
Abstract
Dysregulation of circadian rhythms associates with cardiovascular disorders. It is known that deletion of the core circadian gene Bmal1 in mice causes dilated cardiomyopathy. However, the biological rhythm regulation system in mouse is very different from that of humans. Whether BMAL1 plays a role in regulating human heart function remains unclear. Here we generated a BMAL1 knockout human embryonic stem cell (hESC) model and further derived human BMAL1 deficient cardiomyocytes. We show that BMAL1 deficient hESC-derived cardiomyocytes exhibited typical phenotypes of dilated cardiomyopathy including attenuated contractility, calcium dysregulation, and disorganized myofilaments. In addition, mitochondrial fission and mitophagy were suppressed in BMAL1 deficient hESC-cardiomyocytes, which resulted in significantly attenuated mitochondrial oxidative phosphorylation and compromised cardiomyocyte function. We also found that BMAL1 binds to the E-box element in the promoter region of BNIP3 gene and specifically controls BNIP3 protein expression. BMAL1 knockout directly reduced BNIP3 protein level, causing compromised mitophagy and mitochondria dysfunction and thereby leading to compromised cardiomyocyte function. Our data indicated that the core circadian gene BMAL1 is critical for normal mitochondria activities and cardiac function. Circadian rhythm disruption may directly link to compromised heart function and dilated cardiomyopathy in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The circadian clock, also known as a circadian oscillator, is ubiquitously present in every cell and critical to regulate internal physiological rhythms and behaviors throughout an organism’s life. The body’s circadian clock produces a robust 24-hour rhythmic activity as the Earth rotates (Kang et al., 2009). These intrinsic circadian rhythms are based on transcriptional-translational feedback loops (TTFLs) to maintain organ homeostasis (Dierickx et al., 2018). The core TTFLs are driven by four intrinsic clock genes: two activation factors CLOCK and BMAL1, and two inhibitors Period (PER1, 2, 3) and Cryptochrome (CRY1,2) (Huang et al., 2012; Partch et al., 2014). In mammals, the central circadian clock is located in the suprachiasmatic nucleus of the hypothalamus, while the peripheral circadian clocks are present in peripheral organs and tissues (Balsalobre et al., 1998), including the heart (Maemura et al., 2001) and vasculatures (McNamara et al., 2001; Nonaka et al., 2001).
Recent studies revealed that dysregulation of circadian rhythms associates with metabolic and cardiovascular disorders (Mottillo et al., 2010; Maury et al., 2014). Global Bmal1-deficient mice displayed age-associated dilated cardiomyopathy (DCM), exhibiting left ventricular dilatation and contractile dysfunction (Lefta et al., 2012). This suggests that dysregulation of circadian rhythms may cause gradual abnormality of heart function and finally lead to cardiac dilation and failure. However, whether BMAL1 deficiency causes a similar cardiac phenotype in humans is unknown. And, the molecular mechanisms of BMAL1 deficiency leading to the DCM phenotype is also unclear.
DCM is one of the main causes of heart failure (Maron et al., 2006). So far, more than 50 kinds of single-gene mutations have been found to lead to DCM(Vikhorev et al., 2017). Gene mutations involving sarcomere (Gerull et al., 2002), nuclear envelope (Fatkin et al., 1999; Parks et al., 2008), transcriptional pathways (Pyun et al., 2018), ion channel (Schmitt et al., 2003) and mitochondrial (Song et al., 2015) proteins are the most common genetic causes of DCM. However, the etiology of some idiopathic DCM remains unclear. In this study, by analyzing the published gene expression omnibus (GEO) data (GSE42955), we found that the BMAL1 mRNA level in advanced DCM patients was much higher than that of the normal people. We disrupted BMAL1 gene expression in human embryonic stem cells (hESCs) using genomic editing techniques and further derived human cardiomyocytes by cardiac-specific differentiation. We found hESC-derived cardiomyocytes with BMAL1 knockout (KO) exhibited abnormalities such as disruption of sarcomeres, enlarged cell size, impaired contractility and calcium handling, as well as arrhythmia. Cardiomyocytes from both BMAL1 KO hESCs and Bmal1 KO mice showed an increased mitochondrial fusion and a decline in mitochondrial autophagy, resulted in attenuated mitochondrial oxidative phosphorylation and mitochondrial function. We further discovered that BMAL1 binds to the E-box element in the promoter region of the mitochondrial gene BNIP3. Knocking out BMAL1 caused a decrease in BNIP3 protein expression, which partly induced impaired mitochondrial fission and mitophagy, leading to compromised cardiomyocyte function and the macroscopic phenotype of DCM.
Overall, our results showed that the circadian gene BMAL1 plays a critical role in maintaining normal function of cardiomyocytes and the heart. Circadian rhythm disruption may directly link to compromised heart function and dilated cardiomyopathy in humans.
Results
Bmal1 deficient mice developed dilated cardiomyopathy
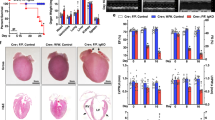
To see whether the circadian clock gene Bmal1 plays a role in cardiac function, we generated mice with global deletion of the Bmal1 gene (Fig. S1) and examined their heart morphology and function over time. Mice with Bmal1 KO showed measurable weight loss and heart rate reduction during their growth (Fig. S2A and S2B). At 32 weeks of age, histological analyses of cross sections of the heart at the level of papillary muscles showed that Bmal1 deficient mice displayed a larger left ventricular cavity plus thinning of left ventricular posterior wall and interventricular septum (Fig. 1A and 1B). The myocyte cross-sectional area was also increased in Bmal1 KO mice when compared to that of the wild type mice (Fig. 1C and 1D), indicating dilation of the myocardium and the ventricle. In addition, transmission electron microscopy showed that Bmal1 KO mice exhibited disorganization and compaction of sarcomeres. The sarcomeric Z-lines were also become condensed and wider (Fig. 1E).

Bmal1 deletion caused dilated cardiomyopathy in mice. (A) Histological analyses of heart sections of wild type and Bmal1 KO mice by H&E staining at 32 weeks of age. Scale bar: 1 mm. (B) Statistics of the average thickness of IVS and LVPW of wild type and Bmal1 KO mice (n = 4). *P < 0.05 and **P < 0.01 versus control by two-tailed Student’s t test. (C) Representative confocal microscopy images of WGA staining of myocardium from wild type and Bmal1 KO mice. (D) Quantification of cell surface area as shown in (C) (n = 59 per group). ****P < 0.0001 versus control by two-tailed Student’s t test. (E) Representative electron micrographs of ventricular cardiomyocytes from wide type and Bmal1 KO mice. Red arrows indicate Z-lines. (F) Representative echocardiography images of 32-weeks-old wild type and Bmal1 KO mice. (G–N) Echocardiographic parameters of heart functions from wild type and Bmal1 KO mice over time (n = 4 per group). 2-way ANOVA with post-hoc tes. Data were represented mean ± SD. *P < 0.05 and **P < 0.01 versus control by 2-way ANOVA with post-hoc test
From 12-weeks of age, we assessed heart functions of wild type and Bmal1 KO mice by echocardiography at four-weeks intervals (Fig. 1F). Bmal1 KO mice showed a significantly increased left ventricular end systolic internal diameter (LVIDs) at 32 weeks compared to that in wild type mice (Fig. 1G). The interventricular septum thickness at end-systole (IVSs) and left ventricular posterior wall thickness at end-systole (LVPWs) also gradually became thinner in Bmal1 KO mice than those in wild type mice (Fig. 1H and 1I). Increased left ventricular internal dimension at end-diastole (LVIDd), decreased interventricular septum thickness at end-diastole (IVSd), and left ventricular posterior wall thickness at end-diastole (LVPWd) were also observed in Bmal1 KO mice (Fig. 1J–L). Further, Bmal1 KO mice showed impaired cardiac contractility with a decreased fraction of shortening (FS) (Fig. 1M) and ejection fraction (EF) (Fig. 1N). Overall, Bmal1 KO led to a dramatic ventricular dilation and a ~50% reduction in cardiac contractility in mice at later age. These results are consistent with previous reports showing Bmal1 deficiency induced age-associated DCM in mice (Lefta et al., 2012).
BMAL1 deficient hESC-derived cardiomyocytes exhibited myofilament sarcomere disruption and increased apoptosis
To further assess the role of BMAL1 in human cardiomyocytes, we knocked out BMAL1 expression in H7 hESCs by CRISPR/Cas9 mediated genome editing (Fig. 2A). Indel mutations were introduced within exon 10 of BMAL1 by CRISPR/Cas9 nickase, which led to frameshifts and creation of a premature stop codon in BMAL1 locus (Fig. S3A–C). Western blot results indicated that BMAL1 protein expression was knocked out in the selected positive clones (Fig. 2B). In order to confirm whether both alleles of the BMAL1 gene were disrupted, we performed TA cloning of the amplified PCR products of the target cutting region and it showed that those positive clones were all homozygous. The two alleles of the clone 18 had the same genetic modification (Fig. S3D), while the others had two different genetic mutations (data not shown). The established BMAL1 KO hESC lines exhibited normal ES-cell-like morphologies and pluripotency nature (Fig. S4). Although BMAL1 KO hESCs spontaneously differentiated into the three germ layers in teratoma assays, an upregulation of the mesodermal, cardiac progenitor, and endodermal genes were observed (Fig. S5).


Phenotypic characterizations of BMAL1 KO hESCs-derived cardiomyocytes. (A) Schematic showing the design for generation of BMAL1 KO hESC cell line by genomic editing with CRISPR/Cas9 technique. The CRISPR/Cas9 cutting site is on exon 10 of human BMAL1 gene. gRNA: guide RNA, PAM: protospacer adjacent motif. (B) Western blot of BMAL1 protein expression in wild type hESCs and BMAL1 KO positive hESC clones. (C) Flowcytometry analyses of cardiac differentiation efficiency of wild type and BMAL1 KO hESCs. Cardiomyocytes were stained with the classic cardiac marker cardiac troponin T (cTnT). (D) Representative transmission electron micrographs of sarcomeric structures in wild type and BMAL1 KO hESC-derived cardiomyocytes. Scales bar: 1 µm. (E) Immunostaining of cTnT and α-Actinin in wild type and BMAL1 KO hESC-derived cardiomyocytes. Scale bars: 50 µm. (F) Quantification of cell size for wild type and BMAL1 KO hESC-derived cardiomyocytes (n = 85 per group). (G) Higher percentage of disorganized sarcomeres for BMAL1 KO hESC-derived cardiomyocytes. (H) Detection of apoptosis by flowcytometry in wild type and BMAL1 KO hESC-derived cardiomyocytes. (I–K) Quantification of the ratio of annexin V−/PI+ cells, annexin V+/PI− cells and annexin V+/PI+ cells. Data were represented as means ± SD. *P < 0.05 and ****P < 0.0001 versus control by two-tailed Student’s t test
We further differentiated these BMAL1 KO hESCs into beating cardiomyocytes (Supplementary Movie 1 and 2) (Lian et al., 2012). The differentiation efficiency regularly reached greater than 90% (Fig. 2C). To evaluate the impact of BMAL1 KO on cardiomyocyte morphology and myofilament structure, immunofluorescence stainings were performed and it showed that BMAL1 KO hESC-cardiomyocytes exhibited abnormal sarcomeric cardiac troponin T (cTnT) and α-Actinin distribution (Fig. 2E). Assessing ultra-structures using transmission electron microscopy further showed that wildtype hESC-cardiomyocytes exhibited well organized sarcomeres and regular Z-lines, while BMAL1 KO hESC-cardiomyocytes displayed severe myofilament disorganization and irregular sarcomeric Z-lines (Fig. 2D). We also assessed the cell size of both widetype mice and BMAL1 KO hESC-cardiomyocytes. The average cell area is significantly larger in BMAL1 KO hESC-cardiomyocytes (wildtype hESC-cardiomyocytes, 3,370 ± 133.6 μm2; BMAL1−/− hESC-cardiomyocytes, 6,965 ± 396.3 μm2, n = 85 per group, P < 0.001) (Fig. 2F). Compared with wild type hESC-cardiomyocytes, the percentage of BMAL1 deficient hESC-cardiomyocytes with disorganized sarcomeres increased significantly (Fig. 2G). In addition, BMAL1 deficiency led to a significant increase in both early apoptosis and late apoptosis according to flowcytometry analysis (Fig. 2H–K). These data indicated that BMAL1 KO had a great impact on myofilament organization and morphology of human cardiomyocytes.
Compromised contractility and calcium handling in BMAL1 KO cardiomyocytes
Our results showed that Bmal1 knockout mice exhibited reduced heart contractility and cardiomyocyte sarcomeric disruption. To assess whether BMAL1 KO has an impact on contractility of human cardiomyocytes, we next measured the contraction force of single BMAL1 KO hESC-derived cardiomyocytes. We found that single BMAL1 KO hESC-cardiomyocytes exhibited weakened contractile force generation per cell movement and irregular beating rhythm when compared with wildtype control hESC-cardiomyocytes (Fig. 3A–C).

Compromised contraction force and abnormal calcium handling in BMAL1 KO hESC-derived cardiomyocytes. (A) Representative traces of video detection for contraction movements from single wild type hESC-derived cardiomyocytes day 35 post differentiation. (B) Representative traces of video detection for contraction movements from single BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (C) Quantification of relative contraction force of single wild type and BMAL1 KO hESC-derived cardiomyocytes (n = 32 per group). (D) Representative Ca2+ line scan images and spontaneous Ca2+ transients in wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (E–J) Quantification of calcium handling parameters in wild type and BMAL1 KO hESC-derived cardiomyocytes. E, transient amplitude (average ΔF/F0). F, peak to peak time. G, ratio of cardiomyocytes with irregular Ca2+ transients. H, decay time. I, transient duration 50. J, time to peak. n = 25 in each group. Data were represented as mean ± SD. *P < 0.05, **P < 0.01 and ****P < 0.0001 versus control by two-tailed Student’s t test
Previous studies on Ca2+ homeostasis showed that calcium handling plays an important role in excitation-contraction coupling and subsequent force generation of cardiomyocytes (Cheng et al., 1993; Bers, 2002; Eisner et al., 2017). Myocardium from failing hearts also showed defective excitation-contraction coupling which contributes to impaired contractility (Gomez et al., 1997; Yano et al., 2000; Piacentino et al., 2003). We then explored the Ca2+ handling property of BMAL1 KO hESC-cardiomyocytes by calcium imaging. The results showed that BMAL1 KO hESC-cardiomyocytes exhibited more irregular Ca2+ transients compared with those of wild type control hESC-cardiomyocytes (Fig. 3D and 3G). Further, BMAL1 KO hESC-cardiomyocytes showed a reduction in Ca2+ transient amplitude, indicating that BMAL1 deficiency weakened myofilament Ca2+ sensitivity (Fig. 3E). BMAL1 KO hESC-cardiomyocytes also showed a slower peak to peak time, indicating a significantly elongated beating activity (Fig. 3F). There was no significant difference in the relative time to peak and Ca2+ decay kinetics between wildtype and BMAL1 KO hESC-cardiomyocytes (Fig. 3H–J). These data indicated that BMAL1 KO impaired contractility and calcium handling of hESC-cardiomyocytes.
Electrophysiological properties of BMAL1 KO hESC-cardiomyocytes
Previous studies revealed that sympathetic nervous system plays a key role in regulating cardiac physiology(Hoover et al., 2004) by modulating the heart rate, contractility (Li et al., 2000; Shan et al., 2010), and structure of myocytes (Kanevskij et al., 2002; O’Connell et al., 2003; Kreipke and Birren, 2015). To evaluate the effect of BMAL1 deficiency on cardiomyocytes to neuro-hormonal regulation, non-invasive microelectrode arrays (MEAs) was utilized to analyze pharmacologic effects of heart-related drugs on hESC-cardiomyocytes. The drugs that can influence sympathetic nervous system, for instance adrenergic catecholamine norepinephrine (NE) and beta-adrenergic receptor agonist metoprolol (Mtl), were used to measure cardiac electrophysiological characteristics. Wildtype or BMAL1 KO hESC-cardiomyocytes day 30 post differentiation were seeded on MEA probe and the monolayer cardiomyocytes on the MEA surface began to beat spontaneously after 24 h in culture. Synchronous and stable traces for electrophysiological parameters were recorded on day 35 (Fig. 4A). Parameters of MEAs used for the evaluation were characterized by beating rate (Fig. 4B). Irregular beating activities were observed in the onset of BMAL1 KO hESC-cardiomyocytes (Fig. 4C). Moreover, baseline electrophysiological parameters showed that the average beating frequency of BMAL1 KO hESC-cardiomyocytes was much lower than that of the wild type hESC-cardiomyocytes (Fig. 4D).

Multi-electrode array analyses of electrophysiology of BMAL1 KO hESC-derived cardiomyocytes. (A) hESC-derived cardiomyocytes day 35 post differentiation cultured on a MEA probe coated with matrigel. Scale bar: 200 μm. (B) Typical field potential traces of hESC-derived cardiomyocytes recorded by MEA. (C) Representative MEA recordings showed field potential traces from wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (D) Quantification of beating frequency of wild type and BMAL1 KO hESC-derived cardiomyocytes in response to epinephrine and metoprolol. Data were represented as mean ± SD. The statistics were from 3 independent experiments. *P < 0.05 and **P < 0.01 versus control by two-tailed Student’s t test
Mitochondrial fission and autophagy were compromised in BMAL1 KO hESC-cardiomyocytes and in the myocardium of Bmal1-deficient mice
Mitochondrial dysfunction is considered to be a critical mechanism underlying cardiovascular diseases (Wallace, 2013; Murphy et al., 2016; Friederich et al., 2018). Abnormal mitochondrial morphology was observed in some mitochondrial diseases and idiopathic cardiomyopathies (McManus et al., 2019) . To see whether BMAL1 ablation causes abnormality in mitochondrial function, we analyzed the ultrastructure of day 35 BMAL1 KO hESC-cardiomyocytes by transmission electron microscopy. Mitochondria of BMAL1 KO hESC-cardiomyocytes were extensively elongated, with abnormal cristae structures and an increased mitochondrial area/perimeter ratio (Fig. 5A–C). The most common intra-mitochondrial cristae malformations were circle cristae, partitioning cristae, swollen cristae, and cristolysis (Fig. S6). These ultrastructural mitochondria morphologies were consistent with previous findings (Jacobi et al., 2015; McManus et al., 2019). Immunostaining of BMAL1 KO hESC-cardiomyocytes with adCox8a-RFP showed that mitochondria were highly hyper-fusion compared to those of wildtype hESC-cardiomyocytes (Fig. 5D, and Supplementary Movie 3 and Movie 4). Likewise, expression level of MFN2, the marker of mitochondrial fusion, was significantly increased in BMAL1 KO hESC-cardiomyocytes (Fig. 5E and 5F). Similar findings were also present in BMAL1 KO mice (Fig. 6A–E). Taken together, these results indicated that Bmal1 deficiency led to hyper-fused mitochondria with abnormal cristae in cardiomyocytes.

BMAL1 KO hESC-derived cardiomyocytes exhibited mitochondrial hyperfusion and compromised mitochondrial autophagy. (A) Representative transmission electron micrographs of mitochondria (red arrows) from wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. Mitochondrial enlargement in BMAL1 KO hESC-derived cardiomyocytes was apparent. (B) Quantification of mitochondrial size in wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (C) Quantification of the ratio of mitochondrial area-perimeter indicated mitochondrial fusion level (enlarged and fused mitochondria possess greater area-perimeter ratio) was upregulated in BMAL1 KO hESC-derived cardiomyocytes (n = 40). (D) Representative confocal images showing RFP-tagged mitochondria (white arrows) in wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. Cox8a-RFP adenovirus was used to tag mitochondria. (E) Western blot analysis of level of Mfn2 proteins regulating mitochondrial dynamics. (F) Quantification of Western blot signals in (E) and normalized to the loading control (α-ACTIN). (G) Transmission electron microscopy examining alterations in mitochondrial autophagy (red arrows) in wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (H) Mitophagy was counted in 10 different transmission electron images of wild type and BMAL1 KO hESC-derived cardiomyocytes day 35, respectively. (I) Mitophagy intensity in wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation analyzed by flowcytometry. (J) Mean intensity of BMAL1 KO hESC-derived cardiomyocytes was markedly decreased. (K) Western blot evaluation of protein expression level of SQSTM1 and LC3A/B-II in wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (L) Quantification of Western blot signals in (I) and normalized to the loading control (α-ACTIN). n = 3 for each group. (M) Real-time respiration measurements of wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (N) Statistics of respiration parameters between wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. n = 4 for each group. Data were represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus control by two-tailed Student’s t test

Mitochondria in cardiomyocytes of Bmal1-deficient mice exhibited increased fusion and decreased mitophagy. (A) Significantly enlarged mitochondria were observed in Bmal1-deficient myocardium comparing to those in the wild type group. Arrows indicate mitochondria. (B and C) Mitochondrial size and area-perimeter ratio were carried out for the quantification of mitochondrial fusion level (enlarged and fused mitochondria possess greater area-perimeter ratio). (D) The protein expression level of MFN2 was calculated by western Blotting analysis evaluation (expressed as the ratio of β-ACTIN). (E) Columns in graphs show protein normalized for β-ACTIN. (F) Less mitophagy take place in Bmal1−/− mice myocardium than in wild type control group. (G) Mitophagy were counted in 12 different transmission electron images of Bmal1−/− mice myocardium and wild type control group, respectively. (H and I) The protein expression level of LC3A/BI/IIand BNIP3 were assessed by Western Blotting analysis evaluation (expressed as the ratio of LC3A/BII and BNIP3 to β-ACTIN). Data represented the mean ± SD. *P < 0.05, **P < 0.01 and ****P < 0.0001 versus wide type by two-tailed Student’s t test
A previous study demonstrated that the heart is one of the sturdiest mitophagic organs (Kanevskij et al., 2002). Mitophagy plays a crucial role in mitochondrial quality control, by removing dysfunctional, damaged, or aging mitochondria (Song et al., 2015; Song et al., 2017). Transmission electron microscopy imaging showed decreased mitophagy events in BMAL1 KO hESC-cardiomyocytes (Fig. 5G and 5H). We thus further examined mitophagy intensity in wildtype and BMAL1 KO hESC-cardiomyocytes using Mtphagy Dye (Iwashita et al., 2017). Consistent with transmission electron microscopy imaging, knocking out BMAL1 resulted in a decline in mitophagy intensity (Fig. 5I and 5J). Mitochondrial localization of the autophagy chaperone protein SQSTM1 can mediate autophagy of damaged mitochondria by binding to autophagosomal microtubule associated protein1 light chain 3 (LC3) (Narendra et al., 2010). We found that mitochondrial SQSTM1 and LC3 II protein level were also decreased in BMAL1 KO hESC-cardiomyocytes (Fig. 5K and 5L). Similar results were found in Bmal1 KO mice (Fig. 6F–I,). In addition, mitochondrial autophagy-associated protein BNIP3 expression was reduced in myocardium of Bmal1-deficient mice (Fig. 6H–L). These data indicated that BMAL1 ablation caused mitochondria abnormalities in and reduced mitophagy of cardiomyocytes. We further found that mitochondrial oxidative metabolisms of BMAL1 KO hESC-cardiomyocytes were significantly declined (Fig. 5M and 5N). Overall, our data showed that BMAL1 loss-of-function induced dysfunction and diminished respiration in mitochondria, suggesting that BMAL1 deletion impacted the dynamic remodeling of the mitochondrial network.
BMAL1 regulates mitophagy in cardiomyocytes partially by binding to the E-box elements in the promoter of BNIP3 gene
According to genome-wide transcript profiling, BMAL1 is estimated to target more than 150 sites in the human genome, including all known clock genes and genes regulating metabolism (Hatanaka et al., 2010) . BMAL1 is a core circadian transcription factor and regulates downstream genes by binding to the E-box elements in their promoters (Ueda et al., 2005). The transcription of Bnip3 gene exhibits a diurnal rhythm in C57 BL/6J livers (Jacobi et al., 2015). We also identified several canonical E-box sequences (CANNTG), and one noncanonical E-box sequence (CAGCTT) in the promoter region of Bnip3 gene (Table. 1). In addition, BMAL1 is able to bind to the promoter region of Bnip3 in osteosarcoma according to ChIP-seq analysis (Rey et al., 2016) (Fig. 7A). Since BNIP3 is a critical player in mitophagy by mediating autophagy of mitochondria and is located in myocardium (Azad et al., 2008; Chaanine et al., 2013; Xiao et al., 2018), we further examined BNIP3 protein expression in wild type and BMAL1 KO hESC-cardiomyocytes at day 35. Western blot confirmed that BNIP3 protein level was reduced in BMAL1 KO hESC-cardiomyocytes (Fig. 7B and 7C). The mRNA expression of BNIP3 in BMAL1 KO day 35 hESC-cardiomyocytes was also decreased (Fig. 7D). We therefore hypothesized that BMAL1 is responsible for the transcriptional activation of BNIP3 by binding to the E-box elements within its promoter region in cardiomyocytes. ChIP-qPCR assays showed that there was a BMAL1 binding-site from 0 to 85 bp in the promoter region of BNIP3 gene (Fig. 7E). We further used dual-luciferase reporter assays to evaluate the transcriptional activation of BMAL1 on BNIP3. As shown in Fig. 7F, wild type BNIP3 promoter facilitated highest transcriptional activity to luciferase gene along with the co-transfection of BMAL1 and CLOCK. However, E-box mutant BNIP3 promoter was not able to sufficiently trigger the transcription of reporter gene with the exogenous overexpression of BMAL1 and CLOCK. These results indicated that BMAL1 regulates the transcription of BNIP3 gene via binding with the E-Box element within its promoter region (Fig. 8).

BNIP3 is controlled by circadian gene BMAL1 via transcription regulation. (A) BMAL1 ChIP-seq showed binding signals on the promoter region of Bnip3 in human U2OS cells (CistromeDB: 71023). The arrow indicates transcription start site (TSS) of the Bnip3 gene. (B) Relative expression of Bnip3 was analyzed in wild type and BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation. (C) Western Blot showed that the expression of BNIP3 in BMAL1 KO hESC-derived cardiomyocytes was significantly lower than that in wild type cardiomyocytes. (D) The mRNA levels of BNIP3 in BMAL1 KO hESC-derived cardiomyocytes day 35 post differentiation decreased significantly. (E) ChIP analysis of Bmal1 binding to the E-box region of BNIP3 in hESC-derived cardiomyocytes at day 35 post differentiation. (F) Dual luciferase reporter assays revealed that transcriptional activation of Bnip3 in HEK 293 cells by BMAL1. Graph represented firefly luciferase expression normalized to renilla luciferase for each group. Data were mean ± SD from three biological replicates. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control by two-tailed Student’s t test

The regulatory mechanism of BMAL1 in mitophagy of cardiomyocyte and DCM. Our study demonstrated that BMAL1 is able to bind to the E-box element in the promoter region of BNIP3 gene and specifically control BNIP3 protein expression, hence directly affecting the BNIP3-mediated mitochondria quality control process. The circadian gene Bmal1 is critical in the maintenance of normal mitochondria activities and cardiac function
Discussion
The circadian oscillation, which is an endogenous time-keeping system, plays a key role in a wide range of physical processes by generation of 24-hour circadian rhythms in gene expression. Circadian clock mal-adjustment contributes to the pathology of various disorders, including insomnia (Flynn-Evans et al., 2017), aging (Tevy et al., 2013), cancer (Stevens, 2009), metabolic syndromes (Bray and Young, 2008), immune system imbalance (Deng et al., 2018), and cardiovascular diseases (Nonaka et al., 2001; Young et al., 2014). According to statistical studies, people with long-term irregular life routine have higher risk of hypertension, inflammation, and heart injury, which becomes more common in contemporary society (Morris et al., 2016). A number of studies reported that mutations in core circadian gene BMAL1 or its partner CLOCK tended to develop cardiovascular diseases. One of the studies suggested ClockΔ19 mice displayed cardiac hypertrophy, impaired contractility, and reduced myogenic responsiveness (Alibhai et al., 2017). And, Clock specific mutant in cardiomyocytes resulted in myocardial contractile dysfunction and bradycardia (Bray et al., 2008). Different with the phenotypes of ClockΔ19 mice, loss of Bmal1 gave rise to age-associated dilated cardiomyopathy, as a result of altered titin isoform component, change of MYH gene expression, and disrupted sarcomere structure (Lefta et al., 2012). Moreover, cardiomyocyte-specific Bmal1 KO mice also gradually developed into dilated heart disease with age, partly through depressed glucose utilization as well as other cytologic mechanism related to metabolic malfunction (Young et al., 2014). It should be noted that these are all results from global BMAL1 KO mice. Further study of cardiac-specific BMAL1 KO animals are valuable for clarifying the precise role of BMAL1 in the heart. However, rodents are nocturnal creatures, being active during the nights and sleeping during the day, which means their biorhythmic systems are opposite from human beings. And species difference between humans and mice may also give rise to different mechanisms in circadian system and cardiovascular diseases. It was actually not clear previously how BMAL1 function in human cardiac system.
In this study, we successfully established BMAL1 KO hESC cell lines and derived human BMAL1-deficient cardiomyocytes to mimic heart disorders associated with circadian malfunction (Young et al., 2001; Takeda and Maemura, 2010; Alibhai et al., 2017; Alibhai et al., 2018). In accordance with phenotypes in Bmal1 KO mice, which displayed age-dependent dilated cardiomyopathy, BMAL1-deficient human cardiomyocytes showed characteristics of DCM including enlarged cell area, disrupted myofilament structure, increased apoptosis, attenuated contraction force, and dysfunction of calcium handling. We also found altered electrical conduction in our BMAL1 KO cardiomyocytes as reported before, including deceased beating rates and irregular contractile rhythms. This suggested that, in addition to working cells, conduction-like cells may also be affected by Bmal1 ablation.
In addition to disarrangement of sarcomere structure, morphological abnormalities of mitochondria in BMAL1 KO cardiomyocytes were also observed by transmission electron microscopy, including mitochondrial swelling, sparse cristae, vacuolization, and abnormal elongation. Mitochondria function was also significantly impaired in BMAL1 KO cardiomyocytes. Mitochondria events, including mitochondrial biogenesis, mitophagy, fusion and fission, are inseparably interconnected and play a significant role in the maintenance of normal mitochondria function. Mitochondrial biogenesis increases mitochondrial content levels to enhance the ability of energy metabolism. Fission process mediated by Drp1 and fusion process mediated by Mfn1, Mfn2, and Opa1 allow constantly remodeling of mitochondrial networks. And, mitophagy mediated by Bnip3/NIX in mammalian cells is a selective degradation process responsible for removal of damaged or aging mitochondria (Lee and Yoon, 2016; Vega and Kelly, 2017). Previous studies have shown that the dynamic balances in mitochondria were strictly regulated by biological rhythms. For example, mitochondrial fission-fusion turnover and metabolic flux is controlled by circadian activity of Drp1 (Schmitt et al., 2018). In mice, circadian oscillation of NAD+ cycle prompted mitochondrial oxidative metabolism (Peek et al., 2013). Disturbed expression of circadian genes within cardiomyocytes leaded to structural and functional impairment of mitochondria(Bray et al., 2008). Our data is in accordance with these previous findings and suggested that BMAL1 is critical for maintenance of normal mitochondrial structure and function.
Mitophagy is required not only to clear dysfunctional mitochondria, but also to adjust mitochondrial numbers for cellular energy needs. Disruption of autophagy or mitophagy has proved to be related to several cardiovascular disorders, such as myocardial infarction, cardiomyopathies, atherosclerosis, and cardiotoxicity (Bravo-San Pedro et al., 2017). In mammals, mitophagy process is mediated by NIX and its regulator BNIP3. Like many other BCL-2 proteins, BNIP3 participates in modulating the permeability of the outer mitochondrial membrane by forming homo- or hetero-oligomers, thus contributing to mitophagy. We found that BMAL1 KO led to a reduced BNIP3 protein level and less mitophagosomes in hESC-derived cardiomyocytes. We also confirmed BMAL1 binds to one of the E-box elements in the promoter of Bnip3 gene. Furthermore, Bnip3 transcription was directly regulated by BMAL1. Bmal1 deletion downregulated BNIP3 protein level, which compromised Bnip3-mediated mitophagy. Our data suggested that Bmal1 ablation reduced BNIP3 expression and impaired normal mitochondrial quality control process, which might gradually induced abnormal cardiomyocyte function and phenotypes of dilated cardiomyopathy (Fig. 8). However, there are many idiopathic DCM cases in the clinic and the causes and detailed mechanism of this intractable disease remain unclear (Takahashi, 2017). Whether dysfunction of the circadian rhythms links to certain idiopathic DCM remains an interesting topic to clarify. Our results in this study provided a direct link between core circadian system and the disease of DCM.
Overall, our results showed that the circadian gene BMAL1 plays a critical role in maintaining normal function of cardiomyocytes and the heart. Dysfunctional BMAL1 mutant protein may eventually lead to DCM in patients. It worth to check for BMAL1 gene mutations using the whole genome sequencing technique and this could contribute to a clearer etiology diagnosis for clinical idiopathic DCM patients in the future.
Materials and methods
Human embryonic stem cells culture and differentiation
The human embryonic stem cell line H7 was cultured in mTeSR (STEMCELL TECHNOLOGIES, #85851 and #85852). HESC-H7 cells were isolated from matrigel by accutase digestion and reseeded on the new matrigel at 2–6 × 105 cells per 12 well in mTeSR. We utilized the protocols originally developed by Lian et al (Lian et al., 2012) to achieve high-purity cardiomyocytes in vitro. Briefly, on the day of cardiac induction (day 0), 6 μmol/L CHIR99021 (http://www.selleckchem.com) was added for 2 days in 1 mL of insulin-minus RPMI + B27 medium. At day 2, cells were recovered in insulin-minus RPMI + B27 for 24 h. At day 3, cells were treated with 5 μmol/L IWR-1 (Sigma) for 2 days to activate Wnt signaling. At day 5, cells were finally switched to RPMI + B27 plus insulin medium. Beating cells were observed at day 7–8 after differentiation. The mature CMs were cultured in in DMEM containing 10% fetal bovine-serum. All these cells needed to be cultured on Matrigel-coated plates (corning #354248) and maintained at 37 °C with 5% CO2.
CRISPR dual nickase editing
BMAL1 gene mutation was corrected back to human embryonic stem cell line 7 (hESC-H7), using low off-target CRISPR dual nickase plasmid (PX462). Guide-RNA sequences (gRNA-A: 5′:AATCCATCTGCTGCCTAATCAGG; gRNA-B: 5′:TTGTCGTAGGATGTGACCGAGGG) were designed at http://crispr.mit.edu/ and cloned as described at http://www.genome-engineering.org/crispr/. The editing efficiencies of CRISPR plasmids were validated with 293T cells using Sanger sequencing. 4.6 × 105 cells of hESC-H7 were electroporated using Neon Transfection System(Thermo Fisher Scientific) with 3 μg of the most efficient plasmids. Successful electroporation cells were puromycin (0.22 μg/mL) selected after 1 day. Single selected positive clones were picked and verified with Sanger sequencing using TA cloning technology to measure allelic frequency. We derived BMAL1 gene mutation cell line during the CRISPR-editing process.
EB formation
Human EB were generated as described previously(Ang et al., 2016). In ultra-low attachment plates (Corning), embryoid bodies can be formed in specific medium (DMEM, 20% FBS, 1% non-essential amino acid, 100 μmol/L BME, 1mmol/L L-glutamine) for 10 days followed by quantitative real-time PCR for evaluating gene expression of endoderm, mesoderm, ectoderm, cardiac progenitor, and mature cardiomyocytes.
Animals
All animal procedures conformed to the National Institutes of Health Guidelines for the Use of Laboratory Animals were approved by Fudan University’s Animal Care and Use Committee guidelines. The germLine Bmal1−/− mice were imported from Jackson Lab (Bunger et al., 2000). Mice were kept on a 14-h:10-h light-dark schedule and had access to food and water. Mice were anaesthetized using 1%–2% isoflurane and euthanized by cervical dislocation.
Flow cytometry
The beating cardiomyocytes were digested with 1 mg/mL collagenase I (Sigma) from petri dishes, and followed by 0.25% Trypsin/EDTA (Gibco) to further isolate the cells. Then the isolated cells were fixed and infiltrated by permeabilization solution (BD Biosciences). The cells were stained for cardiac troponin T (cTnT, Thermo Scientific) for 2 h at 4 °C. The cells were incubated with the secondary antibody for 1 h before washing cells with PBS. Finally, the fluorescence-activated cell detection was performed on the FACSCalibur (BD Biosciences). The antibody information could be found in Table 2. Data was analyzed using FlowJo software.
Mitophagy analysis
Cardiomyocyte mitophagy was detected by Mitophagy Detection Kit (Dojindo). Cardiomyocytes were cultured in 37 °C with 100 nmol/L Mtphagy dyes for 30 min. Cells were wished 2 times with PBS, and then treated with 10 µmol/L CCCP for 8 h prior to induce mitophagy. Cells were suspended into single cells and subsequently subjected to FCM to determine the flourecence intensity at 700 nm.
Mitochondrial morphology
The mitochondria of cardiomyocytes were visualized using adenovirus carrying Cox8a-RFP,which has sequences of mitochondrial matrix localization derived from Cox8a. The differentiated 30-day CMs were digested into single cells and were inoculated in a laser confocal dish at a suitable density for four days with DMEM + 10% FBS. The CMs were then cultured in DMEM + 10% FBS containing Cox8a-RFP for 24 h. After 24 h, the original liquid was discarded and replaced with DMEM + 10% FBS. The mitochondria of cardiomyocytes were photographed within 24 h.
Seahorse bioenergetics analyses
The mitochondrial respiratory capacity of cardiomyocytes was determined by Seahorse XFe96 Analyzer (Agilent Technologies). The differentiated 34-day CMs were plated in 96-well plates at 2 × 104 cells/well at 37 °C for 24 h. The cells were incubated in DMEM containing 0.584 g/L L-glutamine and 5.5 mmol/L D-glucose for 1 h at 37 °C before analysis. 0.5 mg/mL oligomycin, 1 mmol/L FCCP, 1 mmol/L rotenone and 2 mmol/L antimycin A were sequentially added into each assigned well at indicated time point.
Calcium transient
The calcium flux was determined by laser confocal Ca2+ imaging technique to evaluate intracellular calcium transient. The autonomous beating cardiomyocytes labeled by 5 μmol/L Cal-520AM and 0.02% Pluronic F-127 (AAT Bioquest) were detected by LSM-710 laser scanning confocal microscope (Carl Zeiss) with a detection time of 10 milliseconds under the line scanning mode in Zen software. Zen software was used to record calcium transient and MATLAB software (MathWorks) was performed to analysis calcium imaging signals.
Microelectrode array (MEAs) analysis
The cardiomyocytes were digested at 32 days after induction and then inoculated to the surface of the coated MEAs probe. The myocardial cell field potential waveforms were recorded using a MEA2100 system at 35 days post induction. A bright-field signal video was recorded using a heated MC-rack to ensure that the cells were maintained at 37 °C. The data of field potential waveforms were quantitatively analyzed by using the Spike2 7.19 software (CED, UK).
Contraction force measurements
2–3 days before the measurement, the cardiomyocytes were digested and planted on a confocal dish coated with Matrigel. The contractility of cardiomyocytes was detected by using video-based motion edge detection system. Through a Zeiss CFM-500 inversion fluorescence microscope and Video Sarcomere Length software (900B: VSL, Aurora Scientific), beating cardiomyocytes were clearly projected onto the screen. The spontaneous contraction traces of cardiomyocytes were recorded by using data analysis software (FelixGX, PTI). During the testing process, these cells were kept in 37 °C using heating equipment, and only the cardiomyocytes of the digestive single were analyzed.
Plasmid construction
The pGL3 Basic plasmid was digested with two restriction endonucleases Hind III and Nhe I. Subsequently, the 2,000 bp fragment, the 363 bp fragment and the mutated 362 bp fragment of the 5′ upstream sequence of the Bnip3 gene were cloned into the pGL3 basic vector (Promega, Madison, WI). The open reading frame (ORF) of the Clock gene was cloned to pcDNA 3.1 by NotI and Apa 1 endonuclease. The ORF of the BMAL1 gene was cloned to pCMV using BamHI and EcoRI. The plasmid constructed above was added to the DH5α to be transformed and cultured on ampicillin agar plate. The positive clones were selected for amplification, and the plasmid was extracted by using MN large pumping kit, and the concentration was measured. The existence of the CLOCK, BMAL1 and BNIP3 genes were confirmed by sequencing following plasmid extraction.
Dual luciferase reporter assay
The following plasmids were instantly transfected into the HEK293T cells in the 6-well plates by means of LipofectamineTM 2000 regent (Invitrogen,USA) according different distribution combinations. These plasmids included 500 ng of pGL3-BNIP3 plasmid, 100 ng of pRL-TK plasmid, 500 ng pcDNA 3.1, 500 ng pCMV, 500 ng BMAL1 plasmid, and 500 ng CLOCK plasmid. The cells were harvested after 24 h of transfection and the activity of firefly and renilla luciferase was detected by the Dual Luciferase Reporter Gene Assay Kit (Beyotime, China). The firefly luciferase activity was corrected by the renilla luciferase activity.
ChIP
ChIP was performed as described previously (Lan et al., 2007). The whole cell extracts were sonicated and immunoprecipitated with antibody of BMAL1 (CST). Real-time quantitative PCR was used to analyze the precipitated DNA samples.
Statistical analysis
For statistical analysis, all data were expressed using mean ± SD. Student’s t test was used for comparison between the two groups. The statistical significance of differences between the experimental groups and several timepoints was determined using 2-way ANOVA with post-hoc test. P-values less than 0.05 was considered to be statistically different. All experimental results were analyzed using SPASS 17.0. GraphPad Prism 5 and Adobe illustrator software were used for analysis and graphing.
REFERENCES
Alibhai FJ, LaMarre J, Reitz CJ, Tsimakouridze EV, Kroetsch JT, Bolz SS, Shulman A, Steinberg S, Burris TP, Oudit GY et al (2017) Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. J Mol Cell Cardiol 105:24–37
Alibhai FJ, Reitz CJ, Peppler WT, Basu P, Sheppard P, Choleris E, Bakovic M, Martino TA (2018) Female ClockDelta19/Delta19 mice are protected from the development of age-dependent cardiomyopathy. Cardiovasc Res 114:259–271
Ang YS, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu JD, Spencer CI et al (2016) Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell 167(1734–1749):e1722
Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, Gibson SB (2008) Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 4:195–204
Balsalobre A, Damiola F, Schibler U (1998) A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93:929–937
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415:198–205
Bravo-San Pedro JM, Kroemer G, Galluzzi L (2017) Autophagy and mitophagy in cardiovascular disease. Circ Res 120:1812–1824
Bray MS, Young ME (2008) Diurnal variations in myocardial metabolism. Cardiovasc Res 79:228–237
Bray MS, Shaw CA, Moore MW, Garcia RA, Zanquetta MM, Durgan DJ, Jeong WJ, Tsai JY, Bugger H, Zhang D et al (2008) Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol 294:H1036–1047
Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA (2000) Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103:1009–1017
Chaanine AH, Gordon RE, Kohlbrenner E, Benard L, Jeong D, Hajjar RJ (2013) Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ Heart Fail 6:572–583
Cheng H, Lederer WJ, Cannell MB (1993) Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262:740–744
Deng W, Zhu S, Zeng L, Liu J, Kang R, Yang M, Cao L, Wang H, Billiar TR, Jiang J et al (2018) The circadian clock controls immune checkpoint pathway in sepsis. Cell Rep 24:366–378
Dierickx P, Van Laake LW, Geijsen N (2018) Circadian clocks: from stem cells to tissue homeostasis and regeneration. EMBO Rep 19:18–28
Eisner DA, Caldwell JL, Kistamas K, Trafford AW (2017) Calcium and excitation-contraction coupling in the heart. Circ Res 121:181–195
Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De Girolami U et al (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 341:1715–1724
Flynn-Evans EE, Shekleton JA, Miller B, Epstein LJ, Kirsch D, Brogna LA, Burke LM, Bremer E, Murray JM, Gehrman P et al (2017) Circadian phase and phase angle disorders in primary insomnia. Sleep. https://doi.org/10.1093/sleep/zsx16
Friederich MW, Timal S, Powell CA, Dallabona C, Kurolap A, Palacios-Zambrano S, Bratkovic D, Derks TGJ, Bick D, Bouman K et al (2018) Pathogenic variants in glutamyl-tRNA(Gln) amidotransferase subunits cause a lethal mitochondrial cardiomyopathy disorder. Nat Commun 9:4065
Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier H, Labeit S et al (2002) Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 30:201–204
Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ (1997) Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276:800–806
Hatanaka F, Matsubara C, Myung J, Yoritaka T, Kamimura N, Tsutsumi S, Kanai A, Suzuki Y, Sassone-Corsi P, Aburatani H et al (2010) Genome-wide profiling of the core clock protein BMAL1 targets reveals a strict relationship with metabolism. Mol Cell Biol 30:5636–5648
Hoover DB, Ganote CE, Ferguson SM, Blakely RD, Parsons RL (2004) Localization of cholinergic innervation in guinea pig heart by immunohistochemistry for high-affinity choline transporters. Cardiovasc Res 62:112–121
Huang N, Chelliah Y, Shan Y, Taylor CA, Yoo SH, Partch C, Green CB, Zhang H, Takahashi JS (2012) Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science 337:189–194
Iwashita H, Torii S, Nagahora N, Ishiyama M, Shioji K, Sasamoto K, Shimizu S, Okuma K (2017) Live cell imaging of mitochondrial autophagy with a novel fluorescent small molecule. ACS Chem Biol 12:2546–2551
Jacobi D, Liu S, Burkewitz K, Kory N, Knudsen NH, Alexander RK, Unluturk U, Li X, Kong X, Hyde AL et al (2015) Hepatic Bmal1 regulates rhythmic mitochondrial dynamics and promotes metabolic fitness. Cell Metab 22:709–720
Kanevskij M, Taimor G, Schafer M, Piper HM, Schluter KD (2002) Neuropeptide Y modifies the hypertrophic response of adult ventricular cardiomyocytes to norepinephrine. Cardiovasc Res 53:879–887
Kang TH, Reardon JT, Kemp M, Sancar A (2009) Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci U S A 106:2864–2867
Kreipke RE, Birren SJ (2015) Innervating sympathetic neurons regulate heart size and the timing of cardiomyocyte cell cycle withdrawal. J Physiol 593:5057–5073
Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y (2007) Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448:718–722
Lee H, Yoon Y (2016) Mitochondrial fission and fusion. Biochem Soc Trans 44:1725–1735
Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA (2012) Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol 303:H475–485
Li L, Desantiago J, Chu G, Kranias EG, Bers DM (2000) Phosphorylation of phospholamban and troponin I in beta-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol Heart Circ Physiol 278:H769–779
Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP (2012) Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A 109:E1848–1857
Maemura K, Layne MD, Watanabe M, Perrell MA, Nagai R, Lee ME (2001) Molecular mechanisms of morning onset of myocardial infarction. Ann N Y Acad Sci 947:398–402
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart Association et al (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113:1807–1816
Maury E, Hong HK, Bass J (2014) Circadian disruption in the pathogenesis of metabolic syndrome. Diabetes Metab 40:338–346
McManus MJ, Picard M, Chen HW, De Haas HJ, Potluri P, Leipzig J, Towheed A, Angelin A, Sengupta P, Morrow RM et al (2019) Mitochondrial DNA variation dictates expressivity and progression of nuclear DNA mutations causing cardiomyopathy. Cell Metab 29(78–90):e75
McNamara P, Seo SB, Rudic RD, Sehgal A, Chakravarti D, FitzGerald GA (2001) Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature: a humoral mechanism to reset a peripheral clock. Cell 105:877–889
Morris CJ, Purvis TE, Hu K, Scheer FA (2016) Circadian misalignment increases cardiovascular disease risk factors in humans. Proc Natl Acad Sci U S A 113:E1402–1411
Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, Rinfret S, Schiffrin EL, Eisenberg MJ (2010) The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol 56:1113–1132
Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN et al (2016) Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ Res 118:1960–1991
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–1106
Nonaka H, Emoto N, Ikeda K, Fukuya H, Rohman MS, Raharjo SB, Yagita K, Okamura H, Yokoyama M (2001) Angiotensin II induces circadian gene expression of clock genes in cultured vascular smooth muscle cells. Circulation 104:1746–1748
O’Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, Cotecchia S, Rokosh DG, Grossman W, Foster E et al (2003) The alpha(1A/C)- and alpha(1B)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest 111:1783–1791
Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB et al (2008) Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 156:161–169
Partch CL, Green CB, Takahashi JS (2014) Molecular architecture of the mammalian circadian clock. Trends Cell Biol 24:90–99
Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C et al (2013) Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342:1243417
Piacentino V 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR (2003) Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res 92:651–658
Pyun JH, Kim HJ, Jeong MH, Ahn BY, Vuong TA, Lee DI, Choi S, Koo SH, Cho H, Kang JS (2018) Cardiac specific PRMT1 ablation causes heart failure through CaMKII dysregulation. Nat Commun 9:5107
Rey G, Valekunja UK, Feeney KA, Wulund L, Milev NB, Stangherlin A, Ansel-Bollepalli L, Velagapudi V, O’Neill JS, Reddy AB (2016) The Pentose Phosphate Pathway Regulates the Circadian Clock. Cell Metab 24:462–473
Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE (2003) Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299:1410–1413
Schmitt K, Grimm A, Dallmann R, Oettinghaus B, Restelli LM, Witzig M, Ishihara N, Mihara K, Ripperger JA, Albrecht U et al (2018) Circadian control of DRP1 activity regulates mitochondrial dynamics and bioenergetics. Cell Metab 27(657–666):e655
Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR (2010) Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest 120:4388–4398
Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21:273–286
Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd (2017) Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab 26(872–883):e875
Stevens RG (2009) Light-at-night, circadian disruption and breast cancer: assessment of existing evidence. Int J Epidemiol 38:963–970
Takahashi JS (2017) Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet 18:164–179
Takeda N, Maemura K (2010) Circadian clock and vascular disease. Hypertens Res 33:645–651
Tevy MF, Giebultowicz J, Pincus Z, Mazzoccoli G, Vinciguerra M (2013) Aging signaling pathways and circadian clock-dependent metabolic derangements. Trends Endocrinol Metab 24:229–237
Ueda HR, Hayashi S, Chen W, Sano M, Machida M, Shigeyoshi Y, Iino M, Hashimoto S (2005) System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat Genet 37:187–192
Vega RB, Kelly DP (2017) Cardiac nuclear receptors: architects of mitochondrial structure and function. J Clin Invest 127:1155–1164
Vikhorev PG, Smoktunowicz N, Munster AB, Copeland O, Kostin S, Montgiraud C, Messer AE, Toliat MR, Li A, Dos Remedios CG et al (2017) Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci Rep 7:14829
Wallace DC (2013) A mitochondrial bioenergetic etiology of disease. J Clin Invest 123:1405–1412
Xiao C, Wang K, Xu Y, Hu H, Zhang N, Wang Y, Zhong Z, Zhao J, Li Q, Zhu D et al (2018) Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of miR-125b. Circ Res 123:564–578
Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M et al (2000) Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation 102:2131–2136
Young ME, Razeghi P, Cedars AM, Guthrie PH, Taegtmeyer H (2001) Intrinsic diurnal variations in cardiac metabolism and contractile function. Circ Res 89:1199–1208
Young ME, Brewer RA, Peliciari-Garcia RA, Collins HE, He L, Birky TL, Peden BW, Thompson EG, Ammons BJ, Bray MS et al (2014) Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms 29:257–276
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (NSFC No. 81322003, No. 31571527, N.S.; No. 31501098, Q.L.; No. 81500241, C.X.; No. 81870600, C.L.; No. 81570771 and 31871189, RZ.Q.), the Science and Technology Commission of Shanghai Municipality (No. 17XD1400300, No. 17JC1400200), the National Key R&D Program of China 2018YFC2000202, and the Haiju program of National Children’s Medical Center EK1125180102. We apologize to people whose work was relevant to but not cited in this study due to limited space.
ABBREVIATIONS
BMAL1, brain and musle ARNT-like 1; Bnip3, BCL2/adenovirus E1B interacting protein 3; CMs, cardiomyocytes; DCM, cTnT, cardiac muscle Troponin T;dilated cardiomyopathy; EB,embryoid body; hESCs, human embryonic stem cells; IVSs, interventricular septal thickness at end systole; LVIDs, left ventricular internal dimension at end systole; LVPWs, left ventricular posterior wall thickness at end systole; MEAs, multi-electrode arrays; TEM, transmission electron microscopy; WGA, wheat germ agglutinin
COMPLIANCE WITH ETHICS GUIDELINES
Ermin Li, Xiuya Li, Jie Huang, Chen Xu, Qianqian Liang, Kehan Ren, Aobing Bai, Chao Lu, Ruizhe Qian, and Ning Sun declare that they have no conflict of interest.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (Fudan University, China) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 2 (MP4 2997 kb)
Supplementary material 3 (MP4 1999 kb)
Supplementary material 4 (MP4 495 kb)
Supplementary material 5 (MP4 581 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, E., Li, X., Huang, J. et al. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell 11, 661–679 (2020). https://doi.org/10.1007/s13238-020-00713-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-020-00713-x