
ABSTRACT
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver dysfunction and a significant global health problem with substantial rise in prevalence over the last decades. It is becoming increasingly clear that NALFD is not only predominantly a hepatic manifestation of metabolic syndrome, but also involves extra-hepatic organs and regulatory pathways. Therapeutic options are limited for the treatment of NAFLD. Accordingly, a better understanding of the pathogenesis of NAFLD is critical for gaining new insight into the regulatory network of NAFLD and for identifying new targets for the prevention and treatment of NAFLD. In this review, we emphasize on the current understanding of the inter-organ crosstalk between the liver and peripheral organs that contributing to the pathogenesis of NAFLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
NAFLD refers to excess fat accumulation in the liver of patients with no history of alcohol abuse or other causes of secondary hepatic steatosis. Clinically, it represents a complex spectrum of hepatic damage, from simple steatosis and nonalcoholic steatohepatitis (NASH), progressing to fibrosis, and ultimately cirrhosis (Anstee et al., 2011). NAFLD is one of the most common public health problems worldwide. Recent epidemiology studies suggest that NAFLD is present in 12%–38% of the general population and NASH affects 3%–15% (Vernon et al., 2011). In China, NAFLD is becoming a greater health concern, with increasing rates of metabolic disturbances, such as obesity, type 2 diabetes mellitus, and dyslipidemia. The prevalence of NAFLD in patients with type 2 diabetes is 28%–55% and in those with hyperlipidemia is 27%–92% (Fan and Farrell, 2009). NAFLD is regarded as a hepatic manifestation of metabolic syndrome and is strongly associated with obesity and insulin resistance (Stojsavljevic et al., 2014). Although NAFLD is strongly associated with obesity and insulin resistance, its pathogenesis has not been fully elucidated and therapeutic options are limited. Current treatment is focused on the control of the disease process and risk factors. Thus a comprehensive understanding of the pathogenic mechanism of the development of NAFLD is extremely important (Cohen et al., 2011).
The underlying mechanism for the development and progression of NAFLD is complex and multifactorial. Different theories have been proposed; initially, the “two hits” hypothesis was developed to explain the pathogenesis of the NAFLD spectrum (Day and James, 1998). According to this traditional doctrine, the factors that metabolically promote the deposition of triacylglycerides (TAG) in the liver, including a high-fat diet, obesity, and insulin resistance, represent the “first hit” in the pathogenesis of NAFLD. Signaling process such as extracellular cytokines, adipokines, bacterial endotoxin, mitochondrial dysfunction, and/or endoplasmic reticulum (ER) stress provide the second hit for progression to NASH, activating inflammatory cascades and fibrogenesis (Greenberg et al., 1991; Peverill et al., 2014). However, the traditional “two-hit” pathophysiological theory has been challenged as knowledge of the interplay among insulin resistance, adipokines, adipose tissue inflammation, and other less recognized pathogenic factors have recently increased. In particular, it has been suggested that hepatic steatosis represents an epiphenomenon of several distinct injurious mechanisms, rather than a true “first hit” (Bulankina et al., 2009). For this reason, the initial “two-hit” theory explaining the progression from NAFLD to NASH has been evolved into the “multiple parallel hits” hypothesis (Jou et al., 2008; Buzzetti et al., 2016).
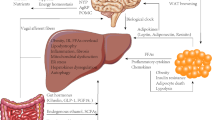
Several lines of evidence suggest that continuous inter-organ crosstalk sustains all processes involved in NAFLD pathogenesis, and crucial roles of the gut, hypothalamus, adipose tissue, and intestine have been suggested. In this review, we emphasize on the current understanding of the inter-organ crosstalk between the liver and peripheral organs that participated in the pathogenesis of NAFLD (Fig. 1).

The “cross-talk” between liver and peripheral organs in the pathogenesis of NAFLD. The impairment of the hypothalamic signaling pathway due to mutations (Leptin receptor and MC4R) by affecting the appetite or inflammation leads to the development of obesity and NAFLD. Dysfunction of adipose tissue in obesity, lipodystrophy or insulin resistance provides a source of excess fat and release of adipokines such as Leptin, Adiponectin, Resistin, and proinflammatory cytokines such as TNF-α and IL-6 that participated in the pathogenesis of NAFLD. In addition, emerging evidence suggests that an altered gut permeability consequently affect circulating levels of molecular such as LPS, FFA, bile acid, and to the release of pro-inflammatory cytokines by the regulation of TLR and FXR further influence the development and progression of NAFLD, recognized as effect of gut–liver axis. In the liver, the dysregulation of lipid de novo lipogenesis and imbalance of lipid influx and efflux causes lipotoxicity and may result in mitochondrial dysfunction, overproduction of ROS and ER stress as well as the consequent activation of inflammatory responses, thus influencing the risk of progression of NAFLD to NASH, as observed in obesity and insulin resistance
Liver and NAFLD
As mentioned above, epidemiological studies have revealed that NAFLD is a specific manifestation of metabolic syndrome and is strongly associated with obesity and insulin resistance. Specifically, hepatic steatosis arises from an imbalance between TAG influx and efflux. During hepatic TAG formation, fatty acids are derived from the diet, de novo lipogenesis (DNL), and adipose tissue via lipolysis. Once in hepatocytes, free fatty acids (FFAs) undergo acyl-CoA synthetic activity to form fatty acyl-CoAs, which further be oxidized in mitochondria via the β-oxidation pathway, re-esterified to TAG, and stored in lipid droplets (LDs) or coupled to apolipoproteins and further secreted as very low-density lipoprotein (VLDL) (Cohen et al., 2011). In obese patients, the increased efflux of FFAs from adipose tissue to the liver may induce defects in the insulin signaling pathway and contribute to insulin resistance (Bandsma et al., 2008; Braliou et al., 2008). In states of insulin resistance, sterol regulatory element binding protein-1c (SREBP-1c), the master regulator of DNL, is over-expressed and DNL is up-regulated (Stefan et al., 2008). Studies in rodent models revealed a peculiar feature of hepatic insulin resistance, in which hepatic glucose metabolism becomes unresponsive to insulin but hepatic lipogenesis continues unabated, recognized as selective hepatic insulin resistance (Brown and Goldstein, 2008, Li et al., 2010b, Cook et al., 2015). Selective hepatic insulin resistance has been proposed to explain the common clinical phenotype of hyperglycemia, hyperlipidemia, and NAFLD in T2D patients (Vatner et al., 2015). Additionally, β-oxidation of FFAs is inhibited in insulin resistance states, thus further promoting hepatic lipid accumulation (Donnelly et al., 2005, Postic and Girard, 2008, Cohen et al., 2011, Lambert et al., 2014). The inhibition of TAG incorporation into new VLDL by blocking micorsomal TAG transport protein (MTTP) and apolipoprotein B (ApoB) causes impaired TAG secretion which induces TAG accumulation in the liver (Amaro et al., 2010, Di Filippo et al., 2014). Hepatic inactivation of diacylgycerol acyltransferase 2 (DGAT2), a key enzyme catalyzing TAG synthesis, also reduces the hepatic TAG content and subsequently increases FFA oxidation, resulting in the worsening of steatohepatitis in mouse models (Yamaguchi et al., 2007).
Lipid droplets (LDs) are dynamic cytoplasmic organelles found ubiquitously in cells. They are linked to many cellular functions, including lipid storage for energy generation and membrane synthesis, viral replication, and protein degradation (Walther and Farese, 2012). Interestingly, connections between LD-associated proteins and NAFLD have been recently identified through genome-wide association studies (GWAS) as well as genomic and proteomic studies (Zhang et al., 2017).
Perilipins (PLINs) were the first type of specific LDs marker proteins identified in 1991 (Greenberg et al., 1991). The PLIN family contains several members, including perilipin 1 (PLIN1), perilipin 2/adipophilin (PLIN2), perilipin 3/Tip47 (PLIN3), perilipin 4 (PLIN4), and perilipin 5/OXPAT (PLIN5). While are not essential for LDs formation, PLINs are important for the regulation of lipid metabolism (Bulankina et al., 2009). PLIN1 is undetectable in normal liver, but is expressed in the liver of humans with NAFLD (Straub et al., 2008). PLIN2 has negative regulatory effects on VLDL lipidation (Chang et al., 2010) and TAG secretion. PLIN2, PLIN3, and PLIN5 levels are elevated in fatty liver of humans and their ablation alleviates steatosis in mouse models (Imai et al., 2007, Wang et al., 2015). Cell death-inducing DFFA-like effector (CIDE) proteins, which are located on LDs and the ER, are involved in fatty liver progression. Cidea and Cidec are responsible for liver steatosis under fasting and obese conditions by mediating the fusion of small and large LDs (Gong et al., 2011, Xu et al., 2016), while Cideb promotes lipid storage under a normal diet by regulating the process of VLDL lipidation and LDs fusion in the liver (Li et al., 2007, Ye et al., 2009)
Patatin-like phospholipid domain-containing protein 3 (PNPLA3) is another ER- and LD-associated protein. It is one of the few examples of a protein that has been validated in several populations to be conclusively shown to be associated with NAFLD, particularly the I148M (rs738409 C/G) variant (Romeo et al., 2008, Anstee, 2015). As a member of the PNPLA family, PNPLA3 is most closely related to PNPLA2 (ATGL), the major cellular TAG lipase. Neither ablation nor overexpression of wild-type PNPLA3 affects the liver fat content in mice, whereas transgenic mice with hepatic specific overexpression of human 148M or PNPLA3 I148M knock-in mice exhibit increased hepatic TAG contents and LD sizes and develop hepatic steatosis (Basantani et al., 2011, Li et al., 2012, Pirazzi et al., 2012, Smagris et al., 2015). Moreover, PNPLA3 I148M affects VLDL secretion in rat hepatoma cells and mouse livers (Pirazzi et al., 2012). These observations suggest two possible mechanisms for the pathogenesis of NAFLD induced by PNPLA3 mutations. First, PNPLA3 may alter lipolysis, not by hydrolysis activity itself, but by the inhibition of other proteins in the family, like ATGL (Smagris et al., 2015). Second, PNPLA3 mutations may reduce the mobilization of TAG on LDs (Pirazzi et al., 2012). Further studies are required to determine the precise mechanisms by which PNPLA3 regulates of hepatic lipid metabolism and determines its association with NASH and fibrosis.
17β-Hydroxysteroid dehydrogenase 13 (17β-HSD13) is a hepatic LD protein associated with NAFLD identified in recent proteomic studies (Su et al., 2014). Another independent study confirmed this result and also indicated a slight upregulation of 17β-HSD13 in patients with NASH without fatty liver (Kampf et al., 2014). In a study of fasted and refed mice, 17β-HSD13 expression was markedly higher on hepatic LDs of mice in the high-fat diet group than on those of mice in the low-fat group (Crunk et al., 2013). Overexpression of 17β-HSD13 in a mouse hepatocyte cell line induced liver steatosis and lipid accumulation. It also lead to increased expression of proteins involved in lipid synthesis, such as mature SREBP-1 and FAS, suggesting that 17β-HSD13 is involved in NAFLD development by promoting lipogenesis (Su et al., 2014).
In recent years, in the pathogenesis of NAFLD, more and more attention has been paid to the hepatokines, which are mainly produced by the liver. NAFLD seems to be associated with altered hepatokines production such as fetuin-A, fibroblast growth factor-21 (FGF-21), selenoprotein P, sex hormone-binding globulin (SHBG), angiopoietin-related growth factor (AGF) and leukocyte derived chemotaxin 2 (LECT2) (Lebensztejn et al., 2016). There is a suggestion that fetuin-A constitutes a link between obesity, insulin resistance and NAFLD, plays a major pathogenic role in metabolic disease (Mori et al., 2011, Pal et al., 2012, Stefan and Haring, 2013). Meanwhile, FGF21 has recently emerged as a novel hormone, leading to beneficial effects on glucose metabolism and lipid homeostasis, in addition to promoting rapid body weight loss in rodents (Li et al., 2010a). A number of publications showed significantly higher serum concentrations of FGF-21 in a population of patients with NAFLD compared to the controls (Yilmaz et al., 2010a, Yilmaz et al., 2010b, Giannini et al., 2013).
In addition, microRNAs (miRNAs), short, noncoding RNAs that regulate gene expression, have been associated with histological features of NAFLD and are readily detected in the circulation. As such, miRNAs are emerging as potentially useful noninvasive markers with which to follow the progression of NAFLD (DiStefano and Gerhard, 2016). Not only the potential mechanistic role of miRNAs involved in the pathogenesis of NAFLD described elsewhere (Gerhard and DiStefano, 2015), but also the multicellular nature and pathophysiological progression of NAFLD suggested that miRNAs may be associated with different disease stages (DiStefano and Gerhard, 2016). In a study of 84 circulating miRNAs measured in 47 NASH patients, 30 individuals with simple steatosis and 19 healthy controls, levels of miR-122, miR-192, and miR-375 were upregulated in patients with NASH compared to those with simple steatosis, and were associated with histological disease severity (Pirola et al., 2015). Furthermore, another non-coding RNA, long non-coding RNAs (lncRNAs) have emerged as important regulatory molecules in the pathogenesis of NAFLD (Li et al., 2015, Chen, 2016). Several fatty liver-related lncRNAs (FLRLs) have been identified to be related to lipogenesis, such as FLRL8, FLRL3 and FLRL7, through proteins in PPAR signaling pathway, such as Fabp5, Lpl and Fads2, indicating their potential regulatory role in lipid metabolism (Chen et al., 2017).
Fat accumulates in the liver of patients with NAFLD mainly in the form of TAG. This accumulation occurs concurrently with an increase in lipotoxicity owing to high levels of FFAs, free cholesterol, and other lipid metabolites. This lipotoxicity is believed to further lead to mitochondrial dysfunction with oxidative stress and the production of reactive oxygen species (ROS) and ER stress-associated mechanisms (Malhotra and Kaufman, 2007, Vonghia et al., 2013, Schneider and Cuervo, 2014, Buzzetti et al., 2016). Mechanistically, alterations in the structure and function of mitochondria contribute to the pathogenesis of NAFLD. Mitochondrial dysfunction may collapse respiratory oxidation with the impairment of fat homeostasis, generation of lipid-derived toxic metabolites, and overproduction of ROS (Begriche et al., 2006). Consequentially, increased ROS causes not only oxidative stress damage, but also the activation of Kupffer cells, whose activation is a key step in the development of NASH (Vonghia et al., 2013). Furthermore, ROS accumulation and related changes in autophagy cause chronic ER stress, which is closely related to the apoptosis of hepatocytes (Malhotra and Kaufman, 2007).
Hypothalamus and NAFLD
The central nervous system is crucial for the regulation of energy metabolism. In particular, the hypothalamus has critical roles in sensing and integrating signals from the periphery tissue and effecting appropriate physiological changes to maintain metabolic homeostasis (Zoccoli et al., 2011). The arcuate nucleus (ARC) provides many physiological roles involved in feeding, metabolism, and cardiovascular regulation (Bouret et al., 2004, Coppari et al., 2005, Sapru, 2013). More specifically, the ARC of hypothalamus is a specific nuclear group to sense different peripheral indicators of metabolic status and integrates responses to afferent information to control food intake and body weight (Schwartz et al., 2000).
The best-characterized peripheral indicator is leptin, which is an adipokine produced primarily in visceral adipocytes. Leptin signaling in the hypothalamus regulates hunger and energy expenditure, which are mediated by a neural circuitry comprising orexigenic and anorectic signals (Kwon et al., 2016). ARC neurons produced anorexigenic neuropeptides pro-opiomelanocortin (POMC), the precursor of α-melanocyte-stimulating hormone (α-MSH), and cocaine-amphetamine-regulated transcript (CART) are both activated by leptin (Cone et al., 1996, Kristensen et al., 1998), whereas orexigenic neuropeptide Y (NPY) and Agouti-related peptide (AgRP) are inhibited by leptin and activated by ghrelin (Broberger et al., 1998, Hahn et al., 1998). Central administration of NPY increases food intake, inhibits the thyroid axis, and decreases sympathetic nervous system outflow to brown adipose tissue, thus lowering energy expenditure. Conversely, stimulation of α-MSH receptors suppresses food intake, activates the thyroid axis, and increases energy expenditure (Wynne et al., 2005).
The spontaneous leptin mutation model ob/ob mice develop severe diabetes with marked hyperglycemia and a propensity to overeat, resulting in obesity and the development of hepatic steatosis (Mayer et al., 1951). A study reported that an obese leptin-deficient girl with hepatic steatosis exhibited rapid improvement after the introduction of recombinant leptin therapy (von Schnurbein et al., 2013). A recent meta-analysis indicated that circulating leptin levels were higher in patients with NAFLD than in controls. Importantly, such increased leptin levels were consistent with the severity of NAFLD, and the association remains significant after the exclusion of pediatric or adolescent populations as well as morbidly obese individuals subjected to bariatric surgery (Polyzos et al., 2016). Leptin action in peripheral tissues involves interaction with specific transmembrane receptors (leptin receptor, LEPR). The observation that LEPR is associated with NAFLD has been pointed by several studies. Among patients with NAFLD, LEPR polymorphisms were found to be associated with lipid metabolism, obesity parameters, and insulin resistance (Aller et al., 2012, Zain et al., 2013). Recent study suggested that the combined effect of variants of LEPR and PNPLA3 conferred increased susceptibility to NAFLD (Zain et al., 2013).
Approximately 20% of ARC NPY neurons innervate the paraventricular nucleus (PVN) (Baker and Herkenham, 1995). Stimulation of this pathway leads to increased food intake through direct stimulation of NPY receptors Y1R and Y5R and through AgRP antagonism of melanocortin receptors MC3R and MC4R in the PVN (Woods et al., 1998; Simpson et al., 2009). Moreover, the administration of α-MSH into the PVN inhibits food intake and the orexigenic effect of NPY administration (Woods et al., 1998). MC4R-KO mice fed with a high-fat diet developed a liver condition similar to human NASH and further progressed to HCC, which is associated with obesity, insulin resistance, and dyslipidemia. These phenotypes seem to result from a loss of function of MC4R in the hypothalamus, rather than in the liver itself (Itoh et al., 2011). Polymorphisms in MC4R are associated with alanine aminotransferase and BMI (Guan et al., 2014), but not with hepatic fat content in population-wide studies (Haupt et al., 2009).
In addition, hypothalamic inflammation was also shown involvement in the regulation of hepatic steatosis in physiological and pathophysiological conditions (Milanski et al., 2012; Valdearcos et al., 2015). Obesity-associated hypothalamic inflammation was first reported in a rat model of diet-induced obesity (De Souza et al., 2005), which was further confirmed by other groups (Zhang et al., 2008, Kleinridders et al., 2009, Milanski et al., 2009, Ozcan et al., 2009, Posey et al., 2009, Holland et al., 2011a). The observation that genetic interventions that disrupt neuronal inflammation can block both obesity and hypothalamic leptin resistance during feeding with a high-fat diet supports such role of inflammation in NAFLD pathogenesis. Several signaling pathways of the innate immune system have been identified as candidate mediators of hypothalamic inflammation during high-fat diet feeding, including toll-like receptor 4 (TLR 4), c-Jun N-terminal kinase (JNK), suppressor of cytokine signaling 3 (SOCS3), and pro-inflammatory cytokines, as well as the induction of ER stress and autophagy defects (De Souza et al., 2005, Zhang et al., 2008, Kleinridders et al., 2009, Milanski et al., 2009, Ozcan et al., 2009, Posey et al., 2009, Holland et al., 2011a).
Adipose and NAFLD
Adipose tissue, in addition to its function as the major storage depot for TAG, is an active endocrine organ that senses metabolic signals and secretes hormones that profoundly influence hepatic lipid metabolism (Berg et al., 2001). Excess adipose tissue in obesity and a lack of adipose tissue in the lipodystrophic state are associated with insulin resistance and NAFLD (Kahn and Flier, 2000).
Obesity, especially visceral adiposity, is a major risk factor for NAFLD in humans. Adipose tissue is a source of FFAs that are delivered to the liver, used for TAG synthesis by hepatocytes, and released into the blood (Diehl et al., 2005). The excess fat storage in obese and insulin-resistant individuals, and increased lipolysis in adipose tissue, are important source of FFA for hepatic TAG formation and storage in the liver of NAFLD patients (Cusi, 2012, Bril et al., 2014).
Additionally, NAFLD is also a typical finding in lipodystrophic patients. Lipodystrophy syndromes represent extreme and opposite ends of the adiposity spectrum related to obesity. A selective loss of body fat is the hallmark of lipodystrophy syndromes associated with an increased prevalence of insulin resistance in the skeletal muscles and the liver, increased plasma TAG levels, and hepatic steatosis (Kahn and Flier, 2000; Agarwal et al., 2004). In patients with lipodystrophy, defective adipose tissue is unable to store even regular amounts of energy. The inability of adipose tissue to store lipid in the form of TAG results in ectopic fat accumulation in aberrant tissues, such as the liver and skeletal muscle. Consequentially, excess TAG deposition in the liver (hepatic steatosis) and skeletal muscle induces NAFLD and peripheral insulin resistance (Agarwal and Garg, 2006). Several genetic mutations are strongly associated with lipodystrophy, including mutations in Agpat2, Pparγ, Lmna, Zmpste24, Akt2, and Bscl2 (Agarwal and Garg, 2006). Among these mutants, AGPAT2 induced lipodystrophy was indicated either by reducing triglyceride accumulation in adipocytes or levels of glycerophospholipids and hence affecting adipocyte function (Agarwal et al., 2004).
As an endocrine organ, adipose tissue is responsive to both peripheral and central metabolic signals by secreting a number of proteins, termed adipokines, to execute a variety of local, peripheral, and central effects (Stojsavljevic et al., 2014). For example, leptin is such a peptide hormone secreted mainly by visceral adipocytes to modulate food intake, body fat composition, insulin sensitivity, thermogenesis, and the immune system via the hypothalamus, as described above (Stojsavljevic et al., 2014).
Another adipokine, adiponectin, is a soluble matrix protein, is produced by visceral adipocytes (Arita et al., 1999). A great number of studies using human, animal, and in vitro models to investigate the pathogenesis and molecular mechanisms have shown that adiponectin influences obesity, insulin resistance, NAFLD, and other components of metabolic syndrome (Hu et al., 1996, Arita et al., 1999, Ryo et al., 2004, Matsuzawa, 2010). Adiponectin circulates in the serum in several oligomeric isoforms whose specific effects have been observed (Schober et al., 2007, Wang et al., 2008). In addition to that adiponectin improves hepatic and peripheral insulin resistance, it also presents some anti-inflammatory and hepato-protective activities (Kadowaki et al., 2006). These effects are partly achieved by enhancing the deacylation of ceramide sphingolipids, independently of AMPK, especially in hepatocytes (Holland et al., 2011b). The anti-inflammatory effects are achieved by blocking the activation of NF-κB, secreting anti-inflammatory cytokines, and inhibiting the release of pro-inflammatory cytokines, such as TNF-α and IL-6 (Tilg and Moschen, 2006). Adiponectin was also shown to have a direct antifibrotic effect (Kamada et al., 2003), which could be mediated by the activation of AMPK (Adachi and Brenner, 2008). Enhanced liver fibrosis has been demonstrated in mice lacking adiponectin (Kamada et al., 2003) whereas the delivery of recombinant adiponectin significantly improves steatohepatitis in mice (Xu et al., 2003). The antioxidant effects of adiponectin are mediated by its receptor AdipoR1 and thus decreased adiponectin levels in obesity may have causal roles in mitochondrial dysfunction and insulin resistance (Kamada et al., 2003). In obese patients, reduced adiponectin and increased leptin levels may result in hepatic steatosis and the activation of inflammation and fibrogenesis (Tsochatzis et al., 2006). Interestingly, adiponectin has been proposed as a good predictor of the necroinflammatory grade and fibrosis in NAFLD (Musso et al., 2005, Handy et al., 2010, Polyzos et al., 2011, Finelli and Tarantino, 2013). Furthermore, serum levels of adiponectin are reduced in obese subjects with type 2 diabetes mellitus and insulin resistance (Statnick et al., 2000, Maeda et al., 2001, Weyer et al., 2001, Spranger et al., 2003, Ozcelik et al., 2013). Replenishment of adiponectin ameliorates insulin resistance and glucose intolerance and decreases the liver triglyceride content in mice (Berg et al., 2001, Fruebis et al., 2001, Yamauchi et al., 2001, Okada-Iwabu et al., 2013). Additionally, resistin, another adipocyte-derived polypeptide, was initially found to be upregulated in obesity and insulin resistance (Holcomb et al., 2000, Steppan et al., 2001). In NAFLD, its levels were also higher than those in controls and were positively correlated with liver inflammation and fibrosis severity, although this result remains controversial (Zou et al., 2005, Pagano et al., 2006, Tsochatzis et al., 2008). In vitro studies have suggested that resistin participates in the progression of inflammation and its pro-inflammatory effects may be mediated by activating the c-Jun-N-terminal kinase (JNK) and NF-κB pathways (Zhang et al., 2010).
Currently, some view that development of NAFLD and insulin resistance may be resulted from imbalance cytokines, namely, increased pro-inflammatory and decreased anti-inflammatory cytokines (Diehl et al., 2005, Day, 2006). For instance, TNF-α is a proinflammatory cytokine that has various biological effects, including metabolic inflammatory and proliferative effects. It exhibits increased expression levels in the liver and adipose tissue of subjects with NAFLD. Data from human and animal studies have indicated that TNF-α has a role in the development of NAFLD and is a predictor of NASH that is correlated with advanced stages (Diehl, 2004, Cai et al., 2005, Jarrar et al., 2008). Furthermore, large quantities of IL-6 are secreted by visceral fat than by subcutaneous fat in obese individuals (Fontana et al., 2007). The role of visceral fat as an independent factor associated with NAFLD is due in large part to its secretion of proinflammatory cytokines (Van der Poorten et al., 2008). Inflammation and fibrosis in NAFLD patients were associated with increased systemic IL-6 (Van der Poorten et al., 2008), which decreased by therapy with Vitamin E in NAFLD patients in a small pilot study (Kugelmas et al., 2003).
Gut and NAFLD
Recently, compelling evidence links the gut microbiome, intestinal barrier integrity, bile acid and NAFLD, indicating that interactions between the liver and the gut, the so-called “gut–liver axis” may play a critical role in NAFLD onset and progression.
Emerging evidence indicates that the human gut microbiota is involved in the development of obesity and related complications, including NAFLD (Drenick et al., 1982, Backhed et al., 2004, Zhu et al., 2015). Gut microflora may stimulate hepatic fat deposition and promote NASH by several mechanisms (Aron-Wisnewsky et al., 2013, Gkolfakis et al., 2015). Changes to the microbiome regulate gut permeability and increase hepatic exposure to injurious substances that increase hepatic inflammation and fibrosis. The first evidence of an increased intestinal permeability (leaky gut) and tight junction alterations in NAFLD patients compared with healthy subjects was reported in 2009 (Miele et al., 2009). Since then, more studies in humans and mice have confirmed the association between impaired intestinal barrier function and hepatic fibrogenesis and inflammation (Miele et al., 2009, Gabele et al., 2011).
The gut microbiota also regulates immune balance and participates in the development and homeostasis of overall host immunity (Burcelin et al., 2012). The cross-talk between host and bacteria, which depends on Toll-like receptors (TLRs) or NOD-like receptors, is responsible for innate and adaptive immune responses that protect the host and maintain intestinal homeostasis (Compare et al., 2012). TLRs recognize highly conserved microbial molecules called “pathogen-associated molecular patterns” (PAMPs) or damage-associated molecular patterns (DAMPs) and initiate a signaling cascade leading to the activation of pro-inflammatory genes, such as TNF-α, IL-6, IL-8, and IL-12 (Pisetsky, 2011, Alisi et al., 2012). Lipopolysaccharide (LPS), the most extensively studied PAMP, is a component of the gram-negative bacteria cell membrane and the active component of endotoxin. LPS-TLR-4 signaling activation is related to insulin-resistance and NASH. Studies on TLR-4 null mice have confirmed that TLR-4 is essential for hepatic fat deposition and NASH development (Poggi et al., 2007, Saberi et al., 2009, Henao-Mejia et al., 2012). In addition, the inflammasome, that is composed of leucine-rich-repeat-containing proteins and nucleotide-binding domain (NLRPs) can act as sensors of PAMPs and DAMPs and participate in the activation of lipid peroxidation and ROS production during NAFLD/NASH progression (Thuy et al., 2008, Harte et al., 2010, Henao-Mejia et al., 2012). Moreover, dysbiosis also affects the metabolism of food substrates, by increasing the production of certain short-chain fatty acids and alcohol and depleting choline.
The bile acid 90% excreted by the gallbladder is reabsorbed in the small intestine and is recycled back to the liver through the portal vein and have emerged as relevant signaling molecules that function in the liver to regulate lipid and carbohydrate metabolic pathways as well as energy homeostasis. Bile acids may function as signaling molecules via a variety of receptors, including members of the nuclear receptor superfamily (farnesoid X receptor [FXR; NR1H4], Vitamin D receptor [NR1I1], and pregnane X receptor [NR1I2]) and members of the G-protein-coupled receptor superfamily (TGR5), to regulate their own synthesis as well as other metabolic processes, such as glucose, lipid, and energy homeostasis (Maeda et al., 2001). Specifically, FXR, originally named for its ability to bind to farnesoid, has shown to play a role in the regulation of lipid metabolism (Sinal et al., 2000, Schaap et al., 2014, Carr and Reid, 2015, Mazuy et al., 2015, Fuchs et al., 2016). FXR-KO mice exhibit a proatherogenic lipoprotein profile with markedly elevated serum and hepatic cholesterol and triglycerides levels (Sinal et al., 2000, Arab et al., 2017). The activation of FXR represses hepatic DNL and stimulates fatty acid β-oxidation, limiting hepatic lipid accumulation (Pineda Torra et al., 2003, Watanabe et al., 2004, Savkur et al., 2005, Moore, 2012). FXR can also promote plasma VLDL triglyceride clearance by inducing the expression of ApoCII, an activator of lipoprotein lipase, and suppressing the expression of ApoCIII, an inhibitor of lipoprotein lipase activity (Mazuy et al., 2015, Fuchs et al., 2016). In addition, the gut flora modifies bile acid metabolism and FXR/TGR5 signaling and hence contributes indirectly to the development of NAFLD (Tremaroli et al., 2012).
The colon is a major site of gut bacterial fermentation, yielding high levels of short chain fatty acids (SCFAs, 70–130 mmol/L) (Duncan et al., 2009). The main substrates for the production of SCFAs by the colonic microbiota are dietary carbohydrates that have escaped digestion in the small intestine, collectively referred to as dietary fibre (Psichas et al., 2015). Experiments comparing the feces of obese and lean individuals demonstrated that the level of short-chain fatty acids was higher in the obese whereas residual calories from food were concomitantly reduced (Turnbaugh et al., 2006, Schwiertz et al., 2010). SCFAs therefore have been proposed to contribute to obesity and liver steatosis as they provide approximately 10% of daily caloric consumption and may enhance nutrient absorption by promoting expression of glucagon-like peptide 2 (den Besten et al., 2013, Boursier and Diehl, 2015) (Zhu et al., 2015). However, SCFAs also improve lipid and glucose metabolism and maintain intestinal homeostasis (den Besten et al., 2013, Puertollano et al., 2014, Boursier and Diehl, 2015). Hence, the net effect of SCFAs on NAFLD pathogenesis remains unclear and is likely complex. For example, although total cecal SCFA concentrations of recipient mice given flora from responder versus nonresponder mice were similar in the Leroy study, two branched-chain fatty acids (isobutyrate and isovalerate) were significantly higher in responder-receiver mice (Boursier and Diehl, 2015). Branched-chain fatty acids, which can be de novo synthesized by several gut bacterial species, have been associated with insulin resistance and metabolic disease development (Newgard, 2012, Boursier and Diehl, 2015).
Extracellular vesicles and NAFLD
Exosomes are small membrane-bound extracellular vesicles (EVs) released by various types of cells into biological fluids (Sato et al., 2016). There are two main populations of EVs, namely exosomes and microparticles (MPs), which differ in size, composition, and mechanism of generation. Exosomes are small, 30–100 nm in diameter, and are released by exocytosis as a result of multivesicular bodies fusing with the plasma membrane (Masyuk et al., 2013). EVs have been the topic of great interest in recent years in NAFLD research. Patients with NAFLD or NASH secrete increased levels of microvesicles derived from macrophages and natural killer T cells (Kornek et al., 2012). Another EV study has shown that the expression levels of various proteins within vesicles are enhanced in a mouse model of NAFLD, and that protein expression pattern differs between exosomes and microvesicles (Povero et al., 2014).
EVs are involved in NAFLD pathology because they regulate cell-cell communication and a number of pathophysiological events in various types of cells via horizontal transfer of their cargo including proteins (membrane, cytosolic, and nuclear), RNAs (including mRNAs and microRNAs), and lipids (Yuan et al., 2009, Diehl et al., 2012, Raposo and Stoorvogel, 2013). Notably, released EVs do not only stay in the tissue of origin, but also circulate in the blood stream (Povero et al., 2014). Recent studies have demonstrated that primary and immortalized hepatocytes are capable of producing and releasing both exosomes and MPs (Conde-Vancells et al., 2008, Witek et al., 2009, Pan et al., 2012, Povero et al., 2013). EVs are formed and released during the accumulation of lipotoxic lipids in hepatocytes, which is a key mechanism of liver damage and disease progression in NAFLD (Povero et al., 2014).
In obese individuals, adipocyte-derived exosomes are known to contribute to the development of insulin resistance via activation of adipose-resident macrophages and secretion of proinflammatory cytokines that can result in insulin resistance (Deng et al., 2009). Recent evidence indicated that visceral obese adipocytes shed exosomes that contain mediators capable of activating end-organ inflammatory and fibrotic signaling pathways and these exosomes contain miRNAs capable of regulating end-organ TGF-β and Wnt/β-catenin signaling in obesity-related comorbid conditions (Zhu et al., 2015).
Conclusion
The pathogenesis of NAFLD and its progression is a complex process. Increasing evidence indicates that a number of diverse and parallel processes contribute to the development of NAFLD and liver inflammation. The impairment of the hypothalamic signaling pathway due to mutations or inflammation leads to the development of obesity and NAFLD. Dysfunction of adipose tissue in obesity or lipodystrophy provides a source of excess fat and results in the secretion of multiple factors involved in the pathogenesis of NAFLD. In addition, emerging evidences suggest that an altered gut microbiota can influence the development and progression of NAFLD, possibly via the gut–liver axis. In the liver, the dysregulation of lipid de novo lipogenesis and imbalance of lipid influx and efflux causes lipotoxicity which may further result in mitochondrial dysfunction and ER stress as well as the consequent activation of inflammatory responses, as observed in obesity and insulin resistance. Combined with the rapidly increasing expending field of studies of miRNAs and LncRNA research suggest that the identification and validation these non coding RNA may improve the diagnosis and clinical monitoring of NAFLD progression.
An improved knowledge of the pathogenic “cross-talk” between the liver and extra-hepatic organs will not only help to modulate known risk factors associated with the onset of NAFLD and/or its progression to end-stage liver disease but may also provide insight for the development of new pharmacological treatments for NAFLD.
REFERENCES
Adachi M, Brenner DA (2008) High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology 47:677–685
Agarwal AK, Garg A (2006) Genetic basis of lipodystrophies and management of metabolic complications. Annu Rev Med 57:297–311
Agarwal AK, Barnes RI, Garg A (2004) Genetic basis of congenital generalized lipodystrophy. Int J Obes Relat Metab Disord 28:336–339
Alisi A, Ceccarelli S, Panera N, Nobili V (2012) Causative role of gut microbiota in non-alcoholic fatty liver disease pathogenesis. Front Cell Infect Microbiol 2:132
Aller R, De Luis DA, Izaola O, Gonzalez Sagrado M, Conde R, Pacheco D, Velasco MC, Ovalle HF (2012) Lys656Asn polymorphism of leptin receptor, leptin levels and insulin resistance in patients with non alcoholic fatty liver disease. Eur Rev Med Pharmacol Sci 16:335–341
Amaro A, Fabbrini E, Kars M, Yue P, Schechtman K, Schonfeld G, Klein S (2010) Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology 139:149–153
Anstee QDC (2015) The genetics of nonalcoholic fatty liver disease: spotlight on PNPLA3 and TM6SF2. Semin Liver Dis 35:270–290
Anstee QM, McPherson S, Day CP (2011) How big a problem is non-alcoholic fatty liver disease? BMJ 343:d3897
Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M (2017) Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology 65:350–362
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K et al (1999) Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79–83
Aron-Wisnewsky J, Gaborit B, Dutour A, Clement K (2013) Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin Microbiol Infect 19:338–348
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101:15718–15723
Baker RA, Herkenham M (1995) Arcuate nucleus neurons that project to the hypothalamic paraventricular nucleus: neuropeptidergic identity and consequences of adrenalectomy on mRNA levels in the rat. J Comp Neurol 358:518–530
Bandsma RH, Prinsen BH, van Der Velden Mde S, Rake JP, Boer T, Smit GP, Reijngoud DJ, Kuipers F (2008) Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr Res 63:702–707
Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, Schoiswohl G, Yang K, Kumari M, Gross RW et al (2011) Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res 52:318–329
Begriche K, Igoudjil A, Pessayre D, Fromenty B (2006) Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion 6:1–28
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001a) The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:947–953
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001b) The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:7
Bouret SG, Draper SJ, Simerly RB (2004) Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci 24:2797–2805
Boursier J, Diehl AM (2015) Implication of gut microbiota in nonalcoholic fatty liver disease. PLoS Pathog 11:e1004559
Braliou GG, Verga Falzacappa MV, Chachami G, Casanovas G, Muckenthaler MU, Simos G (2008) 2-Oxoglutarate-dependent oxygenases control hepcidin gene expression. J Hepatol 48:801–810
Bril F, Lomonaco R, Orsak B, Ortiz-Lopez C, Webb A, Tio F, Hecht J, Cusi K (2014) Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology 59:2178–2187
Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T (1998) The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci USA 95:15043–15048
Brown MS, Goldstein JL (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7:95–96
Bulankina AV, Deggerich A, Wenzel D, Mutenda K, Wittmann JG, Rudolph MG, Burger KN, Honing S (2009) TIP47 functions in the biogenesis of lipid droplets. J Cell Biol 185:641–655
Burcelin R, Garidou L, Pomie C (2012) Immuno-microbiota cross and talk: the new paradigm of metabolic diseases. Semin Immunol 24:67–74
Buzzetti E, Pinzani M, Tsochatzis EA (2016) The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65:1038–1048
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11:183–190
Carr RM, Reid AE (2015) FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr Atheroscl Rep 17(4):500
Chang BH, Li L, Saha P, Chan L (2010) Absence of adipose differentiation related protein upregulates hepatic VLDL secretion, relieves hepatosteatosis, and improves whole body insulin resistance in leptin-deficient mice. J Lipid Res 51:2132–2142
Chen Z (2016) Progress and prospects of long noncoding RNAs in lipid homeostasis. Mol Metab 5:164–170
Chen Y, Huang H, Xu C, Yu C, Li Y (2017) Long non-coding RNA profiling in a non-alcoholic fatty liver disease rodent model: new insight into pathogenesis. Int J Mol Sci. doi:10.3390/ijms18010021
Cohen JC, Horton JD, Hobbs HH (2011) Human fatty liver disease: old questions and new insights. Science 332:1519–1523
Compare D, Coccoli P, Rocco A, Nardone OM, De Maria S, Carteni M, Nardone G (2012) Gut–liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis 22:471–476
Conde-Vancells J, Rodriguez-Suarez E, Embade N, Gil D, Matthiesen R, Valle M, Elortza F, Lu SC, Mato JM, Falcon-Perez JM (2008) Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J Proteome Res 7:5157–5166
Cone RD, Lu D, Koppula S, Vage DI, Klungland H, Boston B, Chen W, Orth DN, Pouton C, Kesterson RA (1996) The melanocortin receptors: agonists, antagonists, and the hormonal control of pigmentation. Recent Prog Horm Res 51:287–317; discussion 318
Cook JR, Langlet F, Kido Y, Accili D (2015) Pathogenesis of selective insulin resistance in isolated hepatocytes. J Biol Chem 290:13972–13980
Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC Jr et al (2005) The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab 1:63–72
Crunk AE, Monks J, Murakami A, Jackman M, Maclean PS, Ladinsky M, Bales ES, Cain S, Orlicky DJ, McManaman JL (2013) Dynamic regulation of hepatic lipid droplet properties by diet. PLoS ONE 8:e67631
Cusi K (2012) Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 142(711–725):e716
Day CP (2006) From fat to inflammation. Gastroenterology 130:207–210
Day CPJ, James OF (1998) Steatohepatitis: a tale of two “hits”. Gastroenterology 114:842–845
De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJA, Velloso LA (2005) Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146:4192–4199
den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54:2325–2340
Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y, Wang J, Xiang X, Zhang S, Zhuang X et al (2009) Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58:2498–2505
Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, Peretti N, Dumortier J, Zoulim F, Fontanges T et al (2014) Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol 61:891–902
Diehl AM (2004) Tumor necrosis factor and its potential role in insulin resistance and nonalcoholic fatty liver disease. Clin Liver Dis 8:619-638, x
Diehl AM, Li ZP, Lin HZ, Yang SQ (2005) Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 54:303–306
Diehl P, Fricke A, Sander L, Stamm J, Bassler N, Htun N, Ziemann M, Helbing T, El-Osta A, Jowett JB et al (2012) Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovasc Res 93:633–644
DiStefano JK, Gerhard GS (2016) Circulating microRNAs in nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol 10:161–163
Donnelly KLSC, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Investig 115:1343–1351
Drenick EJ, Fisler J, Johnson D (1982) Hepatic steatosis after intestinal bypass–prevention and reversal by metronidazole, irrespective of protein-calorie malnutrition. Gastroenterology 82:535–548
Duncan SH, Louis P, Thomson JM, Flint HJ (2009) The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol 11:2112–2122
Fan JG, Farrell GC (2009) Epidemiology of non-alcoholic fatty liver disease in China. J Hepatol 50:204–210
Finelli C, Tarantino G (2013) What is the role of adiponectin in obesity related non-alcoholic fatty liver disease? World J Gastroenterol 19:802–812
Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S (2007) Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 56:1010–1013
Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF (2001) Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA 98:2005–2010
Fuchs CD, Traussnigg SA, Trauner M (2016) Nuclear receptor modulation for the treatment of nonalcoholic fatty liver disease. Semin Liver Dis 36:69–86
Gabele E, Dostert K, Hofmann C, Wiest R, Scholmerich J, Hellerbrand C, Obermeier F (2011) DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol 55:1391–1399
Gerhard GS, DiStefano JK (2015) Micro RNAs in the development of non-alcoholic fatty liver disease. World J Hepatol 7:226–234
Giannini C, Feldstein AE, Santoro N, Kim G, Kursawe R, Pierpont B, Caprio S (2013) Circulating levels of FGF-21 in obese youth: associations with liver fat content and markers of liver damage. J Clin Endocrinol Metab 98:2993–3000
Gkolfakis P, Dimitriadis G, Triantafyllou K (2015) Gut microbiota and non-alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int 14:572–581
Gong J, Sun Z, Wu L, Xu W, Schieber N, Xu D, Shui G, Yang H, Parton RG, Li P (2011) Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol 195:953–963
Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C (1991) Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem 266:11341–11346
Guan L, Shang XR, Liu FH, Song JY, Ma J, Wang HJ (2014) Association of INSIG2 rs9308762 with ALT level independent of BMI. J Pediatr Gastroenterol Nutr 58:155–159
Hahn TM, Breininger JF, Baskin DG, Schwartz MW (1998) Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci 1:271–272
Handy JA, Saxena NK, Fu P, Lin S, Mells JE, Gupta NA, Anania FA (2010) Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis via suppressors of cytokine signaling (SOCS-3). J Cell Biochem 110:1195–1207
Harte AL, da Silva NF, Creely SJ et al (2010) Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond) 7:15
Haupt A, Thamer C, Heni M, Tschritter O, Machann J, Schick F, Machicao F, Haring HU, Staiger H, Fritsche A (2009) Impact of variation near MC4R on whole-body fat distribution, liver fat, and weight loss. Obesity (Silver Spring) 17:1942–1945
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ et al (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482:179–185
Holcomb IN, Kabakoff RC, Chan B, Baker TW, Gurney A, Henzel W, Nelson C, Lowman HB, Wright BD, Skelton NJ et al (2000) FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J 19:4046–4055
Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR et al (2011a) Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121:1858–1870
Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM et al (2011b) Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17:55–63
Hu E, Liang P, Spiegelman BM (1996) AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem 271:10697–10703
Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS (2007) Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology 132:1947–1954
Itoh M, Suganami T, Nakagawa N, Tanaka M, Yamamoto Y, Kamei Y, Terai S, Sakaida I, Ogawa Y (2011) Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol 179:2454–2463
Jarrar MH, Baranova A, Collantes R, Ranard B, Stepanova M, Bennett C, Fang Y, Elariny H, Goodman Z, Chandhoke V et al (2008) Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol Ther 27:412–421
Jou J, Choi SS, Diehl AM (2008) Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis 28:370–379
Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116:1784–1792
Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106:473–481
Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H et al (2003) Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 125:1796–1807
Kampf C, Mardinoglu A, Fagerberg L, Hallstrom BM, Edlund K, Lundberg E, Ponten F, Nielsen J, Uhlen M (2014) The human liver-specific proteome defined by transcriptomics and antibody-based profiling. FASEB J 28:2901–2914
Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Bruning JC (2009) MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab 10:249–259
Kornek M, Lynch M, Mehta SH, Lai M, Exley M, Afdhal NH, Schuppan D (2012) Circulating microparticles as disease-specific biomarkers of severity of inflammation in patients with hepatitis C or nonalcoholic steatohepatitis. Gastroenterology 143:448–458
Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang N et al (1998) Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature 393:72–76
Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ (2003) Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology 38:413–419
Kwon O, Kim KW, Kim MS (2016) Leptin signalling pathways in hypothalamic neurons. Cell Mol Life Sci 73:1457–1477
Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ (2014) Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146:726–735
Lebensztejn DM, Flisiak-Jackiewicz M, Bialokoz-Kalinowska I, Bobrus-Chociej A, Kowalska I (2016) Hepatokines and non-alcoholic fatty liver disease. Acta Biochim Pol 63:459–467
Li JZ, Ye J, Xue B, Qi J, Zhang J, Zhou Z, Li Q, Wen Z, Li P (2007) Cideb regulates diet-induced obesity, liver steatosis, and insulin sensitivity by controlling lipogenesis and fatty acid oxidation. Diabetes 56:2523–2532
Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, Zhang H, Pan X, Bao Y, Xiang K et al (2010a) Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol 53:934–940
Li S, Brown MS, Goldstein JL (2010b) Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA 107:3441–3446
Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, Hobbs HH (2012) Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 122:4130–4144
Li P, Ruan X, Yang L, Kiesewetter K, Zhao Y, Luo H, Chen Y, Gucek M, Zhu J, Cao H (2015) A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell Metab 21:455–467
Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N et al (2001) PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 50:2094–2099
Malhotra JD, Kaufman RJ (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9:2277–2293
Masyuk AI, Masyuk TV, Larusso NF (2013) Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J Hepatol 59:621–625
Matsuzawa Y (2010) Adiponectin: a key player in obesity related disorders. Curr Pharm Des 16:1896–1901
Mayer J, Bates MW, Dickie MM (1951) Hereditary diabetes in genetically obese mice. Science 113:746–747
Mazuy C, Helleboid A, Staels B, Lefebvre P (2015) Nuclear bile acid signaling through the farnesoid X receptor. Cell Mol Life Sci 72:1631–1650
Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Masciana R, Forgione A, Gabrieli ML, Perotti G et al (2009) Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49:1877–1887
Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DML, Anhe G, Amaral ME, Takahashi HK et al (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29:359–370
Milanski M, Arruda AP, Coope A, Ignacio-Souza LM, Nunez CE, Roman EA, Romanatto T, Pascoal LB, Caricilli AM, Torsoni MA et al (2012) Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes 61:1455–1462
Moore DD (2012) Nuclear receptors reverse McGarry’s vicious cycle to insulin resistance. Cell Metab 15:615–622
Mori K, Emoto M, Inaba M (2011) Fetuin-A: a multifunctional protein. Recent Pat Endocr Metab Immune Drug Discov 5:124–146
Musso G, Gambino R, Biroli G, Carello M, Faga E, Pacini G, De Michieli F, Cassader M, Durazzo M, Rizzetto M et al (2005) Hypoadiponectinemia predicts the severity of hepatic fibrosis and pancreatic Beta-cell dysfunction in nondiabetic nonobese patients with nonalcoholic steatohepatitis. Am J Gastroenterol 100:2438–2446
Newgard CB (2012) Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 15:606–614
Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M et al (2013) A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 503:493–499
Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Ozcan U (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9:35–51
Ozcelik F, Yuksel C, Arslan E, Genc S, Omer B, Serdar MA (2013) Relationship between visceral adipose tissue and adiponectin, inflammatory markers and thyroid hormones in obese males with hepatosteatosis and insulin resistance. Arch Med Res 44:273–280
Pagano C, Soardo G, Pilon C, Milocco C, Basan L, Milan G, Donnini D, Faggian D, Mussap M, Plebani M et al (2006) Increased serum resistin in nonalcoholic fatty liver disease is related to liver disease severity and not to insulin resistance. J Clin Endocrinol Metab 91:1081–1086
Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S (2012) Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med 18:1279–1285
Pan Q, Ramakrishnaiah V, Henry S, Fouraschen S, de Ruiter PE, Kwekkeboom J, Tilanus HW, Janssen HL, van der Laan LJ (2012) Hepatic cell-to-cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut 61:1330–1339
Peverill W, Powell LW, Skoien R (2014) Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int J Mol Sci 15:8591–8638
Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B (2003) Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol 17:259–272
Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Stahlman M, Taskinen MR, Orho-Melander M, Perman J, Pujia A et al (2012) Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 57:1276–1282
Pirola CJ, Fernandez Gianotti T, Castano GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M, Flichman D, Mirshahi F, Sanyal AJ, Sookoian S (2015) Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut 64:800–812
Pisetsky D (2011) Cell death in the pathogenesis of immune-mediated diseases: the role of HMGB1 and DAMP-PAMP complexes. Swiss Med Wkly 141:w13256
Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, Peiretti F, Verdier M, Juhan-Vague I, Tanti JF et al (2007) C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50:1267–1276
Polyzos SA, Toulis KA, Goulis DG, Zavos C, Kountouras J (2011) Serum total adiponectin in nonalcoholic fatty liver disease: a systematic review and meta-analysis. Metabolism 60:313–326
Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS (2016) Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia 59:30–43
Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC et al (2009) Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 296:E1003–E1012
Postic C, Girard J (2008) Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 118:829–838
Povero D, Eguchi A, Niesman IR, Andronikou N, de Mollerat du Jeu X, Mulya A, Berk M, Lazic M, Thapaliya S, Parola M et al (2013) Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci Signal 6: ra88
Povero D, Eguchi A, Li H, Johnson CD, Papouchado BG, Wree A, Messer K, Feldstein AE (2014) Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS ONE 9:e113651
Psichas A, Sleeth ML, Murphy KG, Brooks L, Bewick GA, Hanyaloglu AC, Ghatei MA, Bloom SR, Frost G (2015) The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int J Obes (Lond) 39:424–429
Puertollano E, Kolida S, Yaqoob P (2014) Biological significance of short-chain fatty acid metabolism by the intestinal microbiome. Curr Opin Clin Nutr Metab Care 17:139–144
Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383
Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH (2008) Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40:1461–1465
Ryo M, Nakamura T, Kihara S, Kumada M, Shibazaki S, Takahashi M, Nagai M, Matsuzawa Y, Funahashi T (2004) Adiponectin as a biomarker of the metabolic syndrome. Circ J 68:975–981
Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM (2009) Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab 10:419–429
Sapru HN (2013) Role of the hypothalamic arcuate nucleus in cardiovascular regulation. Auton Neurosci 175:38–50
Sato K, Meng F, Glaser S, Alpini G (2016) Exosomes in liver pathology. J Hepatol 65:213–221
Savkur RS, Bramlett KS, Michael LF, Burris TP (2005) Regulation of pyruvate dehydrogenase kinase expression by the farnesoid X receptor. Biochem Biophys Res Commun 329:391–396
Schaap FG, Trauner M, Jansen PLM (2014) Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 11:55–67
Schneider JL, Cuervo AM (2014) Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol 11:187–200
Schober F, Neumeier M, Weigert J, Wurm S, Wanninger J, Schaffler A, Dada A, Liebisch G, Schmitz G, Aslanidis C et al (2007) Low molecular weight adiponectin negatively correlates with the waist circumference and monocytic IL-6 release. Biochem Biophys Res Commun 361:968–973
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, Hardt PD (2010) Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 18:190–195
Simpson KA, Martin NM, Bloom SR (2009) Hypothalamic regulation of food intake and clinical therapeutic applications. Arq Bras Endocrinol Metabol 53:120–128
Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ (2000) Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102:731–744
Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, Cohen JC, Hobbs HH (2015) Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 61:108–118
Spranger J, Kroke A, Mohlig M, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF (2003) Adiponectin and protection against type 2 diabetes mellitus. Lancet 361:226–228
Statnick MA, Beavers LS, Conner LJ, Corominola H, Johnson D, Hammond CD, Rafaeloff-Phail R, Seng T, Suter TM, Sluka JP et al (2000) Decreased expression of apM1 in omental and subcutaneous adipose tissue of humans with type 2 diabetes. Int J Exp Diabetes Res 1:81–88
Stefan N, Haring HU (2013) Circulating fetuin-A and free fatty acids interact to predict insulin resistance in humans. Nat Med 19:394–395
Stefan N, Kantartzis K, Haring HU (2008) Causes and metabolic consequences of fatty liver. Endocr Rev 29:939–960
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA (2001) The hormone resistin links obesity to diabetes. Nature 409:307–312
Stojsavljevic S, Gomercic Palcic M, Virovic Jukic L, Smircic Duvnjak L, Duvnjak M (2014) Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 20:18070–18091
Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P (2008) Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 47:1936–1946
Su W, Wang Y, Jia X, Wu W, Li L, Tian X, Li S, Wang C, Xu H, Cao J et al (2014) Comparative proteomic study reveals 17beta-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA 111:11437–11442
Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Konigsrainer A, Maier KP, Bischoff SC, Bergheim I (2008) Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr 138:1452–1455
Tilg H, Moschen AR (2006) Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6:772–783
Tremaroli V, Backhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249
Tsochatzis E, Papatheodoridis GV, Archimandritis AJ (2006) The evolving role of leptin and adiponectin in chronic liver diseases. Am J Gastroenterol 101:2629–2640
Tsochatzis E, Papatheodoridis GV, Hadziyannis E, Georgiou A, Kafiri G, Tiniakos DG, Manesis EK, Archimandritis AJ (2008) Serum adipokine levels in chronic liver diseases: association of resistin levels with fibrosis severity. Scand J Gastroenterol 43:1128–1136
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Valdearcos M, Xu AW, Koliwad SK (2015) Hypothalamic inflammation in the control of metabolic function. Annu Rev Physiol 77:131–160
Van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, London R, Peduto T, Chisholm DJ, George J (2008) Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology 48:449–457
Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, Bears M, Camporez JP, Cline GW, Jurczak MJ et al (2015) Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci USA 112:1143–1148
Vernon G, Baranova A, Younossi ZM (2011) Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 34:274–285
von Schnurbein J, Heni M, Moss A, Nagel SA, Machann J, Muehleder H, Debatin KM, Farooqi S, Wabitsch M (2013) Rapid improvement of hepatic steatosis after initiation of leptin substitution in a leptin-deficient girl. Horm Res Paediatr 79:310–317
Vonghia L, Michielsen P, Francque S (2013) Immunological mechanisms in the pathophysiology of non-alcoholic steatohepatitis. Int J Mol Sci 14:19867–19890
Walther TC, Farese RV Jr (2012) Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 81:687–714
Wang C, Zhao Y, Gao X, Li L, Yuan Y, Liu F et al (2015) Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 61:870–882
Wang Y, Lam KS, Yau MH, Xu A (2008) Post-translational modifications of adiponectin: mechanisms and functional implications. Biochem J 409:623–633
Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113:1408–1418
Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA (2001) Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86:1930–1935
Witek RP, Yang L, Liu R, Jung Y, Omenetti A, Syn WK, Choi SS, Cheong Y, Fearing CM, Agboola KM et al (2009) Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology 136(320–330):e322
Woods SC, Seeley RJ, Porte D Jr, Schwartz MW (1998) Signals that regulate food intake and energy homeostasis. Science 280:1378–1383
Wynne K, Stanley S, McGowan B, Bloom S (2005) Appetite control. J Endocrinol 184:291–318
Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ (2003) The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 112:91–100
Xu W, Wu L, Yu M, Chen FJ, Arshad M, Xia X, Ren H, Yu J, Xu L, Xu D et al (2016) Differential roles of cell death-inducing DNA fragmentation factor-alpha-like effector (CIDE) proteins in promoting lipid droplet fusion and growth in subpopulations of hepatocytes. J Biol Chem 291:4282–4293
Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM (2007) Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45:1366–1374
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N et al (2001) The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7:941–946
Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, Yao Z, Li P (2009) Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab 9:177–190
Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, Ozdogan O, Imeryuz N, Kalayci C, Avsar E (2010a) Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Invest 40:887–892
Yilmaz Y, Yonal O, Kurt R, Ari F, Oral AY, Celikel CA, Korkmaz S, Ulukaya E, Ozdogan O, Imeryuz N et al (2010b) Serum fetuin A/alpha2HS-glycoprotein levels in patients with non-alcoholic fatty liver disease: relation with liver fibrosis. Ann Clin Biochem 47:549–553
Yuan A, Farber EL, Rapoport AL, Tejada D, Deniskin R, Akhmedov NB, Farber DB (2009) Transfer of microRNAs by embryonic stem cell microvesicles. PLoS ONE 4:e4722
Zain SM, Mohamed Z, Mahadeva S, Cheah PL, Rampal S, Chin KF, Mahfudz AS, Basu RC, Tan HL, Mohamed R (2013) Impact of leptin receptor gene variants on risk of non-alcoholic fatty liver disease and its interaction with adiponutrin gene. J Gastroenterol Hepatol 28:873–879
Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135:61–73
Zhang J, Lei T, Chen X, Peng Y, Long H, Zhou L, Huang J, Chen Z, Long Q, Yang Z (2010) Resistin up-regulates COX-2 expression via TAK1-IKK-NF-kappaB signaling pathway. Inflammation 33:25–33
Zhang X, Wang Y, Liu P (2017) Omic studies reveal the pathogenic lipid droplet proteins in non-alcoholic fatty liver disease. Protein Cell 8:4–13
Zhu L, Baker RD, Baker SS (2015) Gut microbiome and nonalcoholic fatty liver diseases. Pediatr Res 77:245–251
Zoccoli G, Amici R, Silvani A (2011) The hypothalamus and its functions. In: Narcolepsy: pathophysiology, diagnosis, and treatment. Springer, New York, pp 191–203
Zou CC, Liang L, Hong F, Fu JF, Zhao ZY (2005) Serum adiponectin, resistin levels and non-alcoholic fatty liver disease in obese children. Endocr J 52:519–524
ACKNOWLEDGEMENTS
The authors thank Dr. Yongjian Liu (Nanjing Medical University) for his critical reading and useful suggestions. This work was supported by the National Natural Science Foundation of China (Grant Nos. 81471079, 31271268, and 81271828); the National Basic Research Program (973 Program) (No. 2012CB517503 and 2013CB530600); and a Technology/Development Fund of Nanjing Medical University (2016NJMU004).
ABBREVIATIONS
17β-HSD13, 17β-hydroxysteroid dehydrogenase 13; α-MSH, α-melanocyte-stimulating hormone; AdipoR1, adiponectin receptor 1; AGF, angiopoietin-related growth factor; AgRP, agouti-related peptide; AGPAT2, 1-acylglycerol-3-phosphate O-acyltransferase 2; Akt2, protein kinase B; AMPK, AMP-activated protein kinase; ApoB, apolipoprotein B; ApoCII, apolipoprotein CII; ApoCIII, apolipoprotein CIII; ARC, arcuate nucleus; ATGL, adipose triglyceride lipase; BMI, body mass index; Bscl2, berardinelli-seip congenital lipody-strophy 2; CART, cocaine-amphetamine-regulated transcript; CIDE, cell death-inducing DFFA-like effector; DAMPs, damage-associated molecular patterns; DFFA, DNA fragmentation factor subunit alpha; DGAT2, diacylgycerol acyltransferase 2; DIO, diet-induced obesity; DNL, de novo lipogenesis; ER, endoplasmic reticulum; EVs, extracellular vesicles; FAS, fatty acid synthase; FFAs, free fatty acids; FGF21, fibroblast growth factor 21; FLRL, fatty liver-related lncRNA; FXR, farnesoid X receptor; GLP-2, glucagon-like peptide 2; GWAS, genome-wide association studies; HCC, hepatocellular carcinoma; HFD, high fat diet; IL-6, interleukin- 6; IR, insulin resistant; HCC, hepatocellular carcinoma; HFD, high fat diet; JNK, c-Jun N-terminal kinase; LDs, lipid droplets; LECT2, leukocyte derived chemotaxin 2; LEPR, leptin receptor; Lmna, Lamin A; lncRNAs, long non-coding RNAs; LPL, lipoprotein lipase; LPS, lipopolysaccharide; MC3R, melanocortin 3 receptor; MC4R, melanocortin 4 receptor; miRNAs, microRNAs; MPs, microparticles; MTTP, microsomal triglyceride transfer protein; NAFLD, non-alcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NF-kB; nuclear factor kappa B; NLRPs, nucleotide-binding domain; NR1H4, nuclear receptor FXRα; NR1I1, Vitamin D receptor; NR1I2, pregnane X receptor; NPY, neuropeptide Y; PAMPs, pathogen-associated molecular patterns; PLINs, perlipins; PLIN1, perlipin 1; PLIN2, perlinpin 2; PLIN3, perlipin 3; PLIN4, perlipin 4; PLIN5, perlipin 5; PNPLA3, patatin-like phospholipid domain containing protein 3; POMC, anorexigenic neuropeptides pro-opiomelanocortin; Pparγ, peroxisome proliferator activated receptor γ; PVN, paraventricular nucleus; ROS, reactive oxygen species; SCFAs, short-chain fatty acids; SHBG, sex hormone-binding globulin; SOCS3, suppressor of cytokine signalling 3; SREBP-1c, sterol regulatory element binding protein-1c; T2DM, type 2 diabetes mellitus; TAG, triacylglycerides; TGR5, the G-protein-coupled receptor superfamily; TLR 4, toll-like receptor 4; TLRs, toll-like receptors; TNF-α, tumor necrosis factor-α; VLDL, very low-density lipoprotein.
COMPLIANCE WITH ETHICS GUIDELINES
Xu Zhang, Xuetao Ji, Qian Wang, and John Zhong LI declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, X., Ji, X., Wang, Q. et al. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell 9, 164–177 (2018). https://doi.org/10.1007/s13238-017-0436-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-017-0436-0