
Abstract
Gene expression labeling and conditional manipulation of gene function are important for elaborate dissection of gene function. However, contemporary generation of pairwise dual-function knockin alleles to achieve both conditional and geno-tagging effects with a single donor has not been reported. Here we first developed a strategy based on a flipping donor named FoRe to generate conditional knockout alleles coupled with fluorescent allele-labeling through NHEJ-mediated unidirectional targeted insertion in zebrafish facilitated by the CRISPR/Cas system. We demonstrated the feasibility of this strategy at sox10 and isl1 loci, and successfully achieved Cre-induced conditional knockout of target gene function and simultaneous switch of the fluorescent reporter, allowing generation of genetic mosaics for lineage tracing. We then improved the donor design enabling efficient one-step bidirectional knockin to generate paired positive and negative conditional alleles, both tagged with two different fluorescent reporters. By introducing Cre recombinase, these alleles could be used to achieve both conditional knockout and conditional gene restoration in parallel; furthermore, differential fluorescent labeling of the positive and negative alleles enables simple, early and efficient real-time discrimination of individual live embryos bearing different genotypes prior to the emergence of morphologically visible phenotypes. We named our improved donor as Bi-FoRe and demonstrated its feasibility at the sox10 locus. Furthermore, we eliminated the undesirable bacterial backbone in the donor using minicircle DNA technology. Our system could easily be expanded for other applications or to other organisms, and coupling fluorescent labeling of gene expression and conditional manipulation of gene function will provide unique opportunities to fully reveal the power of emerging single-cell sequencing technologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Complex genome modification techniques utilizing site-specific knockin (KI) to achieve gene expression labeling by protein tags or gene inactivation or reactivation via Cre/loxP system have advanced remarkably since the emergence of engineered endonucleases such as TALENs and the CRISPR/Cas system (Zu et al., 2013; Auer et al., 2014; Shin et al., 2014; Li et al., 2015; Hoshijima et al., 2016; Sugimoto et al., 2017; Burg et al., 2018; Luo et al., 2018; Li et al., 2019, 2020). These technical developments are particularly important in organisms lacking embryonic stem cell-based approaches, including zebrafish (Danio rerio). Li et al. reported a fluorescent gene-tagging method which introduced a fluorescent reporter into the last intron via nonhomologous end joining (NHEJ)-mediated targeted insertion in zebrafish using the CRISPR/Cas system (Li et al., 2015), taking advantage of its higher KI efficiency than homologous recombination (HR) (Auer et al., 2014). Recently, we and others further improved the technique and established “two-in-one” dual-function KI strategies to achieve conditional knockout (CKO) coupled with in-frame fusion of double fluorescent reporters to label two different alleles (positive/normal vs. negative/defective), facilitated by specially designs of dual-cassette donors (Li et al., 2019, 2020). However, generation of conditional rescue alleles through these new approaches has not been reported, and a method for simultaneously generating CKO and conditional rescue allele pairs is also not available. Furthermore, a method which could simultaneously generate fluorescent labeling of both positive and negative conditional alleles in the same target gene through a single experiment has not been established.
Unlike HR, integration of donor vectors into the host genome through the NHEJ pathway theoretically could happen in two directions, though KI in only one particular direction is useful for the previously reported methods. We sought to take full advantage of high-efficient targeted insertion and designed a versatile flipping donor vector called FoRe (forward and reverse), and its improved version called Bi-FoRe, to achieve pairwise and dual-function genome modifications which couple fluorescent labeling of gene expression with conditional manipulation of gene function, in both positive and negative states, via NHEJ-mediated targeted insertion. Two functional components, the positive Forward component and the negative Reverse component, were designed in each of these vectors to maintain and disrupt target gene functions, respectively. The two components were arranged in an opposite orientation, so that positive/normal and negative/defective alleles could be generated through forward and reverse integrations, respectively, within a single KI experiment. In addition, to achieve allele-labeling, two different fluorescent reporter genes, tdTomato and EGFP, were included and fused in-frame with the Forward component and the Reverse component, respectively. Furthermore, the two components were flanked by oppositely oriented lox66 and lox71 sequences, so that CKO and conditional rescue could be achieved through Cre recombinase-induced inversion of the two components in the positive/normal and negative/defective alleles, respectively. The feasibility of the FoRe and the Bi-FoRe strategies were evaluated at the sox10 and isl1 loci and demonstrated at the sox10 locus, respectively.
As retention of the bacterial sequence from the donor vector within the KI allele may interfere with the expression or function of the two functional components, we constructed the Bi-FoRe donor vector using a minicircle plasmid whose bacterial backbone is flanked by minimal attB/attP sites and could be removed by phiC31 integrase, either in vitro or in vivo. We successfully achieved efficient in vivo elimination of the plasmid backbone through injection of phiC31 mRNA into embryos bearing KI alleles derived from the insertion of the original full length Bi-FoRe donor. We also constructed a backbone-free minicircle Bi-FoRe donor by in vitro removal of the bacterial sequence using minicircle DNA production technology before injection of the donor vector into embryos and demonstrated its high efficiency in the generation of KI alleles.
Results
Rationale and design of the bidirectional dual-function FoRe donor to generate positive and negative conditional alleles coupled with allele-labeling through targeted insertion
To generate both positive and negative conditional alleles in parallel within one experiment, and simultaneous tagging with different fluorescent reporter genes, we designed a bidirectional KI donor consisting of two major functional components, positive and negative, in a back-to-back opposite orientation, to create and label positive/normal alleles and negative/defective alleles, respectively (Fig. S1). In addition, a lox66 site and a lox71 site were included in each of these two components, respectively, also in an opposite orientation (Albert et al., 1995), to enable Cre-dependent conditional inversion of the positive allele to achieve CKO effect and of the negative allele to achieve conditional rescue effect, respectively (Fig. S1). The two floxed components were placed downstream of a highly efficient CRISPR/Cas9 target site from the human EMX1 (hEMX1) gene to facilitate in vivo linearization of the donor vector (Fig. S1; Table S1). Theoretically, this donor vector could be introduced into the CRISPR/Cas9 site in the target gene through targeted insertion in either orientation via the NHEJ pathway activated by the CRISPR/Cas9 system (Fig. S1) (Lin et al., 2014). Insertion leading to a positive allele is considered forward integration while insertion leading to a negative allele is considered reverse integration. Accordingly, the two components were designated as the Forward (Fo) component and the Reverse (Re) component, and the donor was thus named FoRe, and the region covering the two components was called the FoRe cassette. The Forward component was designed to maintain the correct expression and function of the target gene after forward integration of the FoRe donor into an intron target site or conditional inversion of the reverse-integrated donor by Cre recombinase. It contains the downstream intron sequence of the target site, including the splicing acceptor, as well as the full downstream coding sequence of the target gene, followed by a tdTomato reporter gene, separated by an in-frame 2A peptide coding sequence, to label the expression of the positive (functionally normal) allele. An SV40 poly-A signal (pA) was inserted after these coding sequences and used to terminate transcription (Fig. S1). The Reverse component was designed to disrupt the function of the target gene after reverse integration into the target locus or conditional inversion of the forward-integrated donor by Cre recombinase, and contains the downstream intron sequence of the target site, including the splicing acceptor, and only part of downstream in-frame coding sequence of the target gene, followed by an EGFP reporter gene, separated by an in-frame 2A peptide coding sequence, and a BGH pA to label the expression of the negative (mutant/defective) allele (Fig. S1). The Reverse component (including the lox71 site) was cloned downstream to the Forward component (including the lox66 site) in an opposite direction to form the FoRe cassette (Fig. S1) (Albert et al., 1995; Araki et al., 2002; Araki et al., 2006; Carney and Mosimann, 2018). Cre-induced recombination between the opposite lox66 and lox71 sites would result in the inversion of the FoRe cassette as well as generation of a wild-type loxP site and a mutant lox72 site flanking in an opposite orientation (Fig. S1), leading to either conditional disruption of the target gene from the forward-integrated KI allele or conditional rescue of the target gene from the reverse-integrated KI allele.
When a FoRe donor was co-injected into one-cell stage zebrafish embryos together with the CRISPR/Cas system, the donor could be linearized and integrated into the intron target site in two directions. Upon forward integration, the KI allele serves as a positive conditional allele, where the Forward component will be transcribed and the coding sequence will be spliced with the upstream endogenous transcript, ensuring normal expression and function of the target gene as well as its fluorescent labelling by the tdTomato reporter, under normal conditions. In the presence of Cre recombinase, the FoRe cassette could be inverted and the defective Reverse component expressed, leading to disruption of the target gene as well as fluorescent labelling switch from tdTomato to EGFP (Fig. S1). On the other hand, through integration in the reverse direction, the KI allele would serve as a negative conditional allele. Under normal conditions, the Reverse component would be transcribed, together with its upstream plasmid backbone sequence, and theoretically correct splicing would lead to production of a truncated/defective protein and disruption of target gene function, as well as labelling of the negative conditional allele by the EGFP reporter. Restoration of target gene function could be achieved by Cre-dependent inversion of the reverse-integrated FoRe cassette in this negative conditional allele and subsequent correct expression of the Forward component, thus achieving conditional gene rescue (Fig. S1). By this strategy, positive and negative conditional allele pairs could be generated through forward and reverse KI, respectively, contemporarily within one experiment and using only one donor vector. Furthermore, tdTomato-labeling of functionally normal positive alleles and EGFP-labeling of functionally defective negative alleles could also be achieved at the same time, which can easily be used to trace and distinguish individual live embryos bearing different genotypes by fluorescent signals prior to the emergence of any visible phenotype.
Efficient generation and characterization of the sox10 positive conditional FoRe alleles
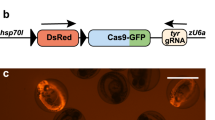
We first evaluated our FoRe donor strategy at the zebrafish sox10 locus (Fig. 1A), which is primarily expressed in otic vesicle cells and neural crest cells (Dutton et al., 2001a, b; Geng et al., 2013). We identified a highly efficient CRISPR/Cas9 target site at the third intron of the sox10 locus (Fig. S2A; Table S1). After co-injection of the sox10 FoRe donor with Cas9 mRNA, sox10 gRNA, and hEMX1 gRNA into one-cell stage embryos, 40.9% (38/93) of the embryos showed tdTomato fluorescent signal in the otic vesicle and pigment cells, derivatives of neural crest cells, at 36 hpf (hours post-fertilization) indicating efficient forward insertion of the donor vector (Fig. S2B; Table S2). Unfortunately, only a few (3/93) embryos showed limited EGFP signal from the same experiment, suggesting either inefficient insertion in the reverse direction or inefficient/incorrect transcription and/or splicing of the Reverse component. Founder embryos showing broad red fluorescence patterns (29/38) (Fig. S2B) were raised to adulthood and screened for germline transmission by outcross with wild-type zebrafish. 57.1% (4/7) of these founders gave rise to offspring showing correct red fluorescent signals, recapitulating the expression pattern of sox10 in otic vesicle and pigment cells, as well as trunk neural crest cells (Figs. 1B and S2C; Table S3). Junction PCR results using pairs of primers spanning the 5′ junction and 3′ junction, as well as further sequencing results, confirmed expected NHEJ-mediated forward KI events at the sox10 locus in the embryos positive for red fluorescent signals (Fig. S2D). The offspring from founder #2 were used for the subsequent experiments and the corresponding forward positive conditional FoRe allele was named sox1066-FoR-ReG−71. Unfortunately, we failed to obtain any heritable green fluorescent labeling of sox10 expression, indicative of reverse insertion, from germline screening of 25 founder fish derived from either EGFP-positive or EGFP-negative embryos. We injected Cre mRNA into one-cell stage F1 embryos from founder #2, and observed the expected switch of the fluorescent signals from red to green (Fig. S2C), indicating successful inversion of the FoRe cassette in response to the Cre recombinase. The resulting inverted mutant allele was named sox10P-ReG-FoR−72. 5′ junction PCR and sequencing results further confirmed Cre-mediated recombination of this sox10 positive conditional FoRe allele in the F1 embryos (Fig. S2E and data not shown). Note that the 3′ junction will not change after Cre-mediated recombination (Fig. 1A).

Generation and evaluation of fluorescent reporter-tagged conditional knockout alleles at the zebrafish sox10 locus and mosaic tracing analysis of sox10 expressing cells. (A) Schematic diagram of the KI strategy of sox10 FoRe donor consisting of two components in opposite orientations (highlighted by red shadow for the Forward component for maintaining the function of sox10, and green shadow for the Reverse component for disrupting the function of sox10). The sox10 CRISPR/Cas target site is shown as a dark blue box, and the hEMX1 target site is shown as a light blue box. (B) Images of a 48 hpf F1 embryo from germline transmission screening of the sox10 FoRe donor KI founder. Scale bar, 200 μm. (C) Phenotype analysis of the 48 hpf F2 embryos from the incross of sox10+/66-PoR-ReG−71 heterozygotes (derived from F0 #2) after the injection of 50 pg Cre mRNA at the one-cell stage. The white arrowheads indicate otic vesicles. Detailed sox10 expression in the trunk region can be seen under higher magnification of the boxed areas. Scale bar, 200 μm. (D) Serial lightsheet images of a sox10 mosaic embryo from incross of sox1066-FoR-ReG−71/66-FoR-ReG−71 homozygotes after vegetal pole injection of 10 pg Cre mRNA and recorded from 24 hpf to 36.5 hpf. Some of the tdTomato-positive neural crest cells could migrate to the two sides of the body (as indicated by the white arrowheads), while all of the EGFP-positive cells remained in the middle. Scale bar, 100 μm
To further test the conditional effect of the sox1066-FoR-ReG−71 allele, the F2 progeny from incross of sox10+/66-FoR-ReG−71 heterozygotes were injected with 50 pg Cre mRNA per embryo at the one-cell stage. All the injected embryos showed little tdTomato signal at 48 hpf, suggesting efficient switch of the fluorescent reporter (Fig. 1C). Among these embryos, 26.7% (32/120) showed normal pigmentation without any fluorescent signal at 48 hpf (Fig. 1C, Group I), indicating these embryos were most likely to be sox10+/+. 50% (60/120) of the injected embryos showed EGFP fluorescent signal and slightly fewer pigment cells (Figs. 1C and S3A, Group II), indicating these embryos are likely to be heterozygous for the inverted FoRe allele, i.e., sox10+/P-ReG-FoR−72. 23.3% (28/120) of the injected embryos showed extensive loss of pigmentation (Figs. 1C and S3A, Group III), and these embryos were all positive for EGFP expression, which suggested that they were most likely homozygous for the inverted FoRe allele, i.e., sox10P-ReG-FoR−72/P-ReG-FoR−72. PCR genotyping results confirmed the expected correlation between the genotype and the phenotype in these three groups of embryos (Fig. S3B). To evaluate the effect of the sox10 KI allele in its defective state after Cre-induced recombination at the molecular level, we performed RT-PCR experiments to measure the expression of sox10 on the embryos from incross of the sox10+/66-FoR-ReG−71 heterozygotes with Cre mRNA injection. The results showed significant reduction of functionally normal sox10 transcripts in the homozygous sox10P-ReG-FoR−72/P-ReG-FoR−72 mutant embryos, as expected (Fig. S3C), indicating highly efficient mutagenesis by the sox10P-ReG-FoR−72 allele. These results together demonstrated that our FoRe strategy could efficiently generate fluorescent reporter-tagged positive conditional KI alleles and successfully achieve CKO coupled with simultaneous fluorescent allele-label switching.
Mosaic analysis and tracing of sox10-expressing cells facilitated by the FoRe KI allele
Since cells with different genotypes could be labeled with different fluorescent reporters, our FoRe KI strategy provides a unique opportunity for real-time mosaic analysis as well as cell fate tracing in live embryos. We injected 10 pg Cre mRNA into the vegetal pole of the progeny from incross of sox1066-FoR-ReG−71/66-FoR-ReG−71 homozygotes, then screened chimeras showing both red and green fluorescent signals at 24 hpf and traced neural crest cell fate for 12.5 hours by live imaging under a lightsheet microscope. The results showed that neural crest cells showing only tdTomato expression displayed the ability to migrate to both the left and right sides of the embryo body, whereas cells with only EGFP labeling were clearly lagging behind with most of the cells staying around the midline (Fig. 1D; Movie S1), indicating that neural crest cells homozygous for mutant sox10, as revealed by the EGFP signal, are defective for lateral migration, which is consistent with previous reports (Kelsh and Eisen, 2000; Dutton et al., 2001b). This observation demonstrated the advantage of the capability of our FoRe system for tracing and comparing functionality of cell lineages with different genetic backgrounds in parallel within one individual.
Application of the FoRe strategy at the zebrafish isl1 locus
Since we did not obtain stable reverse integration of the FoRe donor at sox10 locus, we next evaluated our strategy at a second locus isl1, encoding a DNA-binding transcription factor which is involved in multiple developmental processes, including atrial cardiac muscle cell differentiation, neuron differentiation and pancreas development (Sirbu et al., 2008; Witzel et al., 2012; Wilfinger et al., 2013; Caputo et al., 2015; Witzel et al., 2017). We identified a highly efficient CRISPR/Cas9 target site at the third intron of isl1 (Fig. S4A; Table S1). After co-injection of an isl1 FoRe donor, Cas9 mRNA, isl1 gRNA, and hEMX1 gRNA into one-cell stage zebrafish embryos (Fig. S4B), about 16.5% (13/79) of the injected embryos showed tdTomato expression (Fig. S4C; Table S2), but no embryos expressing EGFP were observed, again indicating efficient forward but not reverse insertion. The potential founders showing tdTomato expression were raised to adulthood and germline screening was conducted by outcross with wild-type zebrafish. 60% (3/5) of the founders produced offspring showing correct tdTomato expression in eyes and trunk motor neurons, indicating stable inheritance of the positive allele derived from forward KI of the FoRe donor (Fig. S4D and S4E; Table S4). Junction PCR as well as sequencing results confirmed correct NHEJ-mediated forward KI events at the isl1 locus (Fig. S4F). The offspring from founder #2 were used for the following experiments and the corresponding positive conditional FoRe allele was named as isl166-FoR-ReG−71. We injected 10 pg of Cre mRNA into one-cell stage F1 embryos from the outcross of this founder, and observed the expected switch of the fluorescent signals from red to green in some cells (Fig. S4E), and the knockout allele derived from Cre-induced inversion was named as isl1P-ReG-FoR−72. PCR and sequencing results further confirmed Cre-induced recombination in the injected embryos with EGFP expression (Fig. S4G).
To evaluate the CKO effect of our flipping donor strategy at the isl1 locus, we injected 50 pg Cre mRNA into the animal pole of F2 progeny from incross of the isl1+/66-FoR-ReG−71 F1 heterozygotes at the one-cell stage. All of the injected embryos showed little tdTomato signal, suggesting efficient inversion of the FoRe cassette (Fig. S5A). Among these embryos, 23.6% (22/93) exhibited defective phenotypes such as reduced head and body size, abnormal heart, and curved body axis at 48 hpf, and all these defective embryos were positive for EGFP expression (Fig. S5A, Group III), indicating that these embryos were most likely isl1P-ReG-FoR−72/P-ReG-FoR−72 homozygous mutants. 53.8% (50/93) of the injected embryos developed normally and also showed EGFP fluorescent signal, suggesting these embryos may be isl1+/P-ReG-FoR−72 heterozygous (Fig. S5A, Group II). 22.6% (21/93) of the injected embryos showed normal embryogenesis without any fluorescent signal, indicating that they were likely isl1+/+ wild-type embryos (Fig. S5A, Group I). PCR genotyping results further confirmed the correlation between genotype and phenotype in each group, as expected (Fig. S5B). To measure the expression of the isl166-FoR-ReG−71 allele in its defective state after Cre-induced inversion, we performed RT-PCR experiments on the mutant embryos from incross of the isl1+/66-FoR-ReG−71 heterozygotes with Cre mRNA injection. The results showed that little isl1 transcript was detected in the mutant embryos (Fig. S5C), indicating highly efficient knockout effect of the isl1P-ReG-FoR−72 allele derived from the inversion of the isl166-FoR-ReG−71 allele. Taken together, these results demonstrated the generality and high efficiency of our FoRe KI strategy for generating positive conditional alleles together with genotype labeling effect through forward insertion of the FoRe donor. However, we failed to effectively isolate negative conditional KI alleles which should derive from reverse integration of the FoRe donor (to allow for conditional rescue effect) through pre-selection of founder embryos by EGFP expression, indicating that the design of our FoRe strategy needed further improvement.
Improvement of our FoRe strategy to achieve contemporary generation of positive and negative conditional alleles with fluorescent allele tagging effect
We noticed that EGFP-positive embryos (representing reverse insertion) were much less frequent than those expressing tdTomato (representing forward insertion) after injection of the FoRe donor KI system at both sox10 and isl1 loci. The major difference between forward insertion and reverse insertion with regard to the reporter gene is its relative position and distance to the insertion site in the zebrafish genome, or more precisely, the relationship with its upstream endogenous genomic sequence. Almost no extra sequence is placed before the tdTomato reporter besides the essential target gene sequence in the case of forward insertion; in contrast, in reverse insertion, there is a long exogenous bacterial plasmid backbone sequence (approximately 2,600 bp) lying between the negative Reverse component and its upstream genomic insertion site. We suspect that this exogenous sequence may contain cryptic splicing acceptor(s) which might disturb the correct and/or efficient splicing of the transcript from the Negative component into the upstream endogenous portion of the transcript, therefore leading to reduced EGFP fluorescent signal. To solve this potential problem, we improved the design of our FoRe donor by moving the Reverse component containing two pA signals to upstream of the hEMX1 site, so that this donor linearization site was placed between the two functional components (Fig. 2A). In this manner, the relative position of the EGFP reporter within the targeted genome after reverse integration will be comparable with the position of the tdTomato reporter after forward integration, eliminating potential interference from the plasmid backbone (Fig. 2A). We call this improved vector Bi-FoRe (bidirectional FoRe) donor. Additionally, physical elimination of the plasmid backbone could be further considered, by using minicircle DNA technology for the construction of the donor vector, which contains attB and attP sites flanking the plasmid backbone that could be removed either in vitro or in vivo through phiC31-induced recombination (Kay et al., 2010; Suzuki et al., 2016) (Fig. 2A).

Generation of positive and negative conditional allele pairs at the sox10 locus through the Bi-FoRe strategy. (A) Schematic diagram of the improved KI strategy based on the bidirectional multi-purpose Bi-FoRe donor consisting of two functional components. The Forward component (highlighted by red shadow) is designed to maintain the function of the sox10 gene, and the Reverse component (highlighted by green shadow) is designed to disrupt the sox10 function. The sox10 CRISPR/Cas target site is shown in dark blue, and the hEMX1 target site is shown in light blue, located in the middle of the two functional components in the donor, to facilitate unbiased identification of both forward and reverse integrations of the donor. (B) Z-stack confocal images of a 48 hpf sox10+/Bi−66-FoR-ReG−71 F1 embryo from outcross of founder #3 and a 48 hpf sox10+/Bi−71-ReG-FoR−66−1 F1 embryo from outcross of founder #1, respectively. Scale bar, 200 μm. (C) Junction PCR and direct sequencing results of F1 progeny showing tdTomato expression from outcross of F0 #3 or #4, demonstrating forward insertion of the sox10 Bi-FoRe donor. KI: pooled genomic DNA template of F1 embryos from outcross of F0 #3. WT: pooled genomic DNA template of wild-type embryos. (D) Junction PCR and direct sequencing results of F1 progeny showing EGFP expression from outcross of F0 #1 or #7, demonstrating reverse insertion of the sox10 Bi-FoRe donor. KI: pooled genomic DNA template of the F1 embryos from outcross of F0 #1. WT: pooled genomic DNA template of wild-type embryos. (E) Z-stack confocal images of a 56 hpf sox10Bi−66-FoR-ReG−71/Bi−71-ReG-FoR−66−2 F1 embryo from the cross between founders #3 and #7, showing overlapping expression of tdTomato and EGFP. Scale bar, 200 μm
Generation of bidirectional and conditional Bi-FoRe KI allele pairs at the sox10 locus
We evaluated the Bi-FoRe strategy at the sox10 locus. After co-injection of the sox10 Bi-FoRe donor with Cas9 mRNA, sox10 gRNA, and hEMX1 gRNA into one-cell stage zebrafish embryos, 20.7% (28/135) of the embryos showed tdTomato signal and 42.2% (57/135) showed EGFP signal (Table S2). Interestingly, 16 embryos exhibited mosaic expression of both tdTomato and EGFP signals, indicating that both forward and reverse integrations happened efficiently in the same embryo. Potential founders showing broad fluorescent patterns were raised to adulthood and germline transmission screening was conducted by outcrossing with wild-type zebrafish. Three out of 18 founders (#3, #4, #9) produced offspring showing only tdTomato expression (Fig. 2B; Table S5), with normal phenotype and identical fluorescence pattern to the sox10+/66-FoR-ReG−71 F1 embryos carrying the previous FoRe donor KI allele, as expected. The offspring from founder #3 were used for the following experiments and the corresponding forward positive conditional Bi-FoRe KI allele was named as sox10Bi−66-FoR-ReG−71. Four out of the same 18 founders (#1, #2, #7, #8) produced offspring showing only EGFP expression (Fig. 2B; Table S6), the offspring from founders #1 and #7 were used for the subsequent experiments and the corresponding reverse negative conditional Bi-FoRe KI alleles were named as sox10Bi−71-ReG-FoR−66−1 and sox10Bi−71-ReG-FoR−66−2, respectively. Interestingly, two founders (#5, #6) showed germline transmission of both tdTomato and EGFP expression, demonstrating high KI as well as germline transmission efficiency of our new strategy (Tables S5 and S6). As expected, junction PCR and sequencing results confirmed correct NHEJ-mediated KI events of these alleles at the sox10 locus (Fig. 2C and 2D). Efficient germline transmission of both forward and reverse KI events at the sox10 locus proved the feasibility of our Bi-FoRe donor strategy as a time-saving one-step efficient method to generate multi-purpose labeled genome modifications in pairs. On one hand, sox10Bi−66-FoR-ReG−71 and sox10Bi−71-ReG-FoR−66−1 (and sox10Bi−71-ReG-FoR−66−2) can be considered as a pair of stable positive (or functionally normal) and negative (or mutant/defective) alleles tagged with different fluorescent reporters; on the other hand, they are also a pair of conditional knockout and conditional rescue alleles, each bearing different fluorescent reporters.
Characterization and application of the phenotype and allele-labeling effect of the sox10 Bi-FoRe KI allele pairs
We further characterized and compared the phenotype as well as expression of the positive sox10Bi−66-FoR-ReG−71 and negative sox10Bi−71-ReG-FoR−66−1 (or sox10Bi−71-ReG-FoR−66−2) alleles under normal conditions (without introduction of Cre recombinase), separately or in pairs. To evaluate whether loss-of-function phenotype and correct reporter gene expression can be detected in the zebrafish carrying the negative sox10 Bi-FoRe KI alleles, we crossed founder #1 with founder #7 and obtained sox10+/Bi−71-ReG-FoR−66−1 and sox10+/Bi−71-ReG-FoR−66−2 heterozygotes, as well as sox10Bi−71-ReG-FoR−66−1/Bi−71-ReG-FoR−66−2 compound heterozygous progeny. In contrast to the positive sox10Bi−66-FoR-ReG−71 allele, the embryos derived from the founders bearing the negative sox10Bi−71-ReG-FoR−66−1 or sox10Bi−71-ReG-FoR−66−2 allele showed only green but not red fluorescent signals (Fig. 2B). In addition, some of the green-fluorescent embryos showed partial reduction of pigmentation, while others were completely devoid of pigmentation in their trunk region at 48 hpf (Fig. 3F, upper panels), which largely recapitulated the expected phenotype of sox10 mutation and suggests that they correspond to sox10+/Bi−71-ReG-FoR−66−1 or sox10+/Bi−71-ReG-FoR−66−2 heterozygotes and sox10Bi−71-ReG-FoR−66−1/Bi−71-ReG-FoR−66−2 compound heterozygotes, respectively. This result indicates that either sox10Bi−71-ReG-FoR−66−1 or sox10Bi−71-ReG-FoR−66−2 allele could efficiently disrupt the function of the sox10 gene. To achieve stable and homogeneous expression tagging of both positive/normal and negative/defective sox10 alleles by tdTomato and EGFP within the same embryo, respectively, we crossed founders #3 and #7, and obtained F1 embryos showing both red and green fluorescent signals. These sox10Bi−66-FoR-ReG−71/Bi−71-ReG-FoR−66−2 embryos showed overlapping expression patterns for the two fluorescent reporters as well as partial reduction of pigmentation similar to the sox10+/Bi−71-ReG-FoR−66−2 heterozygous F1 embryos, as expected, again indicating that both the tdTomato reporter in the positive sox10Bi−66-FoR-ReG−71 allele and the EGFP reporter in the negative sox10Bi−71-ReG-FoR−66−2 allele could faithfully label the expression of the sox10 locus, and furthermore, the positive sox10Bi−66-FoR-ReG−71 allele and negative sox10Bi−71-ReG-FoR−66−2 allele could successfully recapitulate normal and defective sox10 function, respectively (Fig. 2E).

Evaluation of conditional manipulation of paired positive and negative conditional Bi-FoRe KI alleles at the sox10 locus. (A) Schematic diagram of the positive conditional allele (Forward KI allele) sox10Bi−66-FoR-ReG−71 before and after Cre-mediated recombination. (B) Z-stack confocal images of F2 embryos obtained from incross of F1 sox10+/Bi−66-FoR-ReG−71 heterozygotes. Upper panel: An embryo without Cre mRNA injection, showing tdTomato expression and normal pigmentation. Lower panel: An embryo after Cre mRNA injection, showing EGFP expression and defects in pigmentation, resembling sox10 mutant phenotype. Scale bar, 200 μm. (C) Junction PCR results of the F2 embryos from incross of sox10+/Bi−66-FoR-ReG−71 after Cre mRNA injection. EGFP+: pooled genomic DNA template of the F2 progeny showing EGFP expression after Cre mRNA injection. WT: pooled genomic DNA template of wild-type embryos. (D) RT-PCR results using the cDNA of the F2 embryos from B with (Knockout) or without (Control) Cre mRNA injection. (E) Schematic diagram of the negative conditional alleles (Reverse KI allele) sox10Bi−71-ReG-FoR−66−1 and sox10Bi−71-ReG-FoR−2 before and after Cre-mediated recombination. (F) Z-stack confocal images of the embryos obtained from a cross of F0 #1 bearing the sox10Bi−71-ReG-FoR−66−1 allele with #7 bearing the sox10Bi−71-ReG-FoR−66−2 allele. Upper panel: F1 embryo without Cre mRNA injection, showing EGFP expression as well as defects in pigmentation, resembling sox10 mutant phenotype. Lower panel: F1 embryo with Cre mRNA injection, showing tdTomato expression and recovery of pigmentation. Scale bar, 200 μm. (G) Junction PCR results of the F1 embryos from the cross of F0 #1 with #7 after Cre mRNA injection. tdTo+: pooled genomic DNA template from the F1 progeny showing tdTomato expression after Cre mRNA injection. WT: pooled genomic DNA template of wild-type embryos. (H) RT-PCR results using the cDNA of the embryos from panel F with (Rescue) or without (Defective control) Cre mRNA injection. The location of RT-PCR primers in D and H is indicated in Fig. S3C. The expected size of the band is 467 bp
To evaluate the potential utility of the double labeling of the positive and negative sox10 allele pair to reveal cellular genotypes at early embryonic stages before the appearance of visible phenotype, we further analyzed the timing of phenotype and reporter gene expression in embryos obtained from the outcross of sox10+/Bi−66-FoR-ReG−71 with wild type fish and those from the cross between #1 and #7 founders, at early developmental stages. Both red and green fluorescent signals could be detected separately in the corresponding embryos before 14 hpf by lightsheet microscopy (data not shown) and were easily observable under general compound fluorescence microscopy at 21 hpf, displaying similar patterns, prior to the emergence of melanocytes (Fig. S6A), while phenotypic difference in pigmentation between the different genotypes (as revealed by fluorescent signals and confirmed by PCR genotyping) was not visible until 28 hpf (Fig. S6B and S6C), consistent with the previous report (Kelsh and Eisen, 2000). Taken together, these results demonstrated that different fluorescent labeling of the positive/normal and negative/defective alleles by our new Bi-FoRe strategy will aid early in vivo discrimination of different genotypes in embryos before or even without the requirements for the onset of any visible phenotype, especially for large numbers of embryos, which is critical for early and accurate dissection of target gene function and molecular mechanisms.
Evaluation of the conditional manipulation of the sox10 Bi-FoRe KI allele pairs
Next, to evaluate the conditional knockout effect of the floxed positive sox10Bi−66-FoR-ReG−71 allele, the F2 progeny from the incross of sox10+/Bi−66-FoR-ReG−71 heterozygotes were injected with 50 pg Cre mRNA per embryo at the one-cell stage (Fig. 3A and 3B). While the un-injected control embryos showed only red fluorescent signals, most injected embryos exhibited only green fluorescent signal, indicating highly efficient switch of reporter gene expression from tdTomato to EGFP, and therefore highly efficient conversion of the functional sox10Bi−66-FoR-ReG−71 allele into the defective sox10Bi-P-ReG-FoR−72 allele. In addition, all EGFP-positive embryos exhibited either partial or complete loss of pigmentation, suggesting they are sox10+/Bi-P-ReG-FoR−72 heterozygotes and sox10Bi-P-ReG-FoR−72/Bi-P-ReG-FoR−72 homozygous mutants, respectively (Fig. 3B). Junction PCR results confirmed the expected Cre-mediated recombination events at the sox10 locus (Fig. 3C), and genotyping results of the embryos devoid of any pigmentation further confirmed that these embryos were indeed homozygous of the Bi-FoRe KI allele sox10Bi−66-FoR-ReG−71 before Cre mRNA injection (Fig. S7A). RT-PCR experiments detected little normal sox10 transcript in the embryos devoid of any pigmentation after Cre mRNA injection (Fig. 3D), confirming that the sox10Bi-P-ReG-FoR−72 is an effective knockout allele.
Compared with the original FoRe strategy, a unique advantage of our Bi-FoRe strategy is the efficient generation of stable negative alleles by germline transmission of the reverse-integrated Bi-FoRe donor. These alleles are functionally defective for the sox10 gene and express an EGFP reporter under normal conditions (i.e., without Cre recombinase), and exhibit conditional rescue as well as fluorescent reporter switch in the presence of Cre recombinase, i.e., they could be converted into functionally normal sox10 alleles, coupled with a switch of the reporter gene expression from EGFP to tdTomato, after Cre-induced inversion of the Bi-FoRe cassette. To evaluate whether conditional rescue of gene expression and function can be achieved in zebrafish carrying the negative sox10Bi−71-ReG-FoR−66−1 and sox10Bi−71-ReG-FoR−66−2 alleles, we crossed the corresponding founder #1 with founder #7 and injected Cre mRNA into some of the one-cell stage progeny (Fig. 3E and 3F). While the un-injected control embryos showed only green fluorescent signal, injected embryos exhibited mostly red fluorescent signal, indicating highly efficient switch from EGFP to tdTomato reporter gene expression, as well as highly efficient conversion of the sox10Bi−71-ReG-FoR−66−1 and sox10Bi−71-ReG-FoR−66−2 alleles into sox10Bi−72-FoR-ReG-P−1 and sox10Bi−72-FoR-ReG-P−2, respectively (Fig. 3F). Furthermore, all of the tdTomato-expressing embryos displayed complete recovery of body pigmentation, indistinguishable from their wild-type siblings (i.e., those embryos without any fluorescent signal), confirming successful and efficient rescue of sox10 function (Fig. 3F). Junction PCR results further confirmed expected Cre-mediated recombination events at the sox10 locus (Fig. 3G). We randomly selected seven injected embryos expressing only tdTomato for genotyping, and results showed that two of these embryos did not contain the wild-type allele, indicating they harbored Bi-FoRe KI alleles from both founder #1 and #7 (Fig. S7B), indicating efficient rescue of the original mutants by the Cre recombinase. We also performed RT-PCR experiments on the un-injected mutant embryos and Cre-injected rescue embryos from Fig. 3F, and the results showed that sox10 expression was largely recovered in the Cre mRNA injected embryos compared with the original un-injected embryos (Fig. 3H). Taken together, our Bi-FoRe KI strategy provides a simple, versatile and efficient method to simultaneously generate both positive and negative conditional alleles to be used for CKO and conditional gene rescue analyses in conjunction with allele-labeling effect using a single donor vector within a single experiment.
Elimination of the plasmid backbone by phiC31-mediated attB/attP recombination in vivo or in vitro
Integration of a plasmid backbone into the eukaryotic genome might induce DNA methylation and silencing of transcription (Suzuki et al., 2016), interfering with the function and inheritance of the KI alleles. We tested two approaches to eliminate the undesirable influence of the bacterial backbone sequence, first by removing the backbone using flanking attB and attP sites through in vivo phiC31-induced excision in the KI embryos, and secondly by replacing the donor vector with a backbone-free minicircle plasmid for the KI experiments. To test the first approach, we injected phiC31 mRNA into offspring from the outcross of sox10+/Bi−66-FoR-ReG−71 or sox10+/Bi−71-ReG-FoR−66−1. Recombination between attB site and attP site via phiC31 results in the removal of their flanked sequence and production of an attR site and an attL site (Lu et al., 2011; Carney and Mosimann, 2018), and the alleles without backbone sequence were referred to as sox10Mini−66-FoR-ReG−71 (Fig. S8A) and sox10Mini−71-ReG-FoR−66−1, respectively. PCR and sequencing results confirmed the phiC31-mediated recombination events in the injected embryos (Fig. S8B and data not shown), and the expression pattern of the fluorescent reporters was comparable with the un-injected embryos (Fig. S8C). These results demonstrated the feasibility of removing the donor backbone by using the phiC31 and attB/attP system in vivo.
In vitro removal of the plasmid backbone before introduction of the donor vector into the embryos through the minicircle system is an alternative approach to minimize the un-wanted influence from the plasmid vector, and furthermore, the relatively smaller size of the minicircle donor vector may improve the efficiency of targeted KI integration. To validate this assumption, we generated a sox10 minicircle Bi-FoRe donor and injected it along with Cas9 mRNA, sox10 gRNA, and hEMX1 gRNA into one-cell stage zebrafish embryos, in equimolar amounts with the full length donor (Fig. 4A). 51.7% (76/147) and 29.9% (44/147) of the injected embryos showed tdTomato and EGFP fluorescent signal, respectively (Fig. 4B; Table S2). Of these, 25 embryos showed both tdTomato and EGFP expression. Therefore, the overall KI efficiency in minicircle donor-injected founder embryos increased to 64.6%, compared with 51.1% for the injection of the full length Bi-FoRe donor containing the full-length plasmid backbone (Table S2), suggesting that minicircle Bi-FoRe donor may be more efficient for generating NHEJ-mediated KI.

Generation of conditional sox10 KI alleles using minicircle donor and summary of our bidirectional KI strategy. (A) Schematic diagram of the KI strategy based on the minicircle Bi-FoRe donor derived from in vitro backbone elimination. (B) Representative z-stack confocal images of two 48 hpf founder embryos after injection of the sox10 minicircle Bi-FoRe donor vector together with the CRISPR/Cas9 system, showing red (representing forward insertion) and green (representing reverse insertion) fluorescent signals, respectively. Scale bar, 200 μm. (C) A graphical summary of the principle and applications of our Bi-FoRe KI strategy
Discussion
In this study, we developed an efficient flipping donor strategy based on dual-functional FoRe and Bi-FoRe donors to simultaneously generate positive and negative conditional alleles, coupled with fluorescent allele tagging/geno-tagging, through NHEJ-mediated targeted insertion facilitated by the CRISPR/Cas system. The expression of the FoRe and Bi-FoRe KI alleles could be revealed in live embryos by the fluorescent reporters fused in-frame with the coding sequence of the target gene, while the function of these alleles could be conditionally manipulated by Cre-induced inversion of the FoRe or Bi-FoRe cassette due to the recombination between the opposite-orientated lox66 and lox71 sites. Paired multifunctional KI alleles, enabling both fluorescent labeling of positive and negative alleles on one hand, and conditional knock out and conditional rescue on the other hand, could be generated efficiently and simultaneously through bidirectional targeted insertion of the same donor vector in one experiment, which could largely simplify the experimental process and save both time and effort compared with current existing methods (Figs. 4C and S9).
The feasibility of our FoRe strategy was demonstrated at the sox10 and isl1 loci in zebrafish. We first generated double fluorescent reporter-labeled and floxed positive KI alleles of sox10 and isl1 genes through forward insertions of the FoRe donors. We then successfully achieved CKO effect as well as fluorescent tag switching through Cre-induced inversion of the FoRe cassette from its positive state to its negative state. We also performed real-time mosaic analysis and cell fate tracing on different genotypes side-by-side within a single embryo derived from conditional manipulation of the sox10 FoRe KI alleles. By this experiment, we observed a clear difference in the migratory capacity between cells normal and defective in sox10 function (Fig. 1D; Movie S1), which demonstrated the power of our method for tissue/organ specific mosaic analysis of different genotypes. However, we failed to isolate the expected negative conditional alleles which should derive from the reverse insertion. Concerned that the long bacterial backbone might interfere with identification of reverse insertion events, we improved the design by locating the backbone sequence within the FoRe cassette between the Forward component and the Reverse component, so both components are equally close to the integration site within the target gene after insertion. We named this improved donor Bi-FoRe, and successfully generated both positive and negative Bi-FoRe KI alleles at the sox10 locus through bidirectional integration of this new donor within a single KI experiment. Based on these allele pairs, we successfully achieved conditional knockout and conditional rescue in parallel, both coupled with switch of fluorescent reporter genes, from positive and negative conditional alleles at the sox10 locus, respectively.
Similar to other organisms, KI in zebrafish could be achieved by either homology-dependent or -independent approaches (Zu et al., 2013; Auer et al., 2014; Shin et al., 2014; Li et al., 2015; Hoshijima et al., 2016; Sugimoto et al., 2017; Burg et al., 2018; Luo et al., 2018; Li et al., 2019; Li et al., 2020). Inclusion of homology arms in the donor allows for precise integration of the donor through HR, while dramatically compromising the KI efficiency. In contrast, donors without homology arms could incorporate into the target site through NHEJ-induced insertion, which is more efficient than HR-mediated KI, though may be less precise due to introduction of indels at the 5′- and 3′- junctions. Since small indels in introns are usually tolerated, targeting introns for donor KI through the NHEJ pathway could make best use of its high efficiency and at the same time bypass the risk of disrupting gene function by imprecision integration. More importantly, NHEJ-mediated KI allows us to simutaneously generate useful insertions occurring in both directions, each displaying different but complementary KI effects, while homology-directed integration is generally unidirectional and thus inappropriate for our Bi-FoRe strategy. Interestingly, germline transmission rate in this study was high at both sox10 and isl1 loci. We believe there are at least two further reasons accounting for the high KI efficiency, in addition to the benefits of NHEJ-mediated targeted insertion. One is that we have chosen Cas9/gRNA target sites showing high indel efficiency. The other is that we pre-selected fluorescence-positive founder (F0) embryos after injection of the targeting system and before rearing for germline transmission screening. The importance of pre-selection for the enrichment of F0 embryos bearing correct insertions has been demonstrated in our previous publication (Li et al., 2019)
One of the major improvements of our Bi-FoRe strategy is the successful utilization of insertions occurring in both directions, due to the special design of our donor vector. Bidirectional KI enables concurrent generation of positive and negative alleles, allowing for in vivo monitoring of gene expression and conditional manipulation of gene function. To our knowledge, this is the first method available for simple and efficient one-step generation of multi-purpose bidirectional genome modifications, including pairwise fluorescent allele labeling and pairwise conditional allele manipulation. At least two important aspects or applications could be derived from the success of reverse integration, in conjunction with the forward insertion. On one hand, concurrent generation and separate labeling of positive and negative allele pairs with different fluorescent reporter genes could allow for simple and early discrimination of live embryos bearing different genotypes before or without the requirements for the appearance of any visible phenotype, which is crucial for timely and accurate dissection of gene function and mechanisms. This is especially important for timely isolation of large numbers of embryos, which is usually required for genome-wide gene expression and function analyses, such as transcriptome analysis through RNA sequencing, either in bulk or at the single-cell level. For example, the fluorescent signal of our sox10 Bi-FoRe KI embryos could be detected at 21 hpf, seven hours before appearance of visible phenotype (Fig. S6), while the initial phenotype of sox10 mutants is undetectable until 28 hpf. Thus, fluorescent allele labeling provides a powerful tool to capture the correct developmental stage initiating target gene expression and the genotype of the embryos as early as possible, facilitating precise characterization and elucidation of the primary and fundamental molecular events regulated by the target gene.
On the other hand, concurrent pairwise generation of conditional alleles from forward and reverse integration provides a unique opportunity for comprehensive and reciprocal elucidation of gene functions as well as molecular mechanisms. Conditional rescue is an important complementary approach for precise and in-depth analysis of gene function in development, regeneration, and disease progression. Time- and tissue-specific gene function restoration helps confirm the results from gene knockouts and allows detailed dissection of gene functions in different cell lineages and biological stages (Ruehle et al., 2013; Santos et al., 2016; Flores et al., 2018). Utilization of both CKO and conditional rescue analyses of the same gene can largely facilitate the elucidation of cell autonomous or non-autonomous functions of the target gene in adjacent cells and tissues, identification of the effector cells/tissues in which expression of the target gene is both necessary and sufficient, as well as elaboration of gene functions within a specific time window (Santos et al., 2016; Flores et al., 2018). Moreover, time- or tissue-specific recovery of gene expression in embryonic or adult animal disease models can contribute to pathological and therapeutic studies of the affected genes, and thus reveal specific timings and target cells/tissues for gene therapy treatments intended for clinical application, which cannot be achieved by conditional knockouts (Guy et al., 2007; Mei et al., 2016). However, despite these uses, conditional rescues are not widely applied in the studies of gene functions, largely due to the relative complexity and extra effort required in allele construction. Our Bi-FoRe donor strategy provides an efficient method to establish both conditional knockout and conditional rescue alleles of the same gene at the same time within a single experiment, which greatly reduces the time and effort involved in donor construction and allele identification, thus opening new possibilities for detailed dissection of gene function in multiple dimensions.
In the Bi-FoRe KI alleles derived from the full length donor, in addition to the necessary functional sequences, a long bacterial plasmid backbone sequence was also inserted into the target locus. It has been reported that exogenous bacterial sequences introduced into the eukaryotic genome may cause alteration of the expression level of endogenous genes (Chen et al., 2001, 2003; Suzuki et al., 2016). To avoid potential deleterious consequences, we adopted the minicircle vector pTUBB3-MC to construct our Bi-FoRe KI donor (Kay et al., 2010; Suzuki et al., 2016), whose backbone sequence is flanked with minimal attB and attP sites and can be removed by phiC31 integrase-induced recombination either in vivo or in vitro. We first successfully removed the backbone of sox10 Bi-FoRe KI alleles by injection of zebrafish codon optimized phiC31 mRNA in zebrafish embryos (Fig. S8), which is consistent with previous reports of efficient in vivo site-specific excision of minimal attB and attP sites (Lister, 2010; Lu et al., 2011). For in vitro backbone removal, we prepared the sox10 minicircle Bi-FoRe donor according to the minicircle production protocol (Lister, 2010; Lu et al., 2011) and used it for founder embryo injection. Our preliminary results showed that efficient and correct KI events were easily detected in minicircle donor injected embryos (Fig. 4A and 4B), and the ratio of mosaic F0 embryos increased by 14% compared with the full length Bi-FoRe donor (Table S2), which suggested that the smaller donor may be more efficient for NHEJ-mediated KI. In summary, our results showed that unfavorable bacterial backbone sequence could be efficiently eliminated either in vivo by phiC31 mRNA injection or in vitro by minicircle DNA donor production. To our knowledge, this is the first report of the application of minicircle DNA technology in zebrafish.
In addition to integration into the target locus, donor vectors might also exhibit off-targeting insertions after injection into zebrafish embryos. Although we cannot exclude this possibility, we did not observe obvious ectopic expressions of fluorescent reporters in the F0 or F1 embryos in our experiments, indicating that random gene-trapping was undetectable. This may be due to the low random insertion efficiency of in vivo linearized donor vectors as previously reported (Auer et al., 2014), and in addition, the requirement for donors to be inserted into appropriate gene regions to ensure its expression. However, “silent” random insertions are still possible, which should be investigated in the future.
Besides what we have presented in this study, our FoRe and Bi-FoRe donor strategy can be further modified for expanded or improved applications. For example, a disease-related mutation could be substituted in the downstream CDS to generate disease models with allele expression tagging, or coding sequences of various protein tags could be fused in-frame with the remaining CDS for biochemical studies. Furthermore, since the CRISPR/Cas system, including Cas9 and Cas12a, has been widely and efficiently applied to many organisms (Mali et al., 2013; Zetsche et al., 2015; Hur et al., 2016; Moreno-Mateos et al., 2017), our bidirectional multi-function KI strategy should be applicable in vivo in other model systems, as well as in vitro in cell culture studies.
Materials and methods
Zebrafish husbandry
All the zebrafish used in this study were maintained at 28.5 °C in the zebrafish facility of Peking University with a 14 h/10 h light/dark cycle. The wild-type strain used was Tübingen (TU).
Donor plasmid construction
To construct the FoRe donor for the sox10 locus targeting the third intron, the hEMX1 target site and the lox66 site were linked by a 111-bp DNA fragment cloned from the pMD18-T vector (TAKARA, 6011) to avoid the disruption of the lox66 sequence due to NHEJ-mediated DSB repair. This hEMX1-linker-lox66 sequence was cloned into the pMD19-T simple vector (TAKARA, 3271). Then, the intron 3 and exon 4 sequence (without stop codon and 3′ UTR) downstream of the sox10 target site (including the splicing acceptor) from the zebrafish genome was amplified and cloned into the above vector downstream to the lox66 site. Then T2A-tdTomato-SV40 pA sequence was fused downstream to the sox10 exon 4 (referred to as “Remaining CDS” in Fig. 1A). These elements aside from the hEMX1 site were referred as the Forward component of the vector. Finally, the lox71 sequence, the intron 3 sequence downstream of the sox10 site and the first 98 bp of the sox10 exon 4 sequence were ligated with P2A-EGFP-BGH pA and cloned into the above vector in reverse orientation downstream of the SV40 pA sequence, as the Reverse component of the vector. For the convenience of cloning/replacing other target gene sequence, KpnI and AvrII sites were introduced upstream and downstream, respectively, to the sox10 sequence in the Forward component in forward direction, while SalI and EcoRV recognition sites were introduced upstream and downstream, respectively, to the sox10 sequence in the Reverse component in reverse direction.
The above sox10 FoRe donor was used as the basic vector to construct the FoRe donor for the isl1 locus and the Bi-FoRe donor for the sox10 locus. To construct the isl1 FoRe donor, the sox10 FoRe donor was digested with KpnI and AvrII and used as the donor backbone. Then, the intron 3 sequence downstream of the isl1 target site and the subsequent remaining downstream CDS without stop codon (referred to as “Remaining CDS” in Fig. S4B) were cloned into the backbone. The plasmid was then digested by SalI and EcoNI and ligated with the intron 3 sequence downstream of the isl1 site and the first 17 bp of isl1 exon 4 to generate the complete isl1 FoRe donor.
To construct the sox10 Bi-FoRe donor, the minicircle plasmid pTUBB3-MC (gift from Juan Belmonte, Addgene plasmid #87112; http://n2t.net/addgene:87112; RRID: Addgene_87112) was used as the backbone after digestion with XmaI and BsrGI (Suzuki et al., 2016). The region spanning the hEMX1 site and the entire Forward component was amplified from the sox10 FoRe donor and ligated into the digested pTUBB3-MC plasmid, together with a BGH pA sequence. Then, the Reverse component from the sox10 FoRe donor, including the sequence spanning the BGH pA through the lox71 site, together with its upstream flanking SV40 pA sequence and downstream 70-bp sequence in the sox10 FoRe donor (for protection from NHEJ-mediated sequence deletion), was amplified and cloned upstream of the hEMX1 site to generate the full length sox10 Bi-FoRe donor. For the production of the sox10 Bi-FoRe minicircle donor vector, the above full length sox10 Bi-FoRe donor was transformed into the ZYCY10P3S2T strain (System Biosciences), then bacteria amplification and plasmid extraction was performed as reported by Kay et al. (2010), except for adjusting the incubation volume to a level suitable for miniprep.
For kits and enzymes used in molecular cloning, initial T-A cloning was performed using Solution I (TAKARA), and the cloning of the subsequent fragments was performed using Gibson assembly according to the manufacture’s protocol (NEB, M5510AA). Restriction enzymes were ordered from NEB, and high-fidelity versions were adopted if available.
Preparation of gRNA and the mRNAs encoding Cas9, Cre and phiC31
The zebrafish codon-optimized Cas9 expression vector pT3TS-nCas9n was linearized by XbaI and used as the template for generating Cas9 mRNA through in vitro transcription using the mMessage mMachine T3 kit (Ambion) (Jao et al., 2013).
The gRNAs were designed by using the CasOT program (http://casot.cbi.pku.edu.cn/) (Xiao et al., 2014). Forward oligonucleotides containing a T7 promoter, gRNA target site and partial gRNA scaffold sequences were designed for gRNA template synthesis through PCR amplification by using the pUC19-scaffold as the template (Chang et al., 2013), together with the universal reverse primer (5′-AAAAAAAGCACCGACTCGGTGCCAC-3′). Then, the gRNAs were synthesized by in vitro transcription with T7 RiboMAX™ Express Large Scale RNA Production System (Promega). The gRNA target sequences are listed in Table S1.
The Cre expression vector pX-T7-Cre (Li et al., 2019) was linearized by XbaI and used as the template for synthesizing Cre mRNA through in vitro transcription using the mMessage mMachine T7 kit (Ambion).
Zebrafish codon optimized phiC31 (zphiC31) coding sequence was chemically synthesized (Ruibiotech Biotechnology, Beijing) and cloned to pT3TS plasmid to generate the pT3TS-zphiC31 plasmid. The plasmid was linearized by XbaI and used as template for in vitro transcription of zphiC31 mRNA with the mMessage mMachine T3 kit (Ambion).
All the RNA products were purified by LiCl precipitation according to the manufacture’s protocol (Ambion).
Microinjection of zebrafish embryos
To evaluate the indel efficiency of each target site, 1 nL of a solution containing 600 ng/μL Cas9 mRNA and 100 ng/μL gRNA was injected into each one-cell stage zebrafish embryo. For knockin experiments, 1 nL of a solution containing 600 ng/μL Cas9 mRNA, 100 ng/μL for each gRNA and 15 ng/μL of full length donor plasmid or 8 ng/μL of the minicircle donor (to ensure approximately equal amount of molecules for different donors) was injected into the animal pole of one-cell stage zebrafish embryos.
For Cre recombinase-induced inversion experiments, 10 pg Cre mRNA was injected into the vegetal pole of one-cell stage embryos to obtain mosaics, and 50 pg Cre mRNA was injected into the animal pole of one-cell stage embryos to achieve efficient Cre-mediated recombination.
For the experiments involving phiC31 function, 50 pg zphiC31 mRNA was injected into the animal pole of one-cell stage embryos to induce recombination of minimal attB and attP in vivo.
Restriction endonuclease assay to evaluate indel efficiencies of the Cas9 target sites
To prepare genomic DNA, five 24 hpf embryos were collected in each tube and lysed by 20 μL 50 mmol/L NaOH solution at 95 °C for 15 min, then neutralized by 2 μL 1 mol/L Tris-HCl (pH = 8.0). Then, 1 μL of the genomic DNA extract was used as the template to PCR amplify the region spanning the target sites using relevant primers (Table S7). The PCR products were then digested by corresponding restriction endonucleases and analyzed by agarose gel electrophoresis. The indel efficiency was estimated by the ratio of undigested products.
Junction PCR and Sanger sequencing
Genomic DNA of 48–72 hpf F1 embryos was extracted by NaOH lysis as described above. Then, genome pool of corresponding embryos (3–5 individuals) was used as template to PCR amplify the 5′ and 3′ junction fragments of the KI alleles using the appropriate primers (Table S7). The PCR products were directly sent for Sanger sequencing.
RNA extraction, cDNA synthesis and RT-PCR analysis
RNA was isolated from 10 embryos at 2–3 dpf (days post-fertilization) using Trizol reagent (Invitrogen) according to the manufacture’s protocol. 1 μg of total RNA was used for cDNA synthesis by reverse transcription using 5X All-In-One RT MasterMix (Applied Biological Materials). 1 μL of the resulting cDNA was used as template for PCR amplification with corresponding primers (Table S7), and gapdh was used as internal reference. The predicted length of the PCR products generated by sRT or iRT primer pairs is 467 bp and 509 bp, respectively.
Imaging and processing
For general imaging, zebrafish embryos were anesthetized with 0.02% tricaine (ethyl 3-aminobenzoate methanesulfonate salt, Sigma), positioned in 3% methylcellulose (Sigma), and imaged under a compound microscope (AXIO Imager Z1; Zeiss) equipped with AxioCam MRm (Zeiss). The images were processed by the AxioVision Rel.4.8 software. Confocal imaging was performed using a LSM 710 confocal microscope (Zeiss) with a 10× or 20× water immersion objective, and Z-stack images were acquired with a 5 to 7 μm resolution and processed by the ZEN 2009 imaging software. Lightsheet imaging was performed using a Lightsheet Z1 microscope (Zeiss) with a 10X objective, and the images were processed by the ZEN 2014 SP1 imaging software. The embryos used for imaging in all the isl1 KI experiments as well as sox10 KI experiments in Fig. S2B and S2C were pretreated with 0.0045% PTU (1-Phenyl-2-thiourea, Sigma) dissolved in fish water for inhibition of the pigmentation.
References
Albert H, Dale EC, Lee E, Ow DW (1995) Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J 7:649–659
Araki K, Araki M, Yamamura KI (2002) Site-directed integration of the cre gene mediated by Cre recombinase using a combination of mutant lox sites. Nucleic Acids Res 30:e103
Araki K, Araki M, Yamamura KI (2006) Negative selection with the Diphtheria toxin A fragment gene improves frequency of Cre-mediated cassette exchange in ES cells. J Biochem 140:793–798
Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F (2014) Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res 24:142–153
Burg L, Palmer N, Kikhi K, Miroshnik ES, Rueckert H, Gaddy E, Cunningham CM, Mattonet K, Lai SL, Marin-Juez R et al (2018) Conditional mutagenesis by oligonucleotide-mediated integration of loxP sites in zebrafish. PLoS Genet 14:e1007754
Caputo L, Witzel HR, Kolovos P, Cheedipudi S, Looso M, Mylona A, van IJcken WF, Laugwitz KL, Evans SM, Braun T et al (2015) The Isl1/Ldb1 complex orchestrates genome-wide chromatin organization to instruct differentiation of multipotent cardiac progenitors. Cell Stem Cell 17:287–299
Carney TJ, Mosimann C (2018) Switch and trace: recombinase genetics in zebrafish. Trends Genet 34:362–378
Chang NN, Sun CH, Gao L, Zhu D, Xu XF, Zhu XJ, Xiong JW, Xi JJ (2013) Genome editing with RNA-guided Cas9 nuclease in Zebrafish embryos. Cell Res 23:465–472
Chen ZY, Yant SR, He CY, Meuse L, Shen S, Kay MA (2001) Linear DNAs concatemerize in vivo and result in sustained transgene expression in mouse liver. Mol Ther 3:403–410
Chen ZY, He CY, Ehrhardt A, Kay MA (2003) Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther 8:495–500
Dutton KA, Dutton JR, Pauliny A, Kelsh RN (2001a) A morpholino phenocopy of the colourless mutant. Genesis 30:188–189
Dutton KA, Pauliny A, Lopes SS, Elworthy S, Carney TJ, Rauch J, Geisler R, Haffter P, Kelsh RN (2001b) Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 128:4113–4125
Flores DJ, Duong T, Brandenberger LO, Mitra A, Shirali A, Johnson JC, Springer D, Noguchi A, Yu ZX, Ebert SN et al (2018) Conditional ablation and conditional rescue models for Casq2 elucidate the role of development and of cell-type specific expression of Casq2 in the CPVT2 phenotype. Hum Mol Genet 27:1533–1544
Geng FS, Abbas L, Baxendale S, Holdsworth CJ, Swanson AG, Slanchev K, Hammerschmidt M, Topczewski J, Whitfield TT (2013) Semicircular canal morphogenesis in the zebrafish inner ear requires the function of gpr126 (lauscher), an adhesion class G protein-coupled receptor gene. Development 140:4362–4374
Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science 315:1143–1147
Hoshijima K, Jurynec MJ, Grunwald DJ (2016) Precise editing of the zebrafish genome made simple and efficient. Dev Cell 36:654–667
Hur JK, Kim K, Been KW, Baek G, Ye S, Hur JW, Ryu SM, Lee YS, Kim JS (2016) Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol 34:807–808
Jao LE, Wente SR, Chen WB (2013) Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci USA 110:13904–13909
Kay MA, He CY, Chen ZY (2010) A robust system for production of minicircle DNA vectors. Nat Biotechnol 28:1287–1289
Kelsh RN, Eisen JS (2000) The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development 127:515–525
Li J, Zhang BB, Ren YG, Gu SY, Xiang YH, Huang C, Du JL (2015) Intron targeting-mediated and endogenous gene integrity-maintaining knockin in zebrafish using the CRISPR/Cas9 system. Cell Res 25:634–637
Li W, Zhang Y, Han B, Li L, Li M, Lu X, Chen C, Lu M, Zhang Y, Jia X et al (2019) One-step efficient generation of dual-function conditional knockout and geno-tagging alleles in zebrafish. Elife 8:e48081
Li J, Li HY, Gu SY, Zi HX, Jiang L, Du JL (2020) One-step generation of zebrafish carrying a conditional knockout-knockin visible switch via CRISPR/Cas9-mediated intron targeting. Sci. China Life Sci. 63:59–67
Lin S, Staahl BT, Alla RK, Doudna JA (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3:e04766
Lister JA (2010) Transgene excision in zebrafish using the phiC31 integrase. Genesis 48:137–143
Lu JJ, Maddison LA, Chen WB (2011) PhiC31 integrase induces efficient site-specific excision in zebrafish. Transgenic Res 20:183–189
Luo JJ, Bian WP, Liu Y, Huang HY, Yin Q, Yang XJ, Pei DS (2018) CRISPR/Cas9-based genome engineering of zebrafish using a seamless integration strategy. FASEB J 32:5132–5142
Mali P, Esvelt K, Church G (2013) Cas9 as a versatile tool for engineering biology. Nat Methods 10:957–963
Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, Feng G (2016) Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530:481–484
Moreno-Mateos MA, Fernandez JP, Rouet R, Vejnar CE, Lane MA, Mis E, Khokha MK, Doudna JA, Giraldez AJ (2017) CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nat Commun 8:2024
Ruehle S, Remmers F, Romo-Parra H, Massa F, Wickert M, Wortge S, Haring M, Kaiser N, Marsicano G, Pape HC et al (2013) Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J Neurosci 33:10264–10277
Santos R, Kawauchi S, Jacobs RE, Lopez-Burks ME, Choi H, Wikenheiser J, Hallgrimsson B, Jamniczky HA, Fraser SE, Lander AD et al (2016) Conditional creation and rescue of nipbl-deficiency in mice reveals multiple determinants of risk for congenital heart defects. PLoS Biol 14:e2000197
Shin J, Chen J, Solnica-Krezel L (2014) Efficient homologous recombination-mediated genome engineering in zebrafish using TALE nucleases. Development 141:3807–3818
Sirbu IO, Zhao X, Duester G (2008) Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev Dyn 237:1627–1635
Sugimoto K, Hui SP, Sheng DZ, Kikuchi K (2017) Dissection of zebrafish shha function using site-specific targeting with a Cre-dependent genetic switch. Elife 6:e24635
Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z et al (2016) In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540:144–149
Wilfinger A, Arkhipova V, Meyer D (2013) Cell type and tissue specific function of islet genes in zebrafish pancreas development. Dev Biol 378:25–37
Witzel HR, Jungblut B, Choe CP, Crump JG, Braun T, Dobreva G (2012) The LIM protein Ajuba restricts the second heart field progenitor pool by regulating Isl1 activity. Dev Cell 23:58–70
Witzel HR, Cheedipudi S, Gao R, Stainier DY, Dobreva GD (2017) Isl2b regulates anterior second heart field development in zebrafish. Sci Rep 7:41043
Xiao A, Cheng ZC, Kong L, Zhu ZY, Lin S, Gao G, Zhang B (2014) CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30:1180–1182
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A et al (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771
Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, Hu Y, Luo Z, Huang P, Wu Q et al (2013) TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods 10:329–331
ACKNOWLEDGEMENTS
We thank China Zebrafish Resource Center (CZRC) and Wenbiao Chen for providing us with the pT3TS-nCas9n plasmid, Jingwei Xiong for providing us with the pUC19-scaffold plasmid, Juan Belmonte for providing the pTUBB3-MC plasmid, Yao Zu for providing us with the pX-T7-Cre plasmid, Dali Li for help on minicircle DNA production, and Wenyuan Li for sharing the isl1 I3 target site. We thank Yuying Gao and Yan Shen for lab management, Yingdi Jia and Jingliang Chen for zebrafish husbandry. We thank Xiaochen Li, Siying Qin and Xuemei Hao from the Core Facilities at the School of Life Sciences of Peking University for assistance with confocal microscopy. We also thank Imaging Core at National Center for Protein Sciences at Peking University, particularly Liqin Fu for invaluable technical help with lightsheet microscopy (Zeiss Lightsheet Z1) and image processing. This work was partially supported by grants from the National Key Research and Development Program of China (2019YFA0802800, 2018YFA0801000, 2016YFA0100500); the National Natural Science Foundation of China (NSFC) (Grant Nos. 81770376, 31871458, 31671500 and 81371264); and the PKU Qidong-SLS Innovation Fund.
AUTHOR CONTRIBUTIONS
Bingzhou Han, Yage Zhang and Bo Zhang conceived and designed experiments; Bingzhou Han, Yage Zhang and Xinli Hu performed donor construction; Bingzhou Han and Yage Zhang performed microinjection and imaging; Bingzhou Han, Yage Zhang, Xuetong Bi and Yang Zhou performed germline transmission screening and RT-PCR; Bingzhou Han, Yage Zhang, Christopher J Krueger, Zuoyan Zhu, Xiangjun Tong and Bo Zhang analyzed the data and wrote the manuscript.
ABBREVIATIONS
Bi-FoRe, bidirectional FoRe; CKO, conditional knockout; CRISPR, clustered regularly interspaced short palindromic repeats; dpf, days post-fertilization; FoRe, forward and reverse; hpf, hours post-fertilization; HR, homologous recombination; KI, knockin; NHEJ, nonhomologous end joining
COMPLIANCE WITH ETHICS GUIDELINES
Bingzhou Han, Yage Zhang, Xuetong Bi, Yang Zhou, Christopher J Krueger, Xinli Hu, Zuoyan Zhu, Xiangjun Tong, and Bo Zhang declare that they have no conflict of interest. All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary movie 1 (MP4 8853 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, B., Zhang, Y., Bi, X. et al. Bi-FoRe: an efficient bidirectional knockin strategy to generate pairwise conditional alleles with fluorescent indicators. Protein Cell 12, 39–56 (2021). https://doi.org/10.1007/s13238-020-00747-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-020-00747-1