
ABSTRACT
As of May 1, 2017, 74 antibody-based molecules have been approved by a regulatory authority in a major market. Additionally, there are 70 and 575 antibody-based molecules in phase III and phase I/II clinical trials, respectively. These total 719 antibody-based clinical stage molecules include 493 naked IgGs, 87 antibody-drug conjugates, 61 bispecific antibodies, 37 total Fc fusion proteins, 17 radioimmunoglobulins, 13 antibody fragments, and 11 immunocytokines. New uses for these antibodies are being discovered each year. For oncology, many of the exciting new approaches involve antibody modulation of T-cells. There are over 80 antibodies in clinical trials targeting T cell checkpoints, 26 T-cell-redirected bispecific antibodies, and 145 chimeric antigen receptor (CAR) cell-based candidates (all currently in phase I or II clinical trials), totaling more than 250 T cell interacting clinical stage antibody-based candidates. Finally, significant progress has been made recently on routes of delivery, including delivery of proteins across the blood-brain barrier, oral delivery to the gut, delivery to the cellular cytosol, and gene- and viral-based delivery of antibodies. Thus, there are currently at least 864 antibody-based clinical stage molecules or cells, with incredible diversity in how they are constructed and what activities they impart. These are followed by a next wave of novel molecules, approaches, and new methods and routes of delivery, demonstrating that the field of antibody-based biologics is very innovative and diverse in its approaches to fulfill their promise to treat unmet medical needs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
This year, 2017, marks the 20th anniversary of the approval by the United States Food and Drug Administration (US FDA) of Rituxan® (rituximab) and Zenapax® (daclizumab), for treatment of B cell malignancies and for use to suppress organ rejection in renal transplants, respectively (Table 1). While two antibodies had previously been approved by the FDA (Table 1), the approval of Rituxan® and Zenapax® in 1997 was a watershed moment in the history of monoclonal antibody (mAb) therapeutics. The reasons are very different for each molecule. Rituxan® has become both a huge medical and commercial success, with indications in B cell malignancies as well as in the treatment of rheumatoid arthritis (RA) (Storz, 2014). Rituxan® is currently the fourth best-selling innovative drug of any kind with 2016 worldwide sales of $8,354 MM (Table 2), about 85% of those sales coming in cancer indications and the other 15% from sales for treatment of RA (La Merie Publishing, 2017). Including Rituxan®, seven of the top ten selling innovative drugs in the world in 2016 were proteins, six of which were antibody- related molecules (Table 2). Zenapax®, on the other hand, was the first humanized antibody to be FDA approved but it never achieved significant commercial success and was eventually withdrawn from the market in 2009. Daclizumab, however, has been approved recently under the tradename Zinbryta® for treatment of relapsing forms of multiple sclerosis (MS).
To date, 74 unique, innovative antibodies and Fc fusion proteins have been approved for treatment of diseases in at least one major market (i.e., US, EU, Japan) (Table 1). Of these, seven have been withdrawn from marketing either due to lack of efficacy, poor toxicity to efficacy profiles, or lack of market interest (Table 1). Of the 74 approved antibody-based molecules, five contain completely murine sequences, nine are mouse-human chimeric antibodies, 26 are humanized, 23 are human antibodies, and 11 are Fc fusions (Table 1). Of the 23 fully human antibodies, 17 are derived from transgenic “humanized” mice and six are derived from human antibody phage display libraries (Table 1). Eight of the Fc fusions are Fc-protein fusions, two are Fc-peptide fusions, and one is an Fc-protein fusion with a tissue-targeting peptide fused to it.
Currently, there are 70 phase III clinical stage candidates, as well as 575 known phase I or phase II antibody-based clinical candidates (Table 3). Thus, as of May 1, 2017, there are at least 719 known antibody and Fc fusion protein clinical-stage candidates (Table 3). Of these, 493 are “naked” IgGs, 13 are “naked” antibody fragments (in both cases, “naked” refers to antibodies that are not antibody-drug conjugates [ADCs], bispecific antibodies, radioimmunotherapeutics, or immunocytokines), 87 are ADCs, 61 are bispecific antibodies, 37 are Fc fusion proteins, 17 are conjugated with radioisotopes either as therapeutics or imaging agents, and 11 are immunocytokines (Table 3 and Fig. 1). It is notable that, with the exception of Fc fusion proteins, most of the non-“naked” antibodies are skewed towards the phase I/II clinical stages, likely due to the more recent development of the various innovative technologies incorporated into those molecules (Table 3).

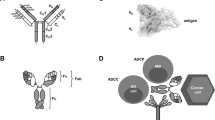
Cartoons of molecules and constructs discussed. (A) IgG monoclonal antibody; (B) FAb fragment; (C) Single chain fragment, variable (scFv); (D) Heterodimeric IgG-based bivalent, bispecific antibody; (E) scFv-based bispecific antibody such as a BiTE (“bispecific T-cell engager”); (F) IgG-scFv-based tetravalent, bispecific antibody; (G) Tetravalent scFv-based antibody called TandAb; (H) IgG-based Immunocytokine (cytokine is denoted by green oval); (I) Tandem scFv-immunocytokine (cytokine is denoted by green oval); (J) Fc-peptide fusion (peptides denoted by squiggled lines); (K) Fc-protein fusion (protein denoted by gray oval); (L) Antibody drug conjugate with three parts (antibody, linker, cytotoxic drug); (M) Chimeric antigen receptor (CAR)-T based antibody (scFvs on surface of recombinant T cell; examples of intracellular domains noted in box)
In addition to these protein antibody-derived clinical stage molecules, there are 145 documented phase I or II clinical stage chimeric antigen receptor (CAR)-T cell or natural killer (NK) cell candidates that incorporate antibodies as their CARs (Table 3 and Fig. 1). Thus, there are at least 864 protein and cell based antibody-derived constructs either approved for medical use or being evaluated for their safety and efficacy in clinical trials.
ANTIBODY TARGETS
The 864 unique antibody-based molecules/cells in development or approved for therapeutic use (Table 3) target 328 unique antigens (Table 4). Because several targets are important for multiple disease areas (e.g., vascular endothelial growth factor [VEGF] as a significant target in both oncology and ophthalmology indications), there are more uses listed than antibodies. Thus, 864 unique molecules are used in 884 different major therapeutic area indications (Table 4), and the 328 unique targets are distributed amongst 351 major uses (Table 5).
About 62% of these protein and recombinant cell-based candidates are directed against targets in oncology (Table 4). Not surprisingly, all 145 of the current CAR-T and CAR-NK candidates are in clinical evaluation for cancer indications. There are, however, preclinical efforts to generate CAR-T cells against viruses and virus-infected cell targets (Sahu et al., 2013; Liu et al., 2015; Hale et al., 2017), so this may change in the near future. Another 19% of the clinical candidates are directed against targets in the immunology therapeutic area (including autoimmune and asthma, but excluding MS) (Table 4). The remaining ca. 19% of antibody-based proteins are divided amongst other therapeutic areas, including cardiovascular and metabolism, neurobiology, bone and muscle disorders, blood disorders, and infectious diseases.
Of the 351 different uses for targets, 222 (~63%) are single-pass membrane bound proteins or cell-bound proteins (e.g., ERBB2 [erb-b2 receptor tyrosine kinase 2; aka Her2], EGFR [epithelial growth factor receptor], ERBB3 [erb-b3 receptor tyrosine kinase 3; aka Her3], MS4A1 [CD20]). Another 12 (~3.4%) are G-coupled protein receptors (GPCRs; e.g., CCR4 [C-C motif chemokine receptor 4], CCR5 [C-C motif chemokine receptor 5], CXCR4 [C-X-C motif chemokine receptor 4]) or other multi-pass (e.g., CD47, STEAP [six-transmembrane epithelial antigen of the prostate] family members) cell surface targets. Additionally, 102 (~29%) are soluble targets (e.g., TNF [tumor necrosis factor-alpha, TNF-α], IL6 [interleukin-6, IL-6], VEGFA [vascular endothelial growth factor A]), and 15 (~4.3%) are infectious disease targets (e.g., respiratory syncytial virus [RSV]-F protein, Bacillus anthracis protective antigen [PA] toxin component, influenza hemagglutinin 2 [HA2; stalk portion], human immunodeficiency virus [HIV] envelop protein gp120) (Table 5).
Cell surface targets in oncology tend to fall into three categories. The first category, which includes about 90 receptors (e.g., CD19, CD20, EPCAM [epithelial cell adhesion molecule, EpCAM], CEACAM5 [carcinoembryonic antigen related cell adhesion molecule 5], MUC1 [mucin 1, cell surface associated]), are essentially “postal addresses” to which killing mechanisms can be targeted directly. These killing mechanisms can include, either individually or in combinations, antibody-dependent cellular cytotoxicity (ADCC) (Ochoa et al., 2017), antibody-dependent cellular phagocytosis (ADCP) (Shi et al., 2015), complement-dependent cytotoxicity (CDC) (Taylor and Lindorfer, 2016), antibody-drug conjugates (ADC) (Tsuchikama and An, 2016; Beck et al., 2017), antibody-induced apoptosis (Sun et al., 2017; Wang et al., 2017), antibody-induced, non-apoptotic programmed cell death (Alduaij et al., 2011), bispecific antibody-redirected killer T or NK cells (Lum and Thakur, 2011; Satta et al., 2013; Suzuki et al., 2015), or CAR-T/CAR-NK cells (Ruella and Gill, 2015; Ruella and June, 2016; Smith et al., 2016). The second group, which overlaps with the first group, are receptors which may be targeted to block ligand binding and signal transduction (Esparis-Ogando et al., 2016; Zhang and Zhang, 2016). The final category are checkpoint modulators, either to block T cell inhibitory pathways or to directly stimulate T or NK cells or macrophages. There are about 20 T-cell related oncology targets in this category.
Of the 328 unique targets for antibody-based drug candidates, the most widely targeted antigen is CD19, which is recognized by 64 clinical candidates, 53 of which are CARs (Table 6). The second most targeted protein is CD3E, found in 32 clinical stage or approved molecules, of which 26 are T cell-redirecting bispecific antibody candidates (Table 6). Thus, the two top targets, CD19 and CD3E, are responsible for the engineered retargeting of T cells, either as CAR-T cells (Ruella and Gill, 2015; Ruella and June, 2016; Smith et al., 2016) or T-cell redirecting bispecific antibodies (Lum and Thakur, 2011; Satta et al., 2013; Suzuki et al., 2015), to kill cancer cells. Of the non-T-cell related targets, the proteins currently most widely targeted are ERBB2 (HER2), EGFR, MS4A1 (CD20), CD22, PDCD1 (PD-1), MSLN (mesothelin), and ERBB3 (Her3), all for cancer indications. The Th17 cytokine, IL17A, to which 14 antibody-related biologics are directed, is currently the top non-oncology target (Table 6). There are 382 unique molecules or recombinant CARs directed against the top 29 targets shown in Table 6, representing about 44% of all of the clinical stage or approved antibody-based molecules/cells; the remaining 482 (~56%) candidates target the remaining 299 unique targets.
The 74 approved mAbs and Fc fusion proteins are directed against 39 unique targets, with TNF (TNF-α) and MS4A1 (CD20) being the most widely targeted, with five antibody-based molecules each (Table 1). The five most valuable targets for approved mAbs and Fc fusion proteins are TNF (TNF-α), VEGF, ERBB2 (HER2), MS4A1 (CD20), and PDCD1 (PD-1) (Table 7). Antibodies against the first four of these targets were approved more than ten years ago, so the market value has built up over time. Remarkably, however, the anti-PD-1 antibodies, Keytruda® and Opdivo®, were approved 2014, making PDCD1 (PD-1) a very fast rising target of value (Table 7). The top ten antibody-based therapeutic targets (Table 7) comprise 85% of the value of the total 39 targets, with the anti-TNF molecules leading the way with a market share of 36% (Table 7).
Based on 2016 sales figures, recombinant proteins comprised seven of the top 10 best selling drugs worldwide (Table 2). Of these seven proteins, five (Humira®, Rituxan®, Remicade®, Avastin®, Herceptin®) are mAbs and one (Enbrel®) is an Fc fusion protein (Table 2). Finally, since January 2014 (the past 3.3 years), antibodies and Fc fusion proteins have comprised 24% (29/121) of innovative United States Food and Drug Administration (US FDA) drug approvals (Fig. 2). This represents the greatest percentage ever since the beginning of the antibody era. Thus, it is clear that mAbs and Fc fusion proteins are making an enormous impact on the pharmaceutical industry, both as novel approaches to treat difficult diseases and meet unmet medical needs, as well as providing an exciting new growth area for the industry.

Small molecule drugs (blue lines), biologics including vaccines (green lines), and monoclonal antibodies/Fc fusion proteins (red lines) approved by the United States Food and Drug Administration from 1997 to May 1, 2017. This information was sourced and extracted from the US FDA website (https://www.fda.gov/drugs/developmentapprovalprocess/druginnovation/ucm537040.htm)
BRIEF OVERVIEW OF ANTIBODY ENGINEERING
Human IgGs have been engineered in a multitude of ways to generate different effects (Strohl and Strohl, 2012), as shown in Table 8. In the earlier days of antibody engineering, the focus was on manipulation of the variable regions to humanize and affinity-mature antibodies, or to generate different types of antibody fragments such as scFvs (Bird et al., 1988; Huston et al., 1988), diabodies (Holliger et al., 1993), PEGylated Fabs (Choy et al., 2002), TandAbs (Kipriyanov et al., 1999), and domain antibodies (Ward et al., 1989). The next wave of antibody engineering focused more on the generation and application of “fit for purpose” antibodies (Strohl, 2011) with tuned Fc functions such as increased ADCC, ADCP, and CDC (Strohl and Strohl, 2012; Brezski and Georgiou, 2016; Sondermann and Szymkowski, 2016; Barnhart and Quigley, 2017), or muted or silenced Fc functions (Labrijn et al., 2008; Vafa et al., 2013; Schlothauer et al., 2016; Lo et al., 2017; Borrok et al., 2017). These functions have been approached using both glyco-engineering strategies such as low or no fucosylation for higher FcγRIIIa binding and increased ADCC (Shields et al., 2002; Ferrara et al., 2006; Malphettes et al., 2010; Golay et al., 2013), higher levels of sialylation for dampened immune responses (Anthony and Ravetch, 2010), or non-glycosylated antibodies for partly subdued Fc effector function (Walker et al., 1989; Nesspor et al., 2012). These and more Fc effector modulations can also be generated with amino acid changes in the lower hinge and Fc (Shields et al., 2001; Lazar et al., 2006; Strohl, 2009; Strohl and Strohl, 2012; Vafa et al., 2013; Brezski and Georgiou, 2016; Sondermann and Szymkowski, 2016; Barnhart and Quigley, 2017).
There are currently three approved antibody-based molecules with modified Fc functionality. These include the CTLA4-Fc fusion proteins, abatacept (Orencia®) and belatacept (Nujolix®), both of which have modified hinges to reduce Fc functionality (Davis et al., 2007), and the humanized anti-C5 mAb, eculizumab (Soliris®), which has an IgG2/IgG4 hybrid Fc to reduce Fc functionality (Rother et al., 2007). All three of these molecules bind to immune system components and the muted Fc design was intended to increase the safety margin.
Additionally, two glyco-engineered antibodies with improved ADCC activities have been approved in at least one major market. The first, mogamulizumab (Poteligeo®), is an afucosylated anti-CCR4 mAb approved in Japan for adult T-cell leukemia/lymphoma that is produced by a cell line with a mutation in the FUT8 (α-1,6-fucosyltransferase) gene (Potelligent® technology from BioWa; Yamane-Ohnuki et al., 2004; Kanda et al., 2006; Malphettes et al., 2010). The second, obinutuzumab (Gazyva®), an anti-MS4A1 (CD20) mAb with low fucose content, has been approved for treatment of chronic lymphocytic leukemia (CLL) (Golay et al, 2013). The low fucose of obinutuzumab is due to the addition to the producing cell line of a β-1,4-N-acetylglucosaminyl-transferase III (GntIII) gene which adds the bisecting N- acetylglucosamine (GlcNAc) that interferes with fucosylation (Glycart GlycoMab® technology acquired by Roche in 2005) (Ferrara et al., 2006). Moreover, there are four glycoengineered (low or no fucose) and one aglycosyl-IgG phase III clinical candidates in the late stage clinical pipeline.
Currently there are no approved IgGs with amino acid-modified increased Fc effector function, although there are two such Fc-modified, increased Fc effector function IgGs in late stage clinical trials, the anti-CD19 mAb, Mor208 (Morphosys, Xencor), in phase II/III clinical trials for treatment of B cell malignancies (NCT02763319), and the anti-ERBB2 (HER2) mAb, margetuximab (Merck, Macrogenics), in phase III clinical trials for breast cancer (NCT02492711).
Moreover, there have been many efforts to modulate aspects of IgG biology beyond just increasing or decreasing Fc effector function (Table 8). The first of these is modulation of half-life via modified interaction of the Fc with the recycling receptor FcRn (Roopenian and Akilesh, 2007; Baker et al., 2009). The most important of these modifications has been the “YTE” mutation (Dall’Aqua et al., 2002; Dall’Aqua et al., 2006) from MedImmune (AstraZeneca), which has been incorporated into a few early stage candidates at this point (Robbie et al., 2013). Other half-life extension mutations of the Fc also have been made, including Xencor’s Xtend technology (Zalevsky et al., 2010), which has been incorporated into at least two early stage clinical candidates, Alexion’s anti-C5 mAb, ALXN5500, and the National Institutes of Health’s (NIH’s) anti-CD4 mAb, VRC01LS.
Several other IgG engineering technologies have been reported that have the potential to modulate the capabilities of existing and future clinical candidates (Table 8). These include protease activated “probody” IgGs for tumor-localized activity (Erster et al., 2012; Desnoyers et al., 2013; Polu and Lowman, 2014), protease-resistant IgGs that are stable in the tumor micro-environment (Kinder et al., 2013), hexameric antibodies with high C1q binding and concomitant CDC effector function (Cook et al., 2016; de Jong et al., 2016), pH-dependent binding (Igawa et al., 2010a; Chaparro-Rogers et al., 2012; Devanaboyina et al., 2013) and sweeping antibodies (Igawa et al., 2013) that improve upon the elimination profile for soluble antigens (Igawa et al., 2016), engineering variable regions for improved solubility and developability (Clark et al., 2014; Seeliger et al., 2015), modulation of the pI or charge of the antibody variable sequences for improved half-life (Igawa et al., 2010b; Li et al., 2014; Datta-Mannan et al., 2015) and/or separation and purification (Sampei et al., 2013), and mutation of protein A binding site for improved purification of a heterobispecific IgG (Tustian et al., 2016). Another area of antibody engineering that is starting to see significant activity is the engineering of IgMs as therapeutics, especially where high avidity effects are desired (Chromikova et al., 2015; Wang et al., 2017b). In a recent example, an anti-TNFRSF10B (DR5) IgM demonstrated 10-fold greater avidity and 1000-fold greater killing effect than a similar IgG (Wang et al., 2017b).
ANTIBODY DRUG CONJUGATES (ADCs)
ADCs target a cytotoxic drug to a tumor to kill cancer cells while lowering the systemic exposure of the active moiety, with the goal of increasing the size of the efficacy/toxicity window of highly toxic anti-tumor drugs (Strohl and Strohl, 2012; Tsuchikama and An, 2016; Beck et al., 2017). ADCs consist of three components, the targeting antibody, the cytotoxic payload, and the linker that couples those two components together (Fig. 1).
With those three components come five considerations for the design and construction of an ADC: First, the targeting antibody must bind to a protein that is found either exclusively on cancer cells or significantly overexpressed on cancer cells as compared with expression on normal tissues. The best targets for ADCs may be oncofetal antigens or targets that may be overexpressed in cancer cells but present in normal tissues at low copy number or in tissues in which the toxicity is tolerable. The cell surface proteins most widely targeted with clinical stage (or approved) ADCs currently are Her2 (five ADCs targeting), CD19 (four ADCs targeting), CD22 (three ADCs targeting), and mesothelin (3 ADCs targeting). CEACAM5, EGFR (wild-type), EGFR (variant III), CD33, and CD70 each have two clinical stage ADCs targeting them. The properties of good ADC targets, as well as descriptions of candidate ADC targets, have been reviewed (Teicher, 2009; Strohl and Strohl, 2012). An interesting strategy being employed by CytomX to increase the tumor specificity of their ADCs is the use of pro-antibodies that possess a peptide sequence covering the paratope, preventing binding to their target until it reaches the tumor microenvironment (TME). Once in the TME, the paratope-shielding peptide is cleaved by matrix metalloproteinases (MMPs), which are in high concentrations in most TMEs, allowing the antibody to bind to targets in that local environment (Desnoyers et al., 2013; Polu and Lowman, 2014). Second, the ADC-directing antibody must be rapidly internalized upon ligation to its targeted receptor. Antibodies that bind cell surface receptors may or may not internalize rapidly, so when isolating the antibody, incorporation of internalization screens into the discovery process is critical (Poul et al., 2000; Zhou et al., 2010). Third, the identity, number, and type of linker attachment sites is a critical issue. In first generation ADCs, the linkers were typically attached to the ɛ-amine of lysine residues (Tsuchikama and An, 2016; Beck et al., 2017). Given that there are about 80 lysine residues in a typical IgG, ten of which can be accessed for chemical coupling (Tsuchikama and An, 2016), the results of such conjugations are highly heterogeneous. Even with optimization, conjugation to lysines results in a drug to antibody ratio (DAR) of about 2–4, with a range of 0–7 (Lazar et al., 2005; Tsuchikama and An, 2016; Beck et al., 2017). There are multiple challenges with heterogeneous ADCs including analytical challenges, batch-to-batch consistencies, the stability of the ADC, and the potential for variable pharmacokinetics if conjugation sites in some antibodies interfere with normal FcRn-mediated recycling (Beck et al., 2017). Site specific conjugation, which has been achieved through a variety of methods and can result in very tight DARs and increased homogeneity (Junutula et al., 2008; Panowski et al., 2014; Perez et al., 2014; Beerli et al., 2015; Ihospice et al., 2015; Siegmund et al., 2016; Thompson et al., 2016; Tsuchikama and An, 2016; Beck et al., 2017), appears to be a significant advancement. New approaches using extension sequences, such as developed by Mersana, can achieve a drug/antibody ratio of 20 (Yurkovetskiy et al., 2015).
Fourth, the stability of the linker can have a huge influence on the efficacy and toxicity of the ADC. In theory, a more stable linker which is only degraded within the lysosome should have the best safety profile. Unfortunately, it is not that simple, as there are cases in which highly stable linkers resulted in safety issues. Some of these may be due to mannose receptor, or potentially also FcγR-mediated binding and internalization of ADCs, which could result in “off-target” toxicity issues (Gorovits and Krinos-Kiorotti, 2013; Beck et al., 2017).
Finally, not all cancer cells within a tumor are target antigen-positive (Singh et al., 2016), thus allowing potential escape of the antigen-negative cells from targeted therapies. It has been demonstrated that membrane permeability of the cytotoxin is a critical factor for potential bystander activity (Li et al., 2016). Thus, design of future ADCs will need to take the chemistry of the resultant ADC into account to optimize bystander effect and efficacy.
There currently are 87 clinical stage ADCs, including three approved ADCs, nine in phase III development, and another 75 in phase I/II clinical development. The three approved ADCs include Mylotarg® (2000, withdrawn in 2010), the CD30-targeting Adcetris®, and the ERBB2 (Her2)-targeting Kadcyla®. These 87 clinical stage ADC molecules are directed against at least 53 different known targets, although a few have not been disclosed, so the actual number may be higher. The most targeted cell surface receptors currently are ERBB2 and CD19 (4 ADCs against each), and CD33, CD22, and MSLN (mesothelin) (3 ADCs against each).
There are 16 known different classes of drugs incorporated into clinical stage ADCs, 11 of which are small molecule classes and five of which are protein-based. The most widely used drug class incorporated into clinical stage ADCs are the auristatins (employed 31 times), followed by the maytansanoids (in 16 ADCs), and benzodiazepines (used in 9 ADCs) (Table 9). Of the biologics, Pseudomonas exotoxin PE38 is incorporated into four ADCs (Table 9).
Even though three ADCs have been approved for therapeutic use, this technology is still relatively early in the developmental cycle and many of the “rules” for optimized ADCs are still being sorted out (Drake and Rabuka, 2015; Beck et al., 2017). More details on the design and construction of ADCs can be found in Tsuchikama and An (2016) and in Beck et al. (2017).
Fc FUSIONS
Fc fusions are fusions of the IgG Fc domain with either a protein or peptide. In theory, the fusion can be to either the C- or N-terminus of the Fc, but most Fc fusions on the market and in clinical development today are N-terminal fusions. The primary reason for generating Fc fusions is to extend the half-life of pharmacologically relevant protein or peptide by using the FcRn-mediated recycling of the Fc (Strohl and Strohl, 2012; Strohl, 2015). Currently, 11 Fc fusion proteins have been approved for therapeutic use (Table 1), three are in phase III clinical trials, and 23 are being evaluated in earlier stage clinical trials (Table 3). Many of the earlier Fc fusions generally were constructed using receptor exodomains in immune pathways (e.g., TNFRSF18 [p75], CD58 [LFA3], CTLA4, IL1R1 [IL-1 receptor]) fused to the Fc to modulate the immune system, either by blocking soluble cytokines or by binding to cells. More recent Fc fusion proteins have become more diverse (Strohl, 2015), with the pharmacologically active “head groups” being blood factors, such as F9 (Factor IX) and F8 (Factor VIII), peptides such as GCG (GLP-1) and a THPO (thrombopoietin) analogue, and an enzyme, such as the tissue non-specific alkaline phosphatase (TNSALP; Millan et al., 2008) in asfotase alfa (Strensiq®) (Hofman et al., 2016).
IMMUNOCYTOKINES
Certain human cytokines such as IL2 have been approved (marketed name, Proleukin®) for systemic delivery and use in severe diseases such as metastatic melanoma and metastatic renal cell carcinoma (Dutcher, 2002). Systemic delivery of the T cell-activating cytokine, IL2, however, brings with it the potential for adverse events. The concept of using antibodies to target cytokines to either tumors or to specific tissues came into fruition around the turn of the century (Penichet and Morrison, 2001; Halin et al., 2002). Since that time, there has been an effort to target IL2, or other cytokines such as IL12 and TNF, to the tumor microenvironment, where the desired activity can take place with reduced adverse systemic effects (Neri and Sondel, 2016). This approach has been actualized by the fusion of cytokines to antibodies to make immunocytokines that may target vasculature associated with tumors (Pasche et al., 2012; Hemmerli and Neri, 2014), tumor cell surface antigens (Klein et al., 2017), or targets that would assist in accumulation in inflamed joints (Hughes et al., 2014). Immunocytokines come in two major formats, cytokine-scFv (or other fragment) fusions which have a short circulating half-life and cytokine-IgG fusions, which retain a long half-life (Neri and Sondel 2016).
There are at least 11 immunocytokines currently being evaluated in clinical trials. Two of these are Darleukin® (fibronectin extra domain B [EDB]-targeting scFv L19-IL2 fusion) and Fibromun® (EDB-targeting scFv L19-TNF fusion), which are both in phase III pivotal clinical trials as combination therapy for malignant melanoma (NCT02938299). Other clinical stage immunocytokines include examples such as Dekavil® (fibronectin extra domain A [EDA] targeting scFv F8-IL-10 fusion in phase II for treatment of RA [NCT02270632]), Teleleukin® (tenascin C alternative splice variant EDA1-targeting scFv F16-IL-2 fusion in phase I for treatment of acute myeloid leukemia [AML; NCT02957032]), RG7461 FAP (fibroblast-activation protein)-IL2 fusion in phase I for treatment of solid tumors [NCT02627274]), and cergutuzumab amunaleukin, an anti-CEA (carcinoembryonic antigen-IgG fused with IL2, currently in phase I clinical trials (NCT02350673) for treatment of solid tumors.
CHECKPOINT MODULATORS
Antibody-directed modulation of immune cell checkpoint receptors has become one of the most exciting and important new areas in antibody therapeutics over the past few years. Most efforts have been focused on T cell checkpoint modulation, but there is increasing interest in B cell, NK cell, and myeloid cell checkpoint modulation as well.
T cell activation is regulated by a series of three signals. The first signal is provided by the interaction of the T cell receptor (TCR) with major histocompatibility complex (MHC, HLA) class I (for CD8 T cells) or MHC (HLA) class II (for CD4 T cells) on antigen presenting cells (APCs). The secondary signal is provided through one of several checkpoint receptors (Table 10), which can either provide a costimulatory signal to activate the T cells, or a blocking signal to dampen T cell response (Topalian et al., 2015). The third signal comes from the production of either pro-inflammatory, T cell-activating cytokines or anti-inflammatory cytokines that would act to reduce T cell response (Chikuma et al., 2017; Schirdewahn et al., 2017).
Cancer cells can express ligands for T cell inhibitory receptors such as PDCD1 (PD-1) (ligand is CD274 [PD- L1]), CTLA-4 (ligands are CD80 and CD86), and HAVCR2 (aka TIM3) (ligand reported to be GAL9) to inhibit T cell activation and cytolytic T cell responses. Ligation of these receptors can lead to T cell anergy or exhaustion, resulting in the inability of the immune system to kill cancer cells. Inhibition of the blocking responses to T cell activation using anti-PDCD1, anti-CTLA4, or anti-CD274 antibodies has proven clinically to result in improved responses for a subset of patients with metastatic melanoma, NSCLC, and potentially other forms of cancer (Achkar and Tarhini, 2017; Kim et al., 2017). Additionally, efforts are ongoing to use combinations of anti-PD1 and anti-CTLA4 antibodies to increase the percentage of patients experiencing durable responses, i.e., “raising the tail of the survival curve” (Harris et al., 2016). Alternatively, several clinical candidates are agonists of T-cell activating receptors such as TNFRSF4 (OX40), CD40, TNFRSF9 (CD137, 4-1BB), TNFRSF18 (GITR), ICOS (CD278), CD27, or CD28 to stimulate T cell responses (Antonia et al., 2016; Table 10).
Additionally, T cell checkpoint pathways are potentially important in infectious diseases, in which T cell exhaustion halts T cells from eliminating viral and bacterial pathogens (Dyck and Mills, 2017). Finally, antibody intervention in T cell checkpoint pathways may play a role in autoimmune diseases, where blocking the activating signals or increasing the blocking signals may result in lowering the T cell activation response (van der Vlist et al., 2016).
Five mAbs and two Fc fusion proteins that target T cell/APC checkpoints have been approved (Table 10). Two more T cell checkpoint inhibitor antibodies are currently in phase III clinical trials and 77 are in phase I/II clinical trials, covering 19 different T cell checkpoint targets. Some of these checkpoint targets are being tested in both immune and oncology related diseases. For example, CD28, CD40, and TNFRSF4 (OX40) antagonists are in early stage clinical trials for treatment of various immune disorders, whereas CD28, CD40, and TNFRSF4 (OX40) agonists are in early stage clinical trials for various cancer indications (Table 10).
Checkpoint ligands expressed on cancer cells also are potentially excellent targets, both because they can block the inhibitory checkpoint interaction as well as targeting the ligand-expressing cancer cells with Fc-active antibodies. For this approach, there are now three approved anti-PD-L1 antibodies and another seven in clinical trials, as well as three clinical stage anti-CD70 (CD27 ligand) mAbs and one CD70-targeting CAR-T cell product in phase I clinical trials., as well as four anti-CD276 (B7H3) antibodies are currently in phase I clinical trials.
B cell transitional checkpoints are centered around B cell homeostasis and the choice of whether the B cell should mature or proceed to apoptosis. This process ensures that B cells expressing autoreactive immunoglobulins are purged (Cancro et al., 2009). Key regulators of B cell maturation that function in B cell checkpoints are TNFSF13B (soluble BLyS, ligands B lymphocyte stimulator; also known as B cell activating factor [BAFF]) and TNFSF13 (APRIL, a proliferation-inducing ligand). TNFSF13B can bind the TNFSF13B receptor (BR3; also known as BAFF-R) to promote B cell survival, and both TNFSF13B and TNFSF13 can bind TNFRSF13B (transmembrane activator-1 and calcium modulator and cyclophilin ligand-interactor, TACI) and TNFRSF17 (B cell maturation antigen, BCMA), both of which result in Ig class switching and T cell-dependent responses (Cancro et al., 2009).
Overexpression of TNFSF13B can lead to autoimmune consequences, such as system lupus erythematosus (SLE) or Sjögren’s syndrome (Cancro et al., 2009). One B cell checkpoint inhibitor (anti-TNFSF13B mAb, Benlysta®) is approved, two more are currently in phase III clinical trials, and three are in phase I/II clinical trials, all targeting the B cell activating factor regulatory pathway.
Another approach that has gained interest in very recent years is the immunomodulation of NK cells. NK cells, as well as CD8 T cells, express a series of inhibitory receptors including KLRC1-form A (NKG2A), TIGIT, CD96, and KIR family members (Carotta, 2016). As an immune defense mechanism, tumor cells express ligands to bind to these receptors to inhibit unwanted activation of NK cells. Currently there are six antibodies in phase I/II clinical trials binding these targets to remove the brake on NK cell activation.
Finally, another checkpoint that regulates the activity of macrophages and their phagocytosis of target cells is the CD47/SIRPA (signal regulatory protein alpha) and CALR (calreticulin)/LRP1 pathway. The CD47/SIRPA ligation is often referred to as the “don’t eat me” signal, whereas CALR/LRP1 ligation is known as the “eat me” signal (McCracken et al., 2015). Blocking of CD47 by antibodies or Fc fusion proteins can lead to an imbalance and a pro-“eat me” response (McCracken et al., 2015). Currently, four anti-CD47 antibodies or Fc fusion proteins are being evaluated in clinical trials for treatment of cancer (Table 10).
ANTIBODY MIXTURES
One approach that has gained interest in recent years is the combination or mixture of antibodies, usually against a single target, included into a single dosage (Raju and Strohl, 2013; Carvalho et al., 2016). Thus far, antibody mixtures are being used mostly for oncology and infectious disease indications. The Danish biotechnology company, Symphogen, has led this space, with four antibody mixtures currently being tested in clinical trials. These include SYM004, a mixture of two anti-EGFR mAbs, SYM013, a mixture of six antibodies against the ERBB (Erb-b2 receptor tyrosine kinase) family of receptors (Ellebaek et al., 2016), SYM015, a mixture of two antibodies targeting MET (cMET), and SYM009, an undisclosed mixture of antibodies partnered with Genentech for an infectious disease target. At least nine other antibody mixtures are being evaluated in clinical trials, all of which are against infectious diseases targets such as Ebola virus, botulinum toxin, and other viruses.
One very interesting new approach in this area that could see significantly greater upside in the coming years is the generation of fully human antibody mixtures, or polyclonal mixtures, in transgenic (tg) cattle (Matsushita et al., 2014, 2015). These may, if found safe and efficacious, at least partially replace “specific” intravenous immunoglobulin (IVIG), which is IgG purified from individuals who have been vaccinated or from convalescing patients who have produced IgGs against a specific target (Llewelyn et al., 1992; Mire et al., 2016). The upside of tg cattle-produced human IgGs is supply, consistency across lots, and the ability to vaccinate the cows with antigens not available for human vaccination due to regulatory and safety considerations. One such polyclonal mixture from tg cattle already being evaluated in clinical trials is SAB-301 (SAB Therapeutics), a polyclonal mixture of human IgGs targeting middle east respiratory (MERS) virus (NCT02788188; Luke et al., 2016).
BISPECIFIC ANTIBODIES
Bispecific antibodies, first conceptualized in 1983 (Milstein and Cuello, 1983), are antibodies that can bind two different antigens simultaneously. There are five fundamental groups of bispecific antibody formats: (i) asymmetric bivalent, bispecific IgG-like antibodies with heterodimeric heavy chains (HCs) (Ridgeway et al., 1996; Merchant et al., 1998; Gunasekaran et al., 2010; Strop et al., 2012; Klein et al., 2012; Labrijn et al., 2013 Von Kreudenstein et al., 2013; Brinkmann and Kontermann, 2017); (ii) tetravalent multispecific antibodies that are comprised of IgGs, with additional binding domains, e.g., scFvs, Fvs, VHH domains, or non-antibody binding scaffolds such as fynomers (Brack et al., 2014; Silacci et al., 2016), fused to either the N- or C-termini of either the heavy or light chains (LCs) (Coloma and Morrison, 1997); (iii) engineered binding domains within the normal IgG structure, such as the “two-in-one” bispecific approach from Genentech (Bostrom et al., 2009; Eigenbrot and Fuh, 2013) and the F-STAR approach of designing novel second binding sites within the CH3 domain (Leung et al., 2015), (iv) engineered antibody fragments linked by short peptide linkers which can be made into bivalent, trivalent, or tetravalent formats addressing two to three targets (Mack et al., 1995; Holliger and Winter, 1997; Kipriyanov et al., 1999; Reusch et al., 2015; Egan et al., 2016). These may be fused to an Fc domain or other half-life extending molecule (Liu et al., 2017); and (v) IgGs that are chemically coupled to generate IgG-IgG conjugates (e.g., Brennan et al., 1985; Garrido et al., 1990). Examples of these five basic formats are shown in Fig. 3. Many variations on these central themes have been reviewed multiple times (Kontermann, 2012; Spiess et al., 2015; Kontermann and Brinkmann, 2015; Ha et al., 2016; Brinkmann and Kontermann, 2017).

Five basic types of bispecific antibodies. (A) Bivalent, bispecific IgG-like antibodies with heteromeric heavy chains (e.g., Triomab, knobs-into-holes (KIH), Duobody, etc); (B) Tetravalent multispecific antibodies comprised of IgGs with other binding domains fused to either the N- or C-termini of either the heavy or light chains (e.g., dual variable domain [DVD], IgG-scFv fusion, Mabtyrin (IgG with non-antibody binding scaffold “centyrin” fused to C-terminal end of heavy chains); (C) IgGs to which additional antigen combining sites have been added within the structure (e.g., two-in-one antibodies, MAT “Modular Antibody Technology” platform from F-Star); (D) Engineered antibody fragments linked by short peptide linkers which can be made into bivalent, trivalent, or tetravalent formats addressing two to three targets (e.g., bispecific T-cell engager (BiTE), Nanobody platform, dual- affinity re-targeting (DART) antibodies, “tandem antibody” structures (TandAbs)); (E) Chemically coupled IgGs
Over the past decade there has been a literal explosion of novel bispecific antibody technologies, approaches, and clinical candidates. Today there are at least 61 bispecific or bifunctional antibodies in clinical trials that are made from at least 24 different bispecific platform technologies (Table 11). These include ten asymmetrical IgG-based platforms (17 bispecific antibodies), five appended IgG platforms (17 bispecific antibodies), a single platform for chemically coupled IgGs (four bispecific antibodies), eight fragment-based platforms (22 bispecific antibodies), and one IgG-based bispecific generated with an unknown platform (Table 11). Two bispecific antibodies have thus far been approved for medical use, both in the field of oncology. The first bispecific antibody of any kind to be approved was catumaxomab (Removab®), a bivalent, trifunctional, hybrid mouse IgG2a – rat IgG2b antibody targeting CD3E with one arm and EPCAM with the other. Catumaxomab, approved in 2009 (only in the European Union) for treatment of malignant ascites, was generated by the three-way fusion of a mouse B-cell, a rat B-cell, and a myeloma cell to form a quadroma cell line (Triomab® technology) (Zeidler et al., 1999). The second bispecific antibody to be approved was the anti-CD3E x anti-CD19 “Bispecific T Cell Engager” (BiTE) MT-103, constructed by linking two scFvs with a five residue (G4S)1 linker (Mack et al., 1995). This BiTE®, now known as blinatumomab (trade name, Blincyto®), was approved in 2014 for treatment of B-cell acute lymphoblastic leukemia (ALL).
The 61 current clinical stage bispecific antibodies are used for a variety of different purposes. For example, 10 of them bind two soluble antigens such as IL13 and IL4 (e.g., SAR156597; NCT02345070), nine bind two receptors on the same cell surface such as EGFR and MET (e.g., JNJ-61186372; NCT02609776), and four bind a cell surface target such as Delta Like Canonical Notch Ligand 4 (DLL4) with one combining site and a soluble ligand such as VEGF with the other (e.g., navicixizumab; NCT02298387). Two current clinical stage bispecific antibodies are biparatopic, i.e., both arms bind to the same receptor, albeit at two different non-overlapping epitopes (e.g., Zymeworks ZW25, which binds two non-overlapping epitopes of ERBB2; NCT02892123).
The most significant use of bispecific antibodies, however, is for T cell redirection, in which one combining site is directed toward a cell surface target on a cancer cell and the other combining site binds CD3E on T cells to redirect those cells to the targeted cancer cell (see below).
T CELL REDIRECTION
Twenty-seven clinical stage bispecific antibodies are immune cell redirection bispecific antibodies. One of these targets FCGR3A (CD16a) for NK cell redirection, while the other 26 bispecific antibodies target CD3E on T cells to redirect the cytotoxic T cells (CTLs) to kill and lyse cancer cells. Of these, 14 are constructed from antibody fragments, seven are asymmetric bispecific IgGs, four are conjugated bispecific IgGs that are used to activate T cells ex corporally (Brennan et al., 1985; Garrido et al., 1990), and two are bispecific IgGs with appended domains (Table 11). The two appended IgGs also utilize an asymmetric Fc format so that only one CD3E-binding arm is present. It is generally accepted that the most potent T cell redirecting antibodies are fragments, with unmodified BiTEs and DARTs (dual-affinity re-targeting antibodies) demonstrating sub-picomolar IC50 values for in vitro killing activities (Moore et al., 2011). Of the two approved antibodies, Blincyto® is a mouse BiTE, while Removab® is an asymmetric rat/mouse IgG. Given that both are “first generation” T cell redirecting, fully mouse antibodies for very different indications, it is difficult to say today which type of platform (fragment vs. IgG-based) will ultimately be the most efficacious for treatment of diseases. The larger IgG-based forms appear to be significantly less potent based on in vitro activities and in vivo preclinical dosing than are fragments (unpublished data). Thus, there is a balance between sheer potency, which can be achieved with small size, and long half-life, which typically brings with it greater size and less potency. Additionally, both the size of the cell surface receptor of the target cells and the epitope to which the antibody binds appear to be critical factors in potency as well (Bluemel et al., 2010). Moreover, the potency of T cell redirected bispecific antibodies depends on the affinity of the arms for each antigen. Typically in the case of bispecific T cell redirection antibodies, the affinity for the cancer cell surface target is much higher (i.e., 10-fold or more) than the affinity for the CD3E chain on T cells (Zhukovsky et al., 2016). In summary, factors that may influence potency in T cell redirected antibodies are size of the antibody, size of the target cell surface protein, epitope on that protein to which the antibody binds, and affinity.
Another area that has not yet been fully investigated with respect to T cell redirection is the role of Fc functionality. The Triomab® platform, on which Removab® is designed, has a highly active Fc domain that interacts with human FcγRs to increase the immune response (Chelius et al., 2010; Hess et al., 2012). On the other hand, most of the current fragment-Fc, asymmetric IgG, or appended IgG platforms have used muted or silenced Fcs so as not to over stimulate the immune system via interactions with myeloid effector cells. Even with the absence of Fc activity, many treatments with T cell redirecting bispecific antibodies are accompanied by cytokine storms that need to be addressed as part of the therapeutic paradigm (Lee et al., 2016). Thus, it seems likely that most T cell redirecting antibodies made in the future will continue to avoid Fc activity in an effort to limit the release of pro-inflammatory cytokines by T cells and other effector cells in the tumor microenvironment.
CAR-T CELLS AND TCR-T CELLS
CARs are anti-tumor targeted antibodies that have been fused genetically to a stalk or linker, a transmembrane domain, and intracellular T cell activation domains that have been borrowed from activation checkpoint receptors such as CD28, TNFRSF9 (CD137), and/or TNFRSF4 (OX40) (Fig. 1M; Figueroa et al., 2015; van der Stegen et al., 2015; Ruella and Gill, 2015; Smith et al., 2016; Ruella and June, 2016; Lim and June, 2017). While the concept of CAR-T cells has been around since the early 1990s (Eshhar et al., 1993), the advancement of technologies required to turn this into a viable “manufacturable” process was only realized in recent years. Thus, similar to bispecific antibody technology, while conceptually old, truly developable CAR-T technology is still relatively young and still developing (Lim and June, 2017).
There are fundamentally two types of CARs. The first is autologous, in which a patient’s T cells are collected by a process known as apheresis, and then either as a whole pool, or a fractioned pool of CD8 T cells, CD4 T cells or possibly both CD4 and CD8 T cells, are transduced with the CARs using either viral vectors such as Lentivirus or transposons such as Sleeping Beauty or PiggyBack (Figueroa et al., 2015; Lim and June, 2017). The recombinant T cells, now armed with CARs targeting a tumor expressed on their surface, are activated and infused back into the patients from which they were derived to kill cancer cells bearing the antigen (Figueroa et al., 2015).
The second major type of CAR is allogeneic, or universal. An “off-the-shelf” cell line is constructed, typically devoid of MHC class I molecules (Ren et al., 2017a) and endogenous T cell receptors (MacLeod et al., 2017; Ren et al., 2017a) to decrease the risk of host vs. graft (rejection) and graft vs. host disease (GvHD), respectively. This universal T cell line also would express CARs for treatment of cancer or possibly viral infections. Thus far, the barriers to generate truly off the-shelf allogeneic cell lines are still quite high, with control of proliferation, continued activation of the cells once they are engrafted, and incorporation of kill switches for safety purposes as critical issues still to be worked out. Nevertheless, significant progress has been made in just the past year suggesting that fully modified allogeneic CAR-T cell therapy is quickly becoming a reality (Ren et al., 2017a, b). To date, there are four generations of autologous CAR-T cell constructs. The first generation typically consisted of the extracellular, cancer cell-targeting scFv fused to the CD8 stalk and transmembrane domain followed by CD247 (aka CD3ζ), which provided the activation signal (Park and Brentjens, 2010; Figueroa et al., 2015; Lim and June, 2017). The first generation CARs possessed ample cytotoxicity but lacked proliferative and survival signals. The second-generation CARs typically linked the exodomain scFv to the transmembrane domain of CD28, TNFRSF9 (CD137, 4-1BB), or TNFRSF4 (OX40) to provide a proliferation signal, followed by CD247 (CD3ζ) to provide the cytolytic activation signal. The third generation CARs have typically linked the targeting scFv to the CD28 transmembrane domain, followed by either the TNFRSF9 (CD137, 4-1BB), or TNFRSF4 (OX40) activation domains, and then CD247 (CD3ζ) (Park and Brentjens, 2010; Figueroa et al., 2015; Smith et al., 2016; Lim and June, 2017). These CARs combined cytolytic activity with both proliferation and survival signals to enhance both their activity and their persistence in the patient’s serum. Fourth generation CARs add new activities such as a suicide mechanism to kill off the CARs in case they become over-proliferative, or utilize T cells that have been conditioned to recognize viral antigens which can be used as “vaccines” to increase the persistence of the CAR-T construct (Chmielewski et al., 2014; Smith et al., 2016; Lim and June, 2017).
There are currently 145 different CAR constructs in clinical trials. As stated earlier, all of the CAR candidates are in phase I or II clinical trials. Almost half (72/145) of the current CARs originated in China, with 67 originating in the US, and 6 originating in Europe. CARs have been generated against 38 different targets, 37 of which are cell-surface proteins on cancer cells and one, WT1, an MHC-displayed peptide target derived from an intracellular antigen (Rafiq et al., 2017). Fifty-three (~37%) clinical CAR candidates are directed against CD19. The next most targeted antigens are GD2 and MSLN (mesothelin) (8 CARs each), ERBB2 (HER2) and CD22 (7 CARs each), and GPC3 (glypican-3) and TNFRSF8 (CD30) (6 CARs each). Most of the current clinical stage CAR constructs are autologous CAR-T constructs generated from αβ T cells (Table 12), but there are a few examples of other formats, including early formats of allogeneic CAR-T cells, autologous CAR γδ T cells, both autologous and allogeneic CAR-NK cells, CAR-NKT cells, and CARs made from TCRs (Table 12).
It is too early to judge the success of the CAR field, although it is clear that this area has generated an enormous amount of interest, as well as funding well exceeding $1B. It is noteworthy that Novartis recently (3/29/17) filed a biologics license application (BLA) to the US FDA for treatment of relapsed and refractory B-cell acute lymphoblastic anemia (B-ALL) with CTL019 (tisagenlecleucel-T), making it the first CAR construct to be submitted for regulatory approval (Kingwell, 2017). Moreover, Kite Pharma announced shortly thereafter (3/21/17) that they had completed their rolling BLA submission for treatment of non-Hodgkin lymphoma (NHL) using KTE-C19 (axicabtagene ciloleucel). If either CTL019 or KTE-C19 is, or both are, approved within the next year, it will mark a huge milestone in this exciting new field.
DELIVERY OF ANTIBODIES TO NOVEL COMPARTMENTS
An area that has been of interest for many years, but has proven challenging, is the targeting of antibodies to compartments into which they do not normally go. These include, for examples, targeting antibodies to the gut via an oral route, to the brain by crossing the blood-brain barrier, or to the cytosolic intracellular compartment. All of these compartments present significant challenges, but in the past few years, significant strides have been made for all of them.
The most advanced tissue-targeted antibody-based product the bone-targeted enzyme replacement-Fc fusion, asfotase alpha (Strensiq®), which was approved by the US FDA for treatment of hypophosphatasia (Hofman et al., 2016). Asfotase alpha (TNSALP-Fc-deca-aspartate fusion protein) is targeted to bone with a deca-aspartate peptide fused to the C-terminus of the Fc (Millan et al., 2008).
The second area of antibody targeting that is represented by clinical candidates is based on the route of delivery to get the antibodies to the desired compartment. At least three orally-delivered, antibody-related proteins targeted to the intestinal tract are currently being evaluated in clinical trials. These include PRX-106 (Protalix®), an anti-TNF plant cell-expressed and delivered Fc fusion protein in phase II clinical trials (NCT02768974) for the treatment of ulcerative colitis (Ilan et al., 2017). The plant cells are thought to protect the Fc fusion while traversing through the stomach. Over the last decade, the mouse anti-CD3E mAb, OKT3, has been evaluated in clinical trials for oral delivery to the gut for treatment of nonalcoholic steatohepatitis (NASH; NCT01205087), with results suggestive of clinical activity (Lalazar et al., 2015). A second anti-CD3E mAb formulated for oral delivery is foralumab (NI-0401; NovImmune, Tiziana Life Sciences), a fully human mAb currently being prepared for phase II clinical trials for oral delivery for the treatment of NASH.
Getting antibodies to cross the blood-brain barrier has been a goal for well over two decades. IgG levels in the human brain are approximately 0.1% of the serum concentration of 9–10 mg/mL (Abbott et al., 2010). This differential is due to the blood-brain barrier (BBB) which effectively keeps antibodies out of the brain. Considering the wealth of potential targets for biologics in the central nervous system (CNS), there has been a great effort to find mechanisms to improve the ability to translocate biologics into the CNS. In recent years, significant progress has been made in getting antibodies to traverse the BBB. Yu et al. (2011) used a bivalent, bispecific antibody binding TFRC (transferrin receptor, CD71) with one arm and BACE1 (β-secretase-1) with the other arm, to demonstrate that low affinity antibodies to TFRC were more efficient at transcytosis than high affinity antibodies. While they only achieved about 12-fold higher accumulations of antibody in the brain over controls, they clearly demonstrated anti-BACE1 pharmacological activity of the antibody, proving that the antibody had accumulated within the brain (Yu et al. 2011). They also generated a bivalent, bispecific antibody targeting human and non-human primate (NHP) TFRC with one arm and human BACE1 with the other arm (Yu et al., 2014). The best variants, which were low-to-moderate affinity antibodies to TFRC, were accumulated 15-fold higher in the brain than control antibodies and they demonstrated in vivo pharmacological activity in NHPs (Yu et al., 2014).
Neiwoehner et al. (2014) compared the efficiency of transcytosis using a tetravalent, bispecific antibody with two arms each binding to TFRC and APP (amyloid-beta, Aβ) to a trivalent, bispecific antibody with only one arm binding TFRC. They found that monovalent binding to TFRC promoted efficient transcytosis whereas bivalent binding to TFRC resulted in shuttling the complex towards lysosomal degradation. They demonstrated a 55-fold improvement in target engagement over the control (Neiwoehner et al. 2014). In contrast to these studies in which monovalent targeting of the transcytotic receptor was optimal, the anti-TMEM30A (α(2,3)-sialoglycoprotein), llama single-domain antibody, FC5 (Abulrob et al., 2005), appeared to be transcytosed more efficiently as a dimer rather than a monomer (Farrington et al., 2014). Recently, FC5 was fused in an scFv format to the N-terminus of the HC of an anti-GRM (glutamate metabotropic receptor 1, mGluR1) antagonist IgG to shuttle it across the BBB (Webster et al., 2016), achieving pharmacological activity with a 10-fold enrichment of the antibody in the brain parenchyma (Webster et al., 2016). Thus, it still appears that there is much to be learned about optimizing antibodies for transcytotic delivery of proteins to the CNS.
William Pardridge and his colleagues have isolated an anti-human INSR (insulin receptor) antibody that can be transcytosed by INSR on endothelial cells lining the vasculature in the brain (Boado et al., 2007). They have used the anti-INSR antibody as a transcytotic carrier to move enzymes across the BBB for CNS enzyme replacement therapy (ERT) (Boado et al., 2012, 2014). These candidates are constructed by fusion of the enzymes to the C-terminus of the BBB-traversing anti-INSR IgG “HIRMAb” (Boado et al., 2012, 2014). AGT-181, which is a tetravalent (two antibody arms and two enzymes) fusion of an anti-INSR antibody and α-L-iduronidase (ALI) (Boado et al., 2012), is being evaluated in phase I clinical trials (NCT02371226) for the treatment of mucopolysaccharidosis I (MPS I; Hurler Syndrome). AGT-181 was recently demonstrated to be taken up by non-human primate brain at 1.2% of injected dose as compared to 0% injected dose of α-L-iduronidase alone (Boado and Pardridge, 2017), demonstrating the pharmacological relevance of the BBB-traversing bispecific antibody. AGT-182, comprised of a fusion of iduronate 2-sulfatase (IDS) to the C-termini of the anti-INSR HCs (Boado et al., 2014), is under phase I clinical testing (NCT02262338) for the treatment of mucopolysaccharidosis II (MPS II; Hunter Syndrome).
The final delivery-related technology that has gotten very interesting in recent years is the delivery of mAbs to the cytosol of cells via pinocytosis and endosomal escape (Marschall et al., 2014; Lönn et al., 2016; Stewart et al., 2016; Lim et al., 2017). Multiple approaches have been taken to get biologically active antibodies into the cytosol of cells, including the use of cell penetrating peptides (Marschall et al., 2014; Lönn et al., 2016; Lim et al., 2017). Just recently, a unique antibody has been generated for the delivery of an IgG to the cytosol of cells via endosomal escape (Choi et al., 2014). This antibody, which has a unique sequence in its light chain variable region, has been matured to increase the proportion of IgG that enters the cytoplasm (Kim et al., 2016). This, and other cell penetration technologies (Marschall et al., 2014; Lönn et al., 2016; Lim et al., 2017) bring hope that one day, antibodies will be used to target cytosolic antigens.
NEW FORMS OF DELIVERY OF ANTIBODY GENES (DNA, RNA, AAVs, ONCOLYTIC VIRUSES)
Traditional forms of delivery for mAbs and Fc fusion proteins has been via either intravenous (IV) or subcutaneous (SC) administration of formulated proteins. Generally, high dose mAbs for oncology indications are limited to IV dosing, whereas low dose antibodies such as adalimumab, golimumab, and ustekinumab can easily be delivered in SC doses. Additionally, in recent years there has been increased interest in intratumoral dosing of antibodies and other biologics for certain types of cancer where the tumor is more accessible (Zeltsman et al., 2016). A novel approach for delivering mAbs and/or Fc fusion proteins is via delivery of the gene or genes that produce them, either as naked DNA, RNA, or by a viral-based vector. This is not an entirely new approach, since studies were done around the turn of the century showing that RNA (Giraud et al., 1999) and viral (Lewis et al., 2002) delivery of IgG genes could result in demonstration of in vivo IgG activity. Nevertheless, there was not much interest until the past few years, when it has become evident that vectored or nucleic acid delivery of IgG could potentially be a significant new approach to deliver antibodies for therapeutic use.
One of the more exciting forms of delivery is the intramuscular injection of adeno-associated viruses (AAVs) encoding antibodies, followed by years of consistently high expression of those antibodies in non-human primates (Fuchs et al., 2016; Greig et al., 2016). It is important to note that AAVs exist in the muscle cells as extrachromosomal elements and do not integrate, which increases the safety of their use for long term expression of antibodies or other proteins (Greig et al., 2016). This suggests that such an approach might be appropriate for delivery of anti-HIV antibodies to help patients either to become cured or, minimally, less reliant on highly active anti-retroviral therapy (HAART) (Schnepp and Johnson, 2014a; Fuchs and Desrosiers, 2016). There are several very promising, potent anti-HIV antibodies in clinical trials currently, some of which have been expressed in vivo using gene-based delivery of antibodies for potential therapeutic use (Schnepp and Johnson, 2014b; Yang and Wang, 2014; Fuchs et al., 2016; Fuchs and Desrosiers, 2016).
Similarly, but with a different twist, AAV-delivered antibodies to the nasal passages of mice have demonstrated excellent prophylaxis against flu virus (Limberis et al., 2013; Balazs et al., 2014; Adam et al., 2014). Since these AAVs enter epithelial cells that are sloughed off over several months, this provides a potentially safe route for delivery of prophylactic anti-flu antibodies that would cover the entire flu season. The potential significance of this approach is that there are several HA-binding and neutralizing antibodies available now that are nearly universal influenza virus inhibitors. These could potentially be used in clinical trials to determine whether or not this prophylactic, pan-influenza nasal delivery approach might be feasible.
Finally, the concept of using oncolytic viruses to deliver anti-tumor or checkpoint modulating antibodies to a tumor is very exciting. Oncolytic viruses have been engineered for years to deliver immune-modulating molecules such as CSF2 (GM-CSF) to the TME (Bommareddy et al., 2017), so it makes sense that they could be engineered to deliver TME modulating antibodies (Du et al., 2014). Several recent examples have demonstrated the potential for various types of oncolytic viruses expressing immune checkpoint inhibitors such as anti-PDCD1 (PD-1), anti-CD274 (PD-L1), and anti-CTLA4 (Du et al., 2014; Kleinpeter et al., 2016; Tanoue et al., 2017), as well as other anti-tumor antibodies (Adelfinger et al., 2015; Liikanen et al., 2016; Fajardo et al., 2017).
SUMMARY
Over the past decade there has been a significant shift from discovery and development of basic antibodies, e.g., naked IgG1 isotype antibodies with no additional engineering other than perhaps humanization and affinity maturation, to more sophisticated forms of antibodies in all kinds of shapes and sizes. These newer forms include Fc-modified, glyco-engineered, bispecific, drug-conjugated, and cell surface expressed antibodies (i.e., CARs) as new weapons to fight difficult to treat diseases. We now see this dramatic shift in the types and numbers of modified antibodies now reaching clinical trial studies. This new phase of antibody drug discovery and development represents an exciting and bold new era that should see antibody-based therapeutics expanding their influence in many types of diseases. In the next few years we will likely see the first regulatory approvals of CAR-T based antibodies and immunocytokines, as well as approvals of additional new bispecific antibodies, new ADCs, Fc engineered antibodies, and glyco-engineered antibodies. Additionally, we should see new advances in targeting antibodies to the CNS and intracellular compartments, as well as nucleic acid or viral-vectored delivery. What an exciting time to be an antibody engineer!
REFERENCES
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25
Abulrob A, Sprong H, Henegouwen E, Van Bergen P, Stanimirovic D (2005) The blood–brain barrier transmigrating single domain antibody: mechanisms of transport and antigenic epitopes in human brain endothelial cells. J Neurochem 95:1201–1214
Achkar T, Tarhini AA (2017) The use of immunotherapy in the treatment of melanoma. J Hematol Oncol 10:88. doi:10.1186/s13045-017-0458-3
Adam VS, Crosariol M, Kumar S, Ge MO, Czack SE, Roy S, Haczku A, Tretiakova A, Wilson JM (2014) Adeno-associated virus 9-mediated airway expression of antibody protects old and immunodeficient mice against influenza virus. Clin Vaccine Immunol 21:1528–1533
Adelfinger M, Bessler S, Frentzen A, Cecil A, Langbein-Laugwitz J, Gentschev I, Szalay AA (2015) Preclinical testing oncolytic vaccinia virus strain GLV-5b451 expressing an anti-VEGF single-chain antibody for canine cancer therapy. Viruses 7:4075–4092
Alduaij W, Ivanov A, Honeychurch J, Cheadle EJ, Potluri S, Lim SH, Shimada K, Chan CHT, Tutt A, Beers SA et al (2011) Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 117:4519–4528
Anthony RM, Ravetch JV (2010) A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol 30(Suppl 1):S9–14
Antonia SJ, Vansteenkiste JF, Moon E (2016) Immunotherapy: beyond anti–PD-1 and anti–PD-L1 therapies. ASCO Educ Book 2016:e450–e458
Baeuerle PA, Reinhardt C, Kufer P (2008) BiTE: a new class of antibodies that recruit T-cells. Drugs Future 33:137–147
Baker K, Qiao S-W, Kuo T, Kobayashi K, Yoshida M, Lencer WI, Blumberg RS (2009) Immune and non- immune functions of the (not so) neonatal Fc receptor, FcRn. Semin Immunopathol 31:223–226
Balazs AB, Ouyang Y, Hong CM, Chen J, Nguyen SM, Rao DS, An DS, Baltimore D (2014) Vectored immunoprophylaxis protects humanized mice from mucosal HIV transmission. Nat Med 20:296–300
Barnhart BC, Quigley M (2017) The role of Fc-FcγR interactions in the anti-tumor activity of therapeutic antibodies. Immunol Cell Biol 95:340–346
Beck A, Goetsch L, Dumontet C, Corvaïa N (2017) Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov 16:315–337
Beerli RR, Hell T, Merkel AS, Grawunder U (2015) Sortase enzyme-mediated generation of site- specifically conjugated antibody drug conjugates with high in vitro and in vivo potency. PLoS ONE 10:e0131177. doi:10.1371/journal.pone.0131177
Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M (1988) Single-chain antigen-binding proteins. Science 242:423–426
Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P (2010) Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother 59:1197–1209
Boado RJ, Pardridge WM (2017) Brain and organ uptake in the rhesus monkey in vivo of recombinant iduronidase compared to an insulin receptor antibody-iduronidase fusion protein. Mol Pharm 14:1271–1277
Boado RJ, Zhang Y, Zhang Y, Pardridge WM (2007) Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol Bioeng 96:381–391
Boado RJ, Hui EK, Lu JZ, Pardridge WM (2012) Glycemic control and chronic dosing of rhesus monkeys with a fusion protein of iduronidase and a monoclonal antibody against the human insulin receptor. Drug Metab Dispos 40:2021–2025
Boado RJ, Hui EK-W, Lu JZ, Pardridge WM (2014) Insulin receptor antibody-iduronate 2-sulfatase fusion protein: pharmacokinetics, anti-drug antibody, and safety pharmacology in rhesus monkeys. Biotechnol Bioeng 111:2317–2325
Bolt S, Routledge E, Lloyd I, Chatenoud L, Pope H, Gorman SD, Clark M, Waldmann H (1993) The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol 23:403–411
Bommareddy PK, Patel A, Hossain S, Kaufman HL (2017) Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol 18:1–15
Borrok MJ, Mody N, Lu X, Kuhn M, Wu H, Dall’Acqu WF, Tsui P (2017) An ‘Fc silenced’ IgG1 format with extended half-life designed for improved stability. J Pharm Sci. doi:10.1016/j.xphs.2016.12.023
Bossi G, Buisson S, Oates J, Jakobsen BK, Hassan NJ (2014) ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol Immunother 63:437–448
Bostrom J, Yu S-F, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, Fuh G (2009) Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 323:1610–1614
Brack S, Attinger-Toller I, Schade B, Mourlane F, Klupsch K, Woods R, Hachemi H, von der Bey U, Koenig- Friedrich S, Bertshinger J, Grabulovski D (2014) A bispecific HER2-targeting FynomAb with superior antitumor activity and novel mode of action. Mol Cancer Ther 13:2030–2039
Brennan M, Davison PF, Paulus H (1985) Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 229:81–83
Brezski RJ, Georgiou G (2016) Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol 40:62–69
Brinkmann U, Kontermann RE (2017) The making of bispecific antibodies. mAbs 9:182–212
Cancro MP, D’Cruz DP, Khamasta MA (2009) The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Investig 119:1066–1073
Capon DJ, Chamow SM, Mordenti J, Marsters SA, Gregory T, Mitsuya H, Byrn RA, Lucas C, Wurm FM, Groopman JE et al (1989) Designing CD4 immunoadhesins for AIDS therapy. Nature 337:525–531
Carotta S (2016) Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol 7:152. doi:10.3389/fimmu.2016.00152
Carvalho S, Levi-Schaffer F, Sela M, Yarden Y (2016) Immunotherapy of cancer: from monoclonal to oligoclonal cocktails of anti-cancer antibodies: IUPHAR Review 18. Br J Pharmacol 173:1407–1424
Chaparro-Riggers J, Liang H, DeVay RM, Bai L, Sutton JE, Chen W, Geng T, Lindquist K, Casas MG, Boustany LM et al (2012) Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem 287:11090–11097
Chelius D, Ruf P, Gruber P, Plöscher M, Liedtke R, Gansberger E, Hess J, Wasiliu M, Lindhofer H (2010) Structural and functional characterization of the trifunctional antibody catumaxomab. mAbs 2:309–319
Chikuma S, Kanamori M, Mise-Omata S, Yoshimura A (2017) Suppressors of cytokine signaling: potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci 108:574–580
Chmielewski M, Hombach AA, Abken H (2014) Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 257:83–90
Choi D-K, Bae J, Shin S-M, Shin J-Y, Kim S, Kim Y-S (2014) A general strategy for generating intact, full- length IgG antibodies that penetrate into the cytosol of living cells. mAbs 6:1402–1414
Choy EH, Hazleman B, Smith M, Moss K, Lisi L, Scott DG, Patel J, Sopwith M, Isenberg DA (2002) Efficacy of a novel PEGylated humanized anti-TNF fragment (CDP870) in patients with rheumatoid arthritis: a phase II double-blinded, randomized, dose-escalating trial. Rheumatol 41:1133–1137
Chromikova V, Mader A, Hofbauer S, Göbl C, Madl T, Gach JS, Bauenfried S, Furtmüller PG, Forthal DN, Mach L et al (2015) Introduction of germline residues improves the stability of anti-HIV mAb 2G12- IgM. Biochim Biophys Acta 1854:1536–1544
Clark RH, Latypov RF, De Imus C, Carter J, Wilson Z, Manchulenko K, Brown ME, Ketchem RR (2014) Remediating agitation-induced antibody aggregation by eradicating exposed hydrophobic motifs. mAbs 6:1540–1550
Coloma MJ, Morrison SL (1997) Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol 15:159–163
Cook EM, Lindorfer MA, van der Horst H, Oostindie S, Beurskens FJ, Schuurman J, Zent CS, Burack R, Parren PW, Taylor RP (2016) Antibodies that efficiently form hexamers upon antigen binding can induce complement-dependent cytotoxicity under complement-limiting conditions. J Immunol 197:1762–1775
Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S (2002) Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 169:5171–5180
Dall’Acqua WF, Kiener PA, Wu H (2006) Properties of human IgG1 engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 281:23514–23524
Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ (2015) Balancing charge in the complementarity determining regions of humanized mAbs without affecting pI reduces non-specific binding and improve pharmacokinetics. mAbs 7:483–493
Davis PM, Abraham R, Xu L, Nadler SG, Suchard SJ (2007) Abatacept binds to the Fc receptor CD64 but does not mediate complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. J Rheumatol 34:2204–2210
de Jong RN, Beurskens FJ, Verploegen S, Strumane K, van Kampen MD, Voorhorst M, Horstman W, Engelberts PJ, Oostindie C, Wang G et al (2016) A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLoS Biol 14:e1002344. doi:10.1371/journal.pbio.1002344
Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EEM, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ et al (2013) Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Trans Med 5:207ra144. doi:10.1126/scitranslmed.3006682
Devanaboyina SC, Lynch SM, Ober RJ, Ram S, Kim D, Puig-Canto A, Breen S, Kasturirangan S, Fowler S, Peng L et al (2013) The effect of pH dependence of antibody–antigen interactions on subcellular trafficking dynamics. mAbs 5:851–859
Drake PM, Rabuka D (2015) An emerging playbook for antibody-drug conjugates: lessons learned from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr Opin Chem Biol 28:174–180
Du T, Shi G, Li YM, Zhang JF, Tian HW, Wei YQ, Deng H, Yu DC (2014) Tumor-specific oncolytic adenoviruses expressing granulocyte macrophage colony-stimulating factor or anti-CTLA4 antibody for the treatment of cancers. Cancer Gene Ther 21:340–348
Dutcher JP (2002) Current status of interleukin-2 therapy for metastatic renal cell carcinoma and metastatic melanoma. Oncology 16(Suppl 13):4–10
Dyck L, Mills KHG (2017) Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. doi:10.1002/eji.201646875
Egan TJ, Diem D, Weldon R, Neumann T, Meyer S, Urech DM (2016) Novel multispecific heterodimeric antibody format allowing modular assembly of variable domain fragments. mAbs 9:68–84
Eigenbrot C, Fuh G (2013) Two-in-one antibodies with dual action Fabs. Curr Opin Chem Biol 17:400–405
Ellebæk S, Brix S, Grandal M, Lantto J, Horak ID, Kragh M, Poulsen TT (2016) Pan-HER—An antibody mixture targeting EGFR, HER2 and HER3 abrogates preformed and ligand-induced EGFR homo- and heterodimers. Int J Cancer 139:2095–2105
Erster O, Thomas JM, Hamzah J, Jabaiah AM, Getz JA, Schoep TD, Hall SS, Ruoslahti E, Daugherty PS (2012) Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. J Control Release 161:804–812
Eshhar Z, Waks T, Gross G, Schindler DG (1993) Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA 90:720–724
Esparis-Ogando A, Montero JC, Arribas J, Ocana A, Pandiella A (2016) Targeting the EGF/HER ligand- receptor system in cancer. Curr Pharm Des 22:5887–5898
Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, June CH, Alemany R (2017) Oncolytic adenoviral delivery of an EGFR-targeting T cell engager improves antitumor efficacy. Cancer Res 77:2052–2063
Farrington GK, Caram-Salas N, Haqqani AS, Brunette E, Eldredge J, Pepinsky B, Antognetti G, Baumann E, Ding W, Garber E et al (2014) A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J 28:4764–4778
Ferrara C, Brünker P, Suter T, Moser S, Püntener U, Umaña P (2006) Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of golgi enzyme localization domain and co-expression of heterologous beta 1,4-N-acetylglucosaminyltransferase III and golgi alpha- mannosidase II. Biotechnol Bioeng 93:851–861
Figueroa JA, Reidy A, Mirandola L, Trotter K, Suvorava N, Figueroa A, Konala V, Aulakh A, Littlefield L, Grizzi F et al (2015) Chimeric antigen receptor engineering: a right step in the evolution of adoptive cellular immunotherapy. Int Rev Immunol 34:54–187
Fuchs SP, Desrosiers RC (2016) Promise and problems associated with the use of recombinant AAV for the delivery of anti-HIV antibodies. Methods Clin Dev 3:16068. doi:10.1038/mtm.2016.68
Fuchs SP, Martinez-Navio Gao G, Desrosiers RC (2016) Recombinant AAV vectors for enhanced expression of authentic IgG. PLoS ONE 11:e0158009. doi:10.1371/journal.pone.0158009
Garrido MA, Valdayo MJ, Winkler DF, Titus JA, Hecht TT, Perez P, Segal DM, Wunderlich JR (1990) Refocusing the immune system to react with human tumors by targeting human lymphocytes with bispecific antibodies. Dev Biol Stand 71:33–42
Giraud A, Ataman-Önal Y, Battail N, Piga N, Brand D, Mandrand B, Verrier B (1999) Generation of monoclonal antibodies to native human immunodeficiency virus type 1 envelope glycoprotein by immunization of mice with naked RNA. J Virol Methods 79:75–84
Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, Klein C, Introna M (2013) Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 122:3482–3491
Gorovits B, Krinos-Fiorotti C (2013) Proposed mechanism of off-target toxicity for antibody-drug conjugates driven by mannose receptor uptake. Cancer Immunol Immunother 62:217–223
Gray KA, Yates B, Seal RL, Wright MW, Bruford EA (2015) Genenames.org: the HGNC resources in 2015. Nucleic Acids Res 43(Database issue):D1079–D1085. doi:10.1093/nar/gku1071
Greenwood J, Clark M, Waldmann H (1993) Structural motifs involved in human IgG antibody effector functions. Eur J Immunol 23:1098–1104
Greig JA, Calcedo R, Grant RL, Peng H, Medina-Jszek CA, Ahonkhai O, Qin Q, Roy S, Tretiakova AP, Wilson JM (2016) Intramuscular administration of AAV overcomes pre-existing neutralizing antibodies in rhesus macaques. Vaccine 34:6323–6329
Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, Sweet H, Foltz IN, Wittekind M, Yan W (2010) Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem 285:19637–19646
Ha J-H, Kim J-E, Kim Y-S (2016) Immunoglobulin Fc heterodimer platform technology: from design to applications in therapeutic antibodies and proteins. Front Immunol. doi:10.3389/fimmu.2016.00394
Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, Scharenberg AM, Rawlings DJ, Wagner TA (2017) Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol Ther 25:570–579
Halin C, Rondini S, Nilsson F, Berndt A, Kosmehl H, Zardi L, Neri D (2002) Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat Biotechnol 20:264–269
Harris SJ, Brown J, Lopez J, Yap TA (2016) Immuno-oncology combinations: raising the tail of the survival curve. Cancer Biol Med. doi:10.20892/j.issn.2095-3941.2016.0015
Hawkins RE, Russel SJ, Winter G (1992) Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol 226:889–896
Hemmerle T, Neri D (2014) The antibody-based targeted delivery of interleukin-4 and 23 to the tumor neovasculature eradicates tumors in three mouse models of cancer. Int J Cancer 134:467–477
Hernandez-Hoyos G, Sewell T, Bader R, Bannink J, Chenault RA, Daugherty M, Dasovich M, Fang H, Gottschalk R, Kumer J et al (2016) MOR209/ES414, a novel bispecific antibody targeting PSMA For the treatment of metastatic castration-resistant prostate cancer. Mol Cancer Ther 15:2155–2165
Hess J, Ruf P, Lindhofer H (2012) Cancer therapy with trifunctional antibodies: linking innate and adaptive immunity. Future Oncol 8:73–85
Hofman C, Seefried L, Jakob F (2016) Asfotase alfa: enzyme replacement for the treatment of bone disease in hypophosphatasia. Drugs Today (Barc) 52:271–285
Holliger P, Winter G (1997) Diabodies: small bispecific antibody fragments. Cancer Immunol Immunother 45:128–130
Holliger P, Prospero T, Winter G (1993) “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA 90:6444–6448
Hughes C, Sette A, Seed M, D’Acquisto F, Manzo A, Vincent TL, Lim NH, Nissim A (2014) Targeting of viral interleukin-10 with an antibody fragment specific to damaged arthritic cartilage improves its therapeutic potency. Arthritis Res Ther 16:R151. doi:10.1186/ar4613
Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotný M, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, Oppermann H (1988) Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci USA 85:5879–5883
Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y et al (2010a) Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 28:1203–1207
Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y et al (2010b) Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 23:385–392
Igawa T, Maeda A, Haraya K, Tachibana T, Iwayanagi Y, Mimoto F, Higuchi Y, Ishii S, Tamba S, Hironiwa N et al (2013) Engineered monoclonal antibody with novel antigen-sweeping activity in vivo. PLoS One 8:e63236
Igawa T, Haray K, Hattori K (2016) Sweeping antibody as a novel therapeutic antibody modality capable of eliminating soluble antigens from circulation. Immunol Rev 270:132–151
Ihospice F, Delphine B, Belmant C, Patrick D, Aristeidis C, Fischer E, Laurent G, Bonnafus C, Viaud N, Represa A et al (2015) Site-specific conjugation of monomethyl auristatin E to anti-CD30 antibodies improves their pharmacokinetics and therapeutic index in rodent models. Mol Pharm 12:1863–1871
Ilan Y, Gingis-Velitski S, Ben Ya’aco A, Shabbat Y, Zolotarov L, Almon E, Shaaltiel Y (2017) A plant cell- expressed recombinant anti-TNF fusion protein is biologically activity in the gut and alleviates immune- mediated hepatitis and colitis. Immunobiol 222:544–551
Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W, Zhang W, Tuaillon N, Rainey J, Barat B et al (2010) Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol 399:436–449
Jones PT, Dear PH, Foote J, Neuberger M, Winter G (1986) Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321:522–525
Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS et al (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26:925–932
Kanda Y, Yamane-Ohnuki N, Sakai N, Yamano K, Nakano R, Inoue M, Misaka H, Iida S, Wakitani M, Konno Y, Yano K, Shitara K, Hosoi S, Satoh M (2006) Comparison of cell lines for stable production of fucose- negative antibodies with enhanced ADCC. Biotechnol Bioeng 94:680–688
Kim J-S, Choi D-K, Shin J-Y, Shin S-M, Park S-W, Cho H-S, Kim Y-S (2016) Endosomal acidic pH-induced conformational changes of a cytosol-penetrating antibody mediate endosomal escape. J Controlled Rel 235:165–175
Kim BJ, Kim JH, Kim HS (2017) Survival benefit of immune checkpoint inhibitors according to the histology in non-small-cell lung cancer: a meta-analysis and review. Oncotarget. doi:10.18632/oncotarget.17214
Kinder M, Greenplate AR, Grugan K, Bannish G, Perpetua M, Jordan RE, Strohl WR, Brezski RJ (2013) Engineered protease-resistant antibodies with selectable cell-killing functions. J Biol Chem 288:30843–30854
Kingwell K (2017) CAR T therapies drives into new terrain. Nat Rev Drug Discov 16:301–304
Kipriyanov SM, Moldenhauer G, Schuhmacher J, Cochlovius B, von der Lieth C-W, Matys ER, Little M (1999) Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J Mol Biol 293:41–56
Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, Brinkmann U, Kettenberger H, Regula JT, Schaefer W (2012) Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 4:653–663
Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M et al (2017) Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology 6:e1277306
Kleinpeter P, Fend L, Thioudellet C, Geist M, Sfrontato N, Koerper V, Fahrner C, Schmitt D, Gantzer M, Remy-Ziller C et al (2016) Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death-1 (PD-1) allows their intratumoral delivery and an improved tumor- growth inhibition. Oncoimmunology 5:e1220467
Konkar A, Suckow A, Hummer T, Chodorge M, Celeste A, Hornigold D, Naylor J, Jenkinson L, Feigh M, Agoram B et al (2016) MEDI4166: a PCSK9 Ab-GLP-1 fusion molecule that elicits robust antidiabetic and antihyperlipidaemic effects in rodents and non-human primates EASD. Abstract #1107
Kontermann RE (2012) Dual targeting strategies with bispecific antibodies. Mabs 4:182–197
Kontermann RE, Brinkmann U (2015) Bispecific antibodies. Drug Discov Today 20:838–847
La Merie Publishing (2017) 2016 sales of recombinant therapeutic antibodies & proteins. Weikersheim 97990, Germany
Labrijn AF, Aalberse RC, Schuurman J (2008) When binding is enough: nonactivating antibody formats. Curr Opin Immunol 20:479–485
Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ et al (2013) Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci USA 110:5145–5150
Lalazar G, Mizrahi M, Turgeman I, Adar T, Ya’acov AB, Shabat Y, Nimer A, Hemed N, Zolotarovya L, Lichtenstein Y et al (2015) Oral administration of OKT3 Mab to patients with NASH, promotes regulatory T-cell induction, and alleviates insulin resistance: results of a phase IIa blinded placebo- controlled trial. J Clin Immunol 35:399–407
Lazar AC, Wang L, Blättler WA, Amphlett G, Lambert JM, Zhang W (2005) Analysis of the composition of immunoconjugates using size-exclusion chromatography coupled to mass spectrometry. Rapid Commun Mass Spectrom 19:1806–1814
Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, Veilmetter J, Carmichael DF, Hayes RJ, Dahiyat BI (2006) Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA 103:4005–4010
Lee KJ, Chow V, Weissman A, Tulpule S, Aldoss I, Akhtari M (2016) Clinical use of blinatumomab for B- cell acute lymphoblastic leukemia in adults. Ther Clin Risk Manag 12:1301–1310
Leung KM, Batey S, Rowlands R, Isaac SJ, Jones P, Drewett V, Carvalho J, Gaspar M, Weller S, Medcalf M, Wydro MM, Pegram R, Mudde GC, Bauer A, Moulder K, Woisetschläger M, Tuna M, Haurum JS, Sun H (2015) A HER2-specific modified Fc fragment (Fcab) induces antitumor effects through degradation of HER2 and apoptosis. Mol Ther 23:1722–1733
Lewis AD, Chen R, Montefiori DC, Johnson PR, Clark KR (2002) Generation of neutralizing activity against human immunodeficiency virus type 1 in serum by antibody gene transfer. J Virol 76:8769–8775
Li J, Ji J, Holmes LM, Burgin KE, Barton LB, Yu X, Wagner TE, Wei Y (2004) Fusion protein from RGD peptide and Fc fragment of mouse immunoglobulin G inhibits angiogenesis in tumor. Cancer Gene Ther 11:363–370
Li B, Tesar D, Boswell A, Cahaya H, Wong A, Zhang J, Gloria Meng Y, Eigenbrot C, Pantua H, Diao J et al (2014) Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 6:1255–1264
Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, Nicholas ND, Okeley NM, Lyon RP, Benjamin DR, Law C-L (2016) Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res 76:2710–2719
Liikanen I, Tahtinen S, Guse K, Gutmann T, Savola P, Oksanen M, Kanerva A, Hemminki A (2016) Oncolytic adenovirus expressing monoclonal antibody trastuzumab for treatment of HER2-positive cancer. Mol Cancer Ther 15:2259–2269
Lim WA, June CH (2016) The principles of engineering immune cells to treat cancer. Cell 168:724–740
Lim SI, Lukianov CI, Champion JA (2017) Self-assembled protein nanocarrier for intracellular delivery of antibody. J Controlled Rel 249:1–10
Limberis MP, Adam VS, Wong G, Gren J, Kobasa D, Ross TM, Konbinger GP, Tretiakova A, Wilson JM (2013) Intranasal antibody gene transfer in mice and ferrets elicits broad protection against pandemic influenza. Sci Trans Med 5(187):187ra72
Liu L, Patel B, Ghanem MH, Bundoc V, Zheng Z, Morgan RA, Rosenberg SA, Dey B, Berger EA (2015) Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J Virol 89:6685–6694
Liu L, Lam CK, Long V, Widjaja L, Yang Y, Li H, Jin L, Burke S, Gorlatov S, Brown J et al (2017) MGD011, a CD19 x CD3 dual-affinity retargeting bi-specific molecule incorporating extended circulating half-life for the treatment of B-cell malignancies. Clin Cancer Res 23:1506–1518
Llewelyn MB, Hawkins RE, Russell SJ (1992) Discovery of antibodies. BMJ 305:1269–1272
Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes J-M, Zhang Y, Lin YL, Chung S, Dennis MS, Joy Y et al (2017) Effector attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem 292:3900–3909
Lönn P, Kacsinta AD, Cui X-S, Hamil AS, Kaulich M, Gogoi K, Dowdy SF (2016) Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Nat Sci Rep 8(6):32301. doi:10.1038/srep32301
Luke T, Wu H, Zhao J, Channappanavar R, Coleman CM, Jiao J-A, Matsushita H, Liu Y, Postnikova EN, Ork BL et al (2016) Human polyclonal immunoglobulin G from transchromosomic bovines inhibits MERS- CoV in vivo. Sci Trans Med 8:326ra21. doi:10.1126/scitranslmed.aaf1061
Lum LG, Thakur A (2011) Targeting T cells with bispecific antibodies for cancer therapy. BioDrugs 25:365–379
Mack M, Riethmüller G, Kufer P (1995) A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci USA 92:7021–7025
MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, Brown AW, Triggiano MA, Hux JA, Pham CD et al (2017) Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol Ther 25:949–961
Madrenas J, Chau LA, Teft WA, Wu PW, Jussif J, Kasaian M, Carreno BM, Ling V (2004) Conversion of CTLA-4 from inhibitor to activator of T cells with a bispecific tandem single-chain Fc ligand. J Immunol 172:5948–5956
Malphettes L, Freyvert Y, Chang J, Liu PQ, Chan E, Miller JC, Zhou Z, Nguyen T, Tsai C, Snowden AW, Collingwood TN, Gregory PD, Cost GJ (2010) Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol Bioeng 106:774–783
Marschall ALJ, Zhang C, Frenzel A, Schirrmann T, Hust M, Perez F, Dübel S (2014) Delivery of antibodies to the cytosol. Debunking the myths. mAbs 6:943–956
Matsushita H, Sano A, Wu H, J-a Jiao, Kasinathan P, Sullivan EJ, Wang Z, Kuroiwa Y (2014) Triple immunoglobulin gene knockout transchromosomic cattle: bovine lambda cluster deletion and its effect on fully human polyclonal antibody production. PLoS ONE 9:e90383. doi:10.1371/journal.pone.0090383
Matsushita H, Sano A, Wu H, Wang Z, J-a Jiao, Kasinathan P, Sullivan EJ, Kuroiwa Y (2015) Species- specific chromosome engineering greatly improves fully human polyclonal antibody production profile in cattle. PLoS ONE 10:e0130699. doi:10.1371/journal.pone.0130699
McCracken MN, Cha AC, Weissman IL (2015) Molecular pathways: activating T cells after cancer cell phagocytosis from blockade of CD47 “don’t eat me” signals. Clin Cancer Res 21:3597–3601
Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P (1998) An efficient route to human bispecific IgG. Nat Biotechnol 16:677–681
Millan JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikova S, Terkeltaub R, Pleshko Camacho N, McKee MD et al (2008) Enzyme replacement therapy for murine hypophosphatasia. J Bone Min Res 23:777–787
Milstein C, Cuello AC (1983) Hybrid hybridomas and their use in immunohistochemistry. Nature 305:537–540
Mire CE, Geisbert JB, Agans KN, Thi EP, Lee AC, Fenton KA, Geisbert TW (2016) Passive immunotherapy: assessment of convalescent serum against ebola virus makona infection in nonhuman primates. J Infect Dis 214(Suppl 3):S367–S374
Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, Gorlatov S, Veri MC, Aggarwal S, Yang Y et al (2011) Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B- cell lymphoma. Blood 117:4542–4551
Moretti P, Skegro D, Ollier R, Wassmann P, Aebischer C, Laurent T, Schmid-Printz M, Giovannini R, Blein S, Bertschinger M (2013) BEAT® the bispecific challenge: a novel and efficient platform for the expression of bispecific IgGs. BMC Proc 7(Suppl 6):O9. http://www.biomedcentral.com/1753-6561/7/S6/O9
Morrison SL, Johnson MJ, Herzenberg LA, Oi VT (1984) Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci USA 81:6851–6855
Mueller JP, Giannoni MA, Hartman SL, Elliott EA, Squinto SP, Mathis LA, Evans MJ (1997) Humanized porcine VCAM-specific monoclonal antibodies with chimeric IgG2/G4 constant regions block human leukocyte binding to porcine endothelial cells. Mol Immunol 34:441–452
Neri D, Sondel PM (2016) Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol 40:96–102
Nesspor TC, Raju TS, Chin C-N, Vafa O, Brezski RJ (2012) Avidity confers FcγR binding and immune effector fuction to aglycosylated IgG1. J Mol Recognit 25:147–154
Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, Rueger P, Stracke JO, Lau W, Tissot AC et al (2014) Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81:49–60
Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, Inoges S, Melero I, Berraondo P (2017) Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol Cell Biol 95:347–355
Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR (2014) Site-specific antibody drug conjugates for cancer therapy. mAbs 6:34–45
Park JH, Brentjens RJ (2010) Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov Med 9:277–288
Pasche N, Wuhlfard S, Pretto F, Carugati E, Neri D (2012) The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clin Cancer Res 18:4092–4103
Penichet ML, Morrison SL (2001) Antibody–cytokine fusion proteins for the therapy of cancer. J Immunol Methods 248:91–101
Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM (2014) Antibody-drug conjugates: current status and future directions. Drug Disc Today 19:869–981
Polu KR, Lowman HB (2014) Probody therapeutics for targeting antibodies to diseased tissue. Expert Opin Biol Ther 14:1049–1053
Poul M-A, Becerril B, Nielsen UB, Morisson P, Marks JD (2000) Selection of tumor-specific internalizing antibodies form phage libraries. J Mol Biol 301:1149–1161
Queen C, Schneider WP, Selick HE, Payne PW, Landolfi NF, Duncan JF, Avdalovic NM, Levitt M, Junghans RP, Waldmann TA (1989) A humanized antibody that binds to the interleukin 2 receptor. Proc Natl Acad Sci USA 86:10029–10033
Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, Scheinberg DA, Brentjens RJ (2017) Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms tumor 1 antigen. Leukemia. doi:10.1038/leu.2016.373
Raju TS, Strohl WR (2013) Potential therapeutic roles for antibody mixtures. Expert Opin Biol Ther 13:1347–1352
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y (2017a) Multiplex genome editing to generate universal CAR T cells resistant to PD-1 inhibition. Clin Cancer Res 23:2255–2266
Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, Zhao Y (2017b) A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 8:17002–17011
Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I, Eser M, McAleese F, Molkenthin V, Gall FL, Topp M, Little M, Zhukovsky EA (2015) A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. mAbs 7:584–604
Ridgeway JB, Presta LG, Carter P (1996) ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 9:617–621
Robbie GJ, Criste R, Dall’acqua WF, Jensen K, Patel NK, Losonsky GA, Griffen MP (2013) A novel investigational Fc-modified humanized antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother 57:6147–6153
Roopenian DC, Akilesh S (2007) FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7:715–725
Root AR, Cao W, Li B, LaPan P, Meade C, Sanford J, Jin M, O’Sullivan C, Cummins E, Lambert M et al (2016) Development of PF-06671008, a highly potent anti-P-cadherin/anti-CD3 bispecific DART molecule with extended half-life for the treatment of cancer. Antibodies 5:6. doi:10.3390/antib5010006
Rother RP, Rollins SA, Mojcik CJ, Brodsky RJ, Bell L (2007) Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 25:1256–1264
Ruella M, Gill S (2015) How to train your T cell: genetically engineered chimeric antigen receptor T cells versus bispecific T-cell engagers to target CD19 in B acute lymphoblastic leukemia. Expert Opin Biol Ther 15:761–766
Ruella M, June CH (2016) Chimeric antigen receptor T cells for B cell neoplasms: choose the right CAR for you. Curr Hematol Malig Rep 11:368–384
Sahu GK, Sango K, Selliah N, Ma Q, Skowron G, Junghans RP (2013) Anti-HIV designer T cells progressively eradicate a latently infected cell line by sequentially inducing HIV reactivation then killing the newly gp120-positive cells. Virology 446:268–275
Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T et al (2013) Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of Factor VIII cofactor activity. PLoS ONE 8:e57479. doi:10.1371/journal.pone.0057479
Satta A, Mezzanzanica D, Turatti F, Canevari S, Figini M (2013) Redirection of T-cell effector functions for cancer therapy: bispecific antibodies and chimeric antigen receptors. Future Oncol 9:527–539
Schirdewahn T, Grabowski J, Sekyere SO, Bremer B, Wranke A, Lunemann S, Schlaphoff V, Kirschner J, Hardtke S, Manns MP et al (2017) The third signal cytokine IL-12 rather than immune checkpoint inhibitors contribute to the functional restoration of hepatitis D virus-specific T-cells. J Infect Dis 215:139–149
Schlereth B, Fichtner I, Lorenczewski G, Kleindienst P, Brischwein K, da Silva A, Kufer P, Lutterbuese R, Junghahn I, Kasimir-Bauer S et al (2005) Eradication of tumors from a human colon cancer cell line and from ovarian cancer metastases in immunodeficient mice by a single-chain Ep-CAM-/CD3-bispecific antibody construct. Cancer Res 65:2882–2889
Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E (2016) Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Prot Eng Des Sel 29:457–466
Schnepp BC, Johnson PR (2014a) Adeno-associated virus delivery of broadly neutralizing antibodies. Curr Opin HIV AIDS 9:250–256
Schnepp BC, Johnson PR (2014b) Vector-mediated in vivo antibody expression. Microbiol Spect 2(4):AID-0016-2014. doi: 10.1128/microbiolspec.AID-0016-2014
Seeliger D, Schulz P, Litzenburger T, Spitz J, Hoerer S, Blech M, Enenkel B, Studts JM, Garidel P, Karow AR (2015) Boosting antibody developability through rational sequence optimization. mAbs 7:505–515
Sharkey RM, Rossi EA, Chang C-H, Goldenberg DM (2010) Improved cancer therapy and molecular imaging with multivalent, multispecific antibodies. Cancer Biother Radiopharm 25:1–12
Shi Y, Fan X, Deng H, Brezski RJ, Rycyzyn M, Jordan RE, Strohl WR, Zou Q, Zhang N, An Z (2015) Trastuzumab triggers phagocytic killing of high HER2 cancer cells by interaction with FcγRIV on immune cells. J Immunol 194:4379–4386
Shields RL, Namenuk AK, Hog K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, Fox JA, Presta LG (2001) High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 276:6591–6604
Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SH, Presta LG (2002) Lack of fructose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma Rlll and antibody- dependent cellular toxicity. J Biol Chem 277:26733–26740
Siegmund V, Piater B, Zakeri B, Eichhorn T, Fischer F, Deutsch C, Becker S, Toleikis L, Hock B, Betz UAK, Kolmar H (2016) Spontaneous isopeptide bond formation as a powerful tool for engineering site- specific antibody-drug conjugates. Sci Rep 6:39291. doi:10.1038/srep39291
Silacci M, Lembke W, Woods R, Attinger-Toller I, Baenziger-Tobler N, Batey S, Santimaria R, von der Bey U, Koenig-Friedrich S, Zha W, Schlereth B, Locher M, Bertschinger J, Grabulovski D (2016) Discovery and characterization of COVA322, a clinical stage bispecific TNF/IL-17A inhibitor for the treatment of inflammatory diseases. mAbs 8:141–149
Singh AP, Sharma S, Shah DK (2016) Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J Pharmacokinet Pharmacodyn 43:567–582
Smith EJ, Olson K, Haber LJ, Varghese B, Duramad P, Tustian AD, Oyejide A, Kirshner JR, Canova L, Menon J, Principio J, MacDonald D, Kantrowitz J, Papadopoulos N, Stahl N, Yancopoulos GD, Thurston G, Davis S (2015) A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep 5:17943
Smith AJ, Oertle J, Warren D, Prato D (2016) Chimeric antigen receptor (CAR) T cell therapy for malignant cancers: summary and perspective. J Cell Immunol 2:59–68
Sondermann P, Szymkowski DE (2016) Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr Opin Immunol 40:78–87
Spiess C, Zhai Q, Carter PJ (2015) Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 67:95–106
Stewart MP, Sharei A, Ding X, Sayay G, Langer R, Jensen KF (2016) In vitro and ex vivo strategies for intracellular delivery. Nature 538:183–192
Storz U (2014) Rituximab. How approval history is reflected by a corresponding patent filing strategy. mAbs 6:820–837
Strohl WR (2009) Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol 20:685–691
Strohl WR (2011) Isotype selection and Fc engineering: design and construction of fit-for-purpose therapeutic antibodies. In: Wood C (ed) Antibody drug discovery. Imperial College Press, London, pp 147–220
Strohl WR (2015) Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 29:215–239
Strohl WR, Strohl LM (2012). Therapeutic antibody engineering: current and future advances driving the strongest growth area in the pharma industry. Woodhead Publishing Series in Biomedicine No. 11 (Cambridge). ISBN 9781907568374. Published Oct 22, 2012
Strop P, Ho W-H, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T et al (2012) Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol 420:204–219
Sun L-M, Liu Y-C, Li W, Liu S, Liu H-X, Li L-W, Ma R (2017) Nivolumab effectively inhibit platinum- resistant ovarian cancer cells via induction of cell apoptosis and inhibition of ADAM17 expression. Eur Rev Med Pharmacol Sci 21:1198–1205
Suzuki M, Curran KJ, Cheung N-KV (2015) Chimeric antigen receptos and bispecific antibodies to retarget T cells in pediatric oncology. Pediatr Blood Cancer 62:1326–1336
Tanoue K, Shaw AR, Watanabe N, Porter C, Rana B, Gottschalk S, Brenner M, Suzuki M (2017) Armed oncolytic adenovirus expressing PD-L1 mini-body enhances anti-tumor effects of chimeric antigen receptor T-cells in solid tumors. Cancer Res 77:2040–2051
Taylor RP, Lindorfer MA (2016) Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti-tumor monoclonal antibodies. Semin Immunol 28:309–316
Teicher BA (2009) Antibody-drug conjugate targets. Curr Cancer Drug Targets 9:982–1004
Thompson P, Ezeadi E, Hutchinson I, Fleming R, Bezabeh B, Lin J, Mao S, Chen C, Masterson L, Zhong H et al (2016) Straightforward glycoengineering approach to site-specific antibody-pyrrolobenzodiazepine conjugates. ACS Med Chem Lett 7:1005–1008
Throsby M, Logtenberg T, De Kruif J (2015) Generation of binding molecules. US 9145588 B2, Published Sept 29, 2015
Topalian SL, Drake CG, Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27:450–561
Tsuchikama K, An Z (2016) Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell (this issue)
Tustian AD, Endicott C, Adams B, Mattila J, Bak H (2016) Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs 8:828–838
Umaña P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE (1999) Engineered glycoforms of an anti- neuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 17:176–180
Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, Scallon B, Teplyakov A, Malia T, Strohl WR (2013) An engineered silent Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 65:114–126
van der Stegen SJC, Hamieh M, Sadelain M (2015) The pharmacology of second-generation chimeric antigen recpetors. Nat Rev Drug Discov 14:499–509
van der Vlist M, Kuball J, Radstake TRD, Meyaard L (2016) Immune checkpoints and rheumatic diseases: what can cancer immunotherapy teach us? Nat Rev Rheumatol 12:593–604
Von Kreudenstein TS, Escobar-Carbrera E, Lario PI, D’Angelo I, Brault K, Kelly J, Durocher Y, Baardsnes J, Woods RJ, Xie MH, Girod PA, Suits MD, Boulanger MJ, Poon DK, Ng GY, Dixit SB (2013) Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. mAbs 5:646–654
Walker MR, Lund J, Thompson KM, Jefferis R (1989) Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing FcγRI and/or FcγRII receptors. Biochem J 259:347–353
Wang Y, Yan Z, Huang Y, Qiu C, Chen X, Hu Y, Meng Q, Wei J (2017a) Study of natural antibodies against vascular endothelial growth factor receptor 1 in hepatocarcinoma. Am J Cancer Res 7:603–609
Wang B, Kothambawala T, Hinton P, Ng D, Saini A, Baliga R, Keyt B (2017b) Multimeric anti-DR5 IgM antibody displays potent cytotoxicity in vitro and promotes tumor regression in vivo. Abstract no 1702 AACR Ann Meet 2017, April 1–5, 2017
Ward ES, Güssow D, Griffiths AD, Jones PT, Winter G (1989) Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 341:544–546
Webster CI, Caram-Salas N, Haqqani AS, Thom G, Brown L, Rennie K, Yogi A, Costain W, Brunette E, Stanimirovic DB (2016) Brain penetration, target engagement, and disposition of the blood-brain barrier-crossing bispecific antibody antagonist of metabotropic glutamate receptor type 1. FASEB J 30:1927–1940
Wu C, Ting H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu RR, Santora L et al (2007) Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol 25:1290–1297
Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, Wakitani M, Niwa R, Sakurada M, Uchida K et al (2004) Establishment of FUT8 knockout Chinese hamster ovary cells; an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody- dependent cellular cytotoxicity. Biotechnol Bioeng 87:614–622
Yang L, Wang P (2014) Passive immunization against HIV/AIDS by antibody gene transfer. Viruses 6:428–447
Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, Dennis MS (2011) Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Trans Med 3:84ra44
Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, Bien-Ly N, Hersom M, Maloney JA, Meilandt WJ et al (2014) Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Trans Med 6:261ra154. doi:10.1126/scitranslmed.3009835
Yurkovetskiy AV, Yin M, Bodyak N, Stevenson CA, Thomas JD, Hammond CE, Qin LL, Zhu B, Gumerov DR, Ter-Ovanesyan E et al (2015) A polymer-based antibody-vinca drug conjugate platform: characterization and preclinical efficacy. Cancer Res 75:3365–3372
Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IWL, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR (2010) Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 28:157–159
Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubel S, Schmitt B, Lindhofer H (1999) Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol 163:1246–1252
Zeltsman M, Mayor M, Jones DR, Adusumilli PS (2016) Surgical immune interventions for solid malignancies. Am J Surg 212:682–690
Zhang X-Y, Zhang P-Y (2016) Receptor tyrosine kinases in carcinogenesis (Review). Oncol Lett 12:3679–3682
Zhou Y, Zou H, Zhang S, Marks JD (2010) Internalizing cancer antibodies from phage libraries selected on tumor cells and yeast displayed tumor antigens. J Mol Biol 404:88–99
Zhukovsky EA, Morse RJ, Maus MV (2016) Bispecific antibodies and CARs: generalized immunotherapeutics harnessing T cell redirection. Curr Opin Immunol 40:24–35
ABBREVIATIONS
AAVs, adeno-associated viruses; ADC, antibody-drug conjugates; ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; ADCs, antibody-drug conjugates; CAR, chimeric antigen receptor; CCR5, C-C motif chemokine receptor; CDC, complement-dependent cytotoxicity; CXCR4, C-X-C motif chemokine receptor 4; EGFR, epithelial growth factor receptor; EpCAM, epithelial cell adhesion molecule; ERBB2, erb-b2 receptor tyrosine kinase 2; GPCRs, G-protein coupled receptors; HIV, human immunodeficiency virus; mAb, monoclonal antibody; MS, multiple sclerosis; NK, natural killer; RA, rheumatoid arthritis; RSV, respiratory syncytial virus; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor
COMPLIANCE WITH ETHICAL GUIDELINES
William R Strohl declares that he has financial interest in Johnson & Johnson, for whom he was recently an employee, but no other potential conflicts of interest. This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell 9, 86–120 (2018). https://doi.org/10.1007/s13238-017-0457-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-017-0457-8