. Presumably, SLC7A11low cancer cells should be sensitive to SLC7A11 inhibition. However, it is also possible that SLC7A11high cancer cells are particularly dependent on SLC7A11, therefore rendering SLC7A11high cancer to be sensitive to SLC7A11 inhibitors too. In addition, it has been shown that tumors with certain oncogenic mutations (such as KRAS mutant tumors (Daher et al., 2019; Lim et al., 2019; Badgley et al., 2020; Hu et al., 2020)) are dependent on SLC7A11-mediated cystine uptake, and therefore such tumors could be particularly sensitive to SLC7A11 inhibitors. Future studies should be directed to further test these hypotheses.
The second question is to identify rational combination strategies with these inhibitors for therapeutic targeting. In this regard, recent studies uncovered that immunotherapy and radiotherapy, two commonly used cancer therapies, can potently induce ferroptosis in cancer cells; further preclinical analyses revealed potent therapeutic efficacy to combine immunotherapy (or radiotherapy) with SLC7A11 inhibitors (IKE, sulfasalazine, sorafenib; class 1 FINs) (Lang et al., 2019; Wang et al., 2019a; Lei et al., 2020; Ye et al., 2020). These studies provide strong justifications to combine these standard-of-cares with SLC7A11 inhibitors in cancer therapy. The rational combination therapies with GLUT or glutaminase inhibitors to treat SLC7A11high cancer remain to be established.
References
Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD (2020) Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol 27:436–447
Artegiani B, van Voorthuijsen L, Lindeboom RGH, Seinstra D, Heo I, Tapia P, Lopez-Iglesias C, Postrach D, Dayton T, Oka R et al (2019) Probing the tumor suppressor function of BAP1 in CRISPR-engineered human liver organoids. Cell Stem Cell 24(927–943):e926
Badeaux AI, Shi Y (2013) Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol 14:211–224
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM et al (2020) Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368:85–89
Bannai S (1986) Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem 261:2256–2263
Bannai S, Ishii T (1988) A novel function of glutamine in cell culture: utilization of glutamine for the uptake of cystine in human fibroblasts. J Cell Physiol 137:360–366
Bannai S, Tsukeda H, Okumura H (1977) Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem Biophys Res Commun 74:1582–1588
Boroughs LK, DeBerardinis RJ (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17:351–359
Cancer Genome Atlas Research, N (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489:519–525
Cancer Genome Atlas Research, N (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543–550
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N et al (2019) The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575:217–223
Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, Dovey CM, Wang J, Locasale JW, Stone E et al (2019) A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep 26(1544–1556):e1548
Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G (2013) BAP1 and cancer. Nat Rev Cancer 13:153–159
Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N (2017) ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 36:5593–5608
Chintala S, Li W, Lamoreux ML, Ito S, Wakamatsu K, Sviderskaya EV, Bennett DC, Park YM, Gahl WA, Huizing M et al (2005) Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc Natl Acad Sci USA 102:10964–10969
Chio IIC, Tuveson DA (2017) ROS in cancer: the burning question. Trends Mol Med 23:411–429
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, Gu W (2019) ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21:579–591
Cobler L, Zhang H, Suri P, Park C, Timmerman LA (2018) xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget 9:32280–32297
Combs JA, DeNicola GM (2019) The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers (Basel) 11:678
Conrad M, Sato H (2012) The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids 42:231–246
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ (2014) Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 13:828–851
Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E, Pouyssegur J, Vucetic M (2019) Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res 79:3877–3890
Dai L, Cao Y, Chen Y, Parsons C, Qin Z (2014) Targeting xCT, a cystine-glutamate transporter induces apoptosis and tumor regression for KSHV/HIV-associated lymphoma. J Hematol Oncol 7:30
De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J, Vermoesen K, Bannai S, Sato H, Michotte Y et al (2011) Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci 31:5792–5803
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS et al (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3:e02523
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13:91–98
Dornier E, Rabas N, Mitchell L, Novo D, Dhayade S, Marco S, Mackay G, Sumpton D, Pallares M, Nixon C et al (2017) Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat Commun 8:2255
Drayton RM, Dudziec E, Peter S, Bertz S, Hartmann A, Bryant HE, Catto JW (2014) Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Cancer Res 20:1990–2000
Eagle H (1955a) Nutrition needs of mammalian cells in tissue culture. Science 122:501–514
Eagle H (1955b) The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J Exp Med 102:37–48
Feng H, Stockwell BR (2018) Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol 16:e2006203
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16:1180–1191
Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, Chen PH, Boroughs LK, Rodriguez MLM, Zhang W et al (2019) LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res 79:3251–3267
Gan B (2019) DUBbing ferroptosis in cancer cells. Cancer Res 79:1749–1750
Gan W, Dai X, Dai X, Xie J, Yin S, Zhu J, Wang C, Liu Y, Guo J, Wang M et al (2020) LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat Cell Biol 22:246–256
Goji T, Takahara K, Negishi M, Katoh H (2017) Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J Biol Chem 292:19721–19732
Green DR, Evan GI (2002) A matter of life and death. Cancer Cell 1:19–30
Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, Ikegami S, Masui K, Gini B, Yang H et al (2017) mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell 67(128–138):e127
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev 17:2648–2663
Hensley CT, Wasti AT, DeBerardinis RJ (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123:3678–3684
Hu K, Li K, Lv J, Feng J, Chen J, Wu H, Cheng F, Jiang W, Wang J, Pei H et al (2020) Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest 130:1752–1766
Huang Y, Dai Z, Barbacioru C, Sadee W (2005) Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res 65:7446–7454
Igney FH, Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2:277–288
Ishii T, Bannai S (1985) The synergistic action of the copper chelator bathocuproine sulphonate and cysteine in enhancing growth of L1210 cells in vitro. J Cell Physiol 125:151–155
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H et al (2011) CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 19:387–400
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–254
Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK et al (2016) An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30:918–930
Ji X, Qian J, Rahman SMJ, Siska PJ, Zou Y, Harris BK, Hoeksema MD, Trenary IA, Heidi C, Eisenberg R et al (2018) xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 37:5007–5019
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520:57–62
Joly JH, Delfarah A, Phung PS, Parrish S, Graham NA (2020) A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose. J Biol Chem 295:1350–1365
Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B et al (2010) Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330:228–231
Kadoch C, Crabtree GR (2015) Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv 1:e1500447
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13:81–90
Kandasamy P, Gyimesi G, Kanai Y, Hediger MA (2018) Amino acid transporters revisited: new views in health and disease. Trends Biochem Sci 43:752–789
Kilberg MS, Shan J, Su N (2009) ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 20:436–443
Kim J, Guan KL (2019) mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol 21:63–71
Koppula P, Zhang Y, Shi J, Li W, Gan B (2017) The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J Biol Chem 292:14240–14249
Koppula P, Zhang Y, Zhuang L, Gan B (2018) Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond) 38:12
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A et al (2019) Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov 9:1673
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H et al (2020) The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res 30:146–162
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W (2012) Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149:1269–1283
Lien EC, Ghisolfi L, Geck RC, Asara JM, Toker A (2017) Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci Signal 10:eaao6604
Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW, Schaffer P et al (2019) Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci USA 116:9433–9442
Liu XX, Li XJ, Zhang B, Liang YJ, Zhou CX, Cao DX, He M, Chen GQ, He JR, Zhao Q (2011) MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett 585:1363–1367
Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, Pearson HB, Fisher OM, Read M, Guerra GR et al (2017) Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun 8:14844
Liu T, Jiang L, Tavana O, Gu W (2019) The Deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res 79:1913–1924
Liu X, Olszewski K, Zhang Y, Lim EW, Shi J, Zhang X, Zhang J, Lee H, Koppula P, Lei G et al (2020) Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol 22:476–486
Lo M, Ling V, Wang YZ, Gout PW (2008) The xc- cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer 99:464–472
Long Y, Tao H, Karachi A, Grippin AJ, Jin L, Chang YE, Zhang W, Dyson KA, Hou AY, Na M et al (2020) Dysregulation of glutamate transport enhances treg function that promotes VEGF blockade resistance in glioblastoma. Cancer Res 80:499–509
Mandal PK, Seiler A, Perisic T, Kolle P, Banjac Canak A, Forster H, Weiss N, Kremmer E, Lieberman MW, Bannai S et al (2010) System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem 285:22244–22253
Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169:381–405
Mao TL, Shih Ie M (2013) The roles of ARID1A in gynecologic cancer. J Gynecol Oncol 24:376–381
Martin L, Gardner LB (2015) Stress-induced inhibition of nonsense-mediated RNA decay regulates intracellular cystine transport and intracellular glutathione through regulation of the cystine/glutamate exchanger SLC7A11. Oncogene 34:4211–4218
Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG (2017) Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6:e27713
Muller PA, Vousden KH (2013) p53 mutations in cancer. Nat Cell Biol 15:2–8
Nagane M, Kanai E, Shibata Y, Shimizu T, Yoshioka C, Maruo T, Yamashita T (2018) Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE 13:e0195151
Nakamura E, Sato M, Yang H, Miyagawa F, Harasaki M, Tomita K, Matsuoka S, Noma A, Iwai K, Minato N (1999) 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J Biol Chem 274:3009–3016
Ogiwara H, Takahashi K, Sasaki M, Kuroda T, Yoshida H, Watanabe R, Maruyama A, Makinoshima H, Chiwaki F, Sasaki H et al (2019) Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell 35(177–190):e178
Okuno S, Sato H, Kuriyama-Matsumura K, Tamba M, Wang H, Sohda S, Hamada H, Yoshikawa H, Kondo T, Bannai S (2003) Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer 88:951–956
Pader I, Sengupta R, Cebula M, Xu J, Lundberg JO, Holmgren A, Johansson K, Arner ES (2014) Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc Natl Acad Sci USA 111:6964–6969
Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM (2016) The integrated stress response. EMBO Rep 17:1374–1395
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23:27–47
Prior IA, Lewis PD, Mattos C (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res 72:2457–2467
Qiao HX, Hao CJ, Li Y, He X, Chen RS, Cui J, Xu ZH, Li W (2008) JNK activation mediates the apoptosis of xCT-deficient cells. Biochem Biophys Res Commun 370:584–588
Rojo de la Vega M, Chapman E, Zhang DD (2018) NRF2 and the hallmarks of cancer. Cancer Cell 34:21–43
Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ et al (2017) Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23:1362–1368
Sato H, Tamba M, Ishii T, Bannai S (1999) Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274:11455–11458
Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S (2004) Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun 325:109–116
Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K, Moriguchi T et al (2005) Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem 280:37423–37429
Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM et al (2017) Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6:e28083
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J (2010) Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465:243–247
Shi J, Vakoc CR (2014) The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 54:728–736
Shin CS, Mishra P, Watrous JD, Carelli V, D’Aurelio M, Jain M, Chan DC (2017) The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun 8:15074
Shin SS, Jeong BS, Wall BA, Li J, Shan NL, Wen Y, Goydos JS, Chen S (2018) Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 7:86
Stewart MD, Li J, Wong J (2005) Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol Cell Biol 25:2525–2538
Stipanuk MH (2004) Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu Rev Nutr 24:539–577
Stipanuk MH, Dominy JE Jr, Lee JI, Coloso RM (2006) Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr 136:1652S–1659S
Stockwell BR, Jiang X (2020) The chemistry and biology of ferroptosis. Cell Chem Biol 27:365–375
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE et al (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171:273–285
Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D (2019) Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis 10:331
Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van ‘t Veer LJ et al (2013) Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24:450–465
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8:579–591
Tsuchihashi K, Okazaki S, Ohmura M, Ishikawa M, Sampetrean O, Onishi N, Wakimoto H, Yoshikawa M, Seishima R, Iwasaki Y et al (2016) The EGF receptor promotes the malignant potential of glioma by regulating amino acid transport system xc(-). Cancer Res 76:2954–2963
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137:413–431
Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y (2004) Role of histone H2A ubiquitination in Polycomb silencing. Nature 431:873–878
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W (2016) Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep 17:366–373
Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, Bosma A, Song JY, Zevenhoven J, Los-de Vries GT et al (2018) An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell 173(1413–1425):e1414
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A et al (2019a) CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569:270–274
Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR, He Q, Zhao S, Zhang GA, Wang Y et al (2019b) Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep 20:e47563
Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, Ding HF, Zhang J, Wang H, Chen X et al (2020a) ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ 27:662–675
Wang Y, Zhao Y, Wang H, Zhang C, Wang M, Yang Y, Xu X, Hu Z (2020b) Histone demethylase KDM3B protects against ferroptosis by upregulating SLC7A11. FEBS Open Biol 10:637–643
Wu Y, Sun X, Song B, Qiu X, Zhao J (2017) MiR-375/SLC7A11 axis regulates oral squamous cell carcinoma proliferation and invasion. Cancer Med 6:1686–1697
Xie L, Song X, Yu J, Guo W, Wei L, Liu Y, Wang X (2011) Solute carrier protein family may involve in radiation-induced radioresistance of non-small cell lung cancer. J Cancer Res Clin Oncol 137:1739–1747
Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H et al (2012) Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 3:883
Yamaguchi I, Yoshimura SH, Katoh H (2020) High cell density increases glioblastoma cell viability under glucose deprivation via degradation of the cystine/glutamate transporter xCT (SLC7A11). J Biol Chem 295:6936–6945
Yang Y, Yee D (2014) IGF-I regulates redox status in breast cancer cells by activating the amino acid transport molecule xC. Cancer Res 74:2295–2305
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156:317–331
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA (2003) The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34:496–502
Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, Ohyama C, Itoh K (2014) Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol 34:3421–3434
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang Y et al (2020) Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol 15:469–484
Yoshikawa M, Tsuchihashi K, Ishimoto T, Yae T, Motohara T, Sugihara E, Onishi N, Masuko T, Yoshizawa K, Kawashiri S et al (2013) xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res 73:1855–1866
Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W et al (2012) Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol 14:276–286
Zhang L, Huang Y, Ling J, Zhuo W, Yu Z, Luo Y, Zhu Y (2018a) Overexpression of SLC7A11: a novel oncogene and an indicator of unfavorable prognosis for liver carcinoma. Fut Oncol 14:927–936
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H et al (2018b) BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20:1181–1192
Zhang Y, Koppula P, Gan B (2019a) Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 18:773–783
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, Stockwell BR (2019b) Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol 226:623
Zhang Y, Zhuang L, Gan B (2019c) BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol Cell Oncol 6:1536845
ACKNOWLEDGEMENTS
We apologize to the colleagues whose relevant work cannot be cited in this review due to space limitations. The research in authors’ lab has been supported by The University of Texas MD Anderson Cancer Center, KC180131 from Department of Defense Kidney Cancer Research Program, R01CA181196, R01CA190370, R01CA244144 from the National Institutes of Health (to BG), CPRIT Research Training Grant (RP170067), and Dr. John J. Kopchick Research Award from The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences (to PK).
AUTHORS’ CONTRIBUTIONS
BG wrote the manuscript with additional support from PK and LZ. PK generated all Fig.s.
ABBREVIATIONS
2DG, 2-deoxyglucose; AAREs, amino acid response elements; AREs, antioxidant response elements; ALOX12, arachidonate 12-lipoxygenase; αKG, α-ketoglutarate; ATF4, activating transcription factor 4; CRISPR, clustered regularly interspaced short palindromic repeats; DUB, deubiquitinase; eIF2α, eukaryotic initiation factor 2α; FDA, Food and Drug Administration; FIN, ferroptosis inducer; GCN2, general control non-derepressible-2; GEMMs, genetically engineered mouse models; GLUT, glucose transporter; GPX4, glutathione peroxidase 4; GSH, reduced glutathione; H2Aub, histone 2A mono-ubiquitination; H2Bub, histone 2B mono-ubiquitination; IKE, imidazole ketone erastin; KEAP1, kelch-like ECH-associated protein-1; KO, knockout; mTORC, mechanistic target of rapamycin complex; NRF2, nuclear factor erythroid 2-related factor 2; PDAC, pancreatic ductal adenocarcinomas; PPP, pentose phosphate pathway; PRC1, polycomb repressive complex 1; PR-DUB, polycomb repressive deubiquitinase; PUFA, polyunsaturated fatty acid; SLC3A2, solute carrier family 3 member 2; SLC7A11, solute carrier family 7 member 11; TCA, tricarboxylic acid; TNBC, triple-negative breast cancer; Tregs, regulatory T-cells; TRP14, thioredoxin-related protein of 14 kDa; Txnrd1, thioredoxin reductase 1; WT, wild-type.
COMPLIANCE WITH ETHICS GUIDELINES
Pranavi Koppula, Li Zhuang, and Boyi Gan declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koppula, P., Zhuang, L. & Gan, B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620 (2021). https://doi.org/10.1007/s13238-020-00789-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-020-00789-5