
Abstract
New oligoetherols with based on cyclotriphosphazene ring were synthesized by functionalization of hexachlorocyclotriphosphazene with glycidol followed by reaction with ethylene glycol and glycerol. Oligoetherols were characterized by IR, 1H-NMR, and MALDI-ToF and hydroxyl number as well as physical properties like density, viscosity and surface tension. The oligoetherols were further converted into polyurethane foams. The rigid foams of enhanced thermal stability and considerably diminished flammability were obtained and their apparent density, water uptake and polymerization shrinkage, thermal conductivity coefficient and thermal stability were determined. The flammability of foams was studied by microcalorimetric methods, horizontal flaming test and oxygen index. The obtained polyurethane foams with incorporated cyclotriphosphazene ring are self-extinguishing.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Polyphosphazenes are inorganic compounds built up of [N = PR2]n units [1, 2]. Both low molecular weight and polymeric polyphosphazenes are known [3]. The linear and cyclic polyphosphazenes were obtained. A cyclic trimeric cyclotriphosphazene is a six-membered ring compound. However, in contrary to aromatic compounds, the ring delocalization of electrons is limited within the cyclotriphosphazene. Cyclotriphosphazenes have stable rings with ca 1.58 nm phosphorus-nitrogen bond length, which is shorter than typical single bond length (1.77 nm) [4]. The symmetry of cyclotriphosphazenes is analogous to that of benzene. One of the most important representatives of cyclotriphosphazenes is hexachlorocyclotriphosphazene (HCCTP, (I)) of quantitative formula (NPCl2)3 (Scheme 1).

Formula of hexachlorocyclotriphosphazene (I)
The hybridization of phosphorus in (I) is sp3, which imposes tetrahedral geometry around phosphorous, while nitrogen atoms are in sp2 hybridization [5,6,7].
HCCTP can be obtained from phosphorus pentachloride and ammonium chloride in chlorobenzene or tetrachloroethane at 120–150 °C as shown in the Scheme 2 [1, 8].

Obtaining of HCCTP
Aside the HCCTP, the octachlorocyclotetraphosphazene is obtained, which can be separated by crystallization or fractional distillation. HHCTP is convenient precursor of inorganic and organic–inorganic polymers [9, 10]. The HHCTP polymerizes upon ring opening reaction 11 (Scheme 3).

Ring-opening polymerization of HCCTP
Such polymers can be modified by reaction with various nucleophiles to give a plethora of polyphosphazenes as shown schematically in Scheme 4 [12,13,14,15,16,17].

Modification of the polymer structure obtained from HCCTP
Phosphorus-attached side groups R can be simple halogens like fluorine, chlorine, bromine, iodine, cyanide or isocyanate, as well as alkoxy, aryloxy, amine, alkyl, aryl groups and also complex carbohydrates, aminoacid esters, oligopeptides, transition metal complexes, porphyrins, phtalocyanines, organosilanes units, up to entire liquid crystals and other biologically active compounds [8, 18,19,20,21,22,23].
The properties of polyphosphazenes depend on kind of substituents. The polyphosphazenes can be linear polymers or crosslinked (chemically, by radiation or metal ion-induced). Generally, polyphosphazenes are non-flammable (they show high oxygen index), resistant against organic solvents or strong acids and are soluble in freons. They are used as low-temperature elastomers or antipyrenes [24, 25], acoustic wave and vibration quenchers, electrolytes in batteries, the composites with polyurethanes in airplane or for medical purposes as drug and anti-coagulant agent carriers [8, 26,27,28].
HHCTP has advantageous thermal properties, like flame resistance and total lack of self-ignition. The thermal resistance of HCCTP and its derivatives makes them valuable flame retardant additives for polymeric materials [29,30,31,32,33,34,35].
Additive flame retardants are added into polymerizing mixtures. They do not react with polymers in contrary to reactive retardants which incorporate into polymers when added into polymerizing mixture or during crosslinking process. The most of reactive retardants incorporate elements like chlorine, bromine, nitrogen, phosphorus or silicon [36, 37]. The most effective flame retardants are those which incorporate phosphorus and nitrogen which act synergistically [16]. The HCCTP is not especially useful due to incorporation of chlorine into polymer. Such modified polymer may release hydrogen chloride upon combustion, which is the high risk side product. In order to use cyclotriphosphazene as flame retardant, it is rather recommended to replace chlorine atoms of HCCTP by other substituents, e.g. by reaction with glycidol (GL). The product of reaction of HCCTP with GL is hexaglycidylocyclotriphosphazene (HGCTP) which was evidenced to be a useful additive antipyren in epoxide resin obtained from diglycidyl ether of bisphenol A [24, 25]. The paper [38] describes the research on the use of this compound in the production of polyurethane insulating coatings. Also a rigid polyurethane foams (PUFs) with decreased flammability were obtained upon addition of other derivative, hexaphenoxycyclo-triphosphazene on expanded graphite as additive flame retardant [39]. HGCTP can be readily obtained from HHCTP and GL in toluene in presence of triethylamine (TEA, Scheme 5) [24]:

Obtaining of HGCTP (II)
HGCTP is non-flammable, non-toxic and thermally resistant compound due to presence of nitrogen and phosphorus. HGCTP is used as flame retardant without influencing the physical properties of polymer. The retardation effect is mostly due to structural properties of the material, which is porous, foamed solid, which makes the gas diffusion barrier and shields the polymer surface against heat and air stream. The novelty of this paper is the incorporation of cyclotriphosphazene ring into polyol, which then is used to obtain polyurethane foam, instead of addition the flame retardant into foaming mixture. We have attempted to functionalize HGCTP further into hydroxyalkyl derivative, the oligoetherol which then was applied to obtain PUFs by reaction with isocyanate in order to test thermal and fire properties of PUFs.
2 Experimental
2.1 Materials
The following materials were used in the work: HHCTP (pure, Sigma-Aldrich, Germany), GL (pure, Sigma-Aldrich, Germany), ethylene glycol (EG, pure, Pol-Aura, Poland), diethylene glycol (DEG, pure, Pol-Aura, Poland), glycerol (GLYC, anal. grade, POCH, Poland), TEA (pure, Fluka, Switzerland), surfactant Silicon L-6900 (pure, Momentive, US), polymeric diphenylmethane 4,4’–diisocyanate (pMDI, Merck, Germany).
2.2 Synthesis of HGCTP
53.28 g GL (0.72 mol), 73.60 g TEA (0.72 mol) and 360 cm3 tetrahydrofuran (THF) were placed in 500 cm3 three-necked round bottom flask equipped with mechanical stirrer, thermometer, and dropping funnel. Then 41.76 g HCCTP (0.12 mol) dissolved in 240 cm3 THF was added dropwise maintaining the temperature of the reaction mixture below 30 °C and vigorous stirring. The mixture was allowed to react at room temperature for 48 h. Afterwards, the white precipitate of triethylammonium chloride was filtered off and volatiles were removed from the liquid phase by rotary evaporation (p = 1067 Pa; t = 20–85 °C). The isolated light yellow liquid was identified as HGCTP by the elemental analysis, epoxide number (EN), 1H NMR and IR spectroscopy. Elemental analysis results were in accordance with formula II: Calc. 37.70% C, 5,24% H, 7,33% N; Found 37,42% C, 5,47% H, 6,97% N; ENcalc. = 1.724 mol of epoxide group/100 g, ENfound = 1.714 mol of epoxide group/100 g; H-NMR, δ (ppm): CH2-CH(O)-CH2O 2.6–2.8, CH2-CH(O)-CH2O 3.3, CH2-CH(O)-CH2O 3.8–4.1; IR: ring oxirane bands at 777 and 863 cm−1, the P-O bands at 1010 and 1050 cm−1 and P = N band at 1257 cm−1.
2.3 Synthesis of oligoetherols
68.76 g HGCTP (0.12 mol) and 44.64 g EG (0.72 mol) were placed in 150 cm3 three-necked round bottom flask equipped with mechanical stirrer, thermometer, and reflux condenser. Then, 2 cm3 TEA was added and the mixture was heated at 70–80 °C with continuous monitoring EN. The heating was discontinued when EN was zero. Further, the TEA was distilled off under reduced pressure (p = 1076 Pa; t = 20 – 120 °C). The product was dark yellow resin.
The syntheses of oligoetherols using DEG (76.32 g) and GLYC (66.24 g, 0.72 mol) instead of EG were performed in analogous way. The resin dark yellow oligoetherols were obtained and characterized as specified above.
2.4 Analytical methods
The reaction of HGCTP with EG, DEG and GLYC was monitored by EN determination using hydrochloric acid in dioxane [40]. The hydroxyl number (HN) was determined by acylation with acetic anhydride in xylene; the anhydride excess was then titrated off with 1.5 M NaOH in presence of phenolphthalein [41]. Elemental analysis for C, H, N, were done with EA 1108, Carlo-Erba analyzer. The 1H NMR spectra of products were recorded at 500 MHz Bruker UltraShield in DMSO-d6 with hexamethyldisiloxane as internal standard. IR spectra were registered on ALPHA FT-IR BRUKER spectrometer by ATR technique. MALDI ToF (Matrix-Associated Laser Desorption Ionization Time of Flight) spectra of post-reaction mixtures were obtained on Voyager-Elite Perceptive Biosystems (US) mass spectrometer working at linear mode with delayed ion extraction, equipped with nitrogen laser working at 352 nm. The method of laser desorption from gold matrix was applied. The samples were diluted with water to 0.5 mg/cm3.
2.5 Physical properties of oligoetherols
Density, viscosity, and surface tension of oligoetherols were determined with pycnometer, Höppler viscometer (typ BHZ, prod. Prüfgeratewerk, Germany) [42] and by the detaching ring method [43], respectively.
2.6 Obtaining the polyurethane foams
Foaming of oligoetherols was performed at 500 cm3 cups at room temperature. The foams were prepared from 10 g of oligoetherol, to which 0.39–0.47 g of surfactant (Silicon L-690) and 0.58–0.65 g of TEA as catalyst and water (2–3%) as blowing agent was added. After the homogenization, the polymeric diphenylmethane 4,4’-diisocyanate was added. The commercial isocyanate containing 30% of trifunctional isocyanates was used. The mixture was vigorously stirred until creaming began. The PUFs were seasoned for 4 days, after which the samples were cut off and subjected to further studies.
2.7 Properties of foams
The apparent density [44], water uptake [45], dimensional stability in 150 °C temperature [46], thermal conductivity coefficient (IZOMET 2104, Slovakia), and compressive strength [47] of PUFs were measured. Apparent density of PUFs was calculated as the ratio of PUF mass to the measured volume of PUF sample in cube of 50 mm edge length. Water volume uptake was measured on cubic samples of 30 mm edge lengths. Dimensional stability was tested on samples of 100 × 100 × 25 mm size. Thermal conductivity coefficient was measured at 20 °C after 48 h of PUF seasoning. The needle was inserted to a depth of 8 cm introduced into cylindrical PUF sample of 8 cm diameter and 9 cm length. Compressive strength was determined using burden causing 10% compression of PUF height related to initial height (along with the PUF growing direction). Thermal resistance of modified foams was determined both by static and dynamic methods. In static method, the foams were heated at 150, 175 and 200 °C with continuous measurement of mass loss and determination of mechanical properties before and after heat exposure. The 100 × 100 × 100 mm cubic samples were used to determine static thermal resistance and compressive strength. In dynamic method, thermal analyses of foams were performed in ceramic crucible at 20–600 °C temperature range, about 100 mg sample, under air atmosphere with Thermobalance TGA/DSC 1 derivatograph, Mettler, with 10 °C/min heating rate. Topological pictures of PUFs were also recorded for cross-sections of PUFs samples cut in direction perpendicular to growing. The pictures were taken with 60-fold enlargement and analysed with MORPHOLOGI G3 (prod. Malvern) using OPTAVIEW—IS ver.4.0 software. Flammability of foams was determined by oxygen index [48] and horizontal test according to norm [49] as follows: the foam samples (150 × 50x13 mm) were weighed, located on horizontal support (wire net of 200 × 80 mm dimensions) and the line was marked at the distance of 25 mm from edge. The sample was set on fire from the opposite edge using Bunsen burner with the blue flame of 38 mm height for 60 s. Then, the burner was removed and time of free burning of foam reaching marked line or cease of flame was measured by stopwatch. After that, the samples were weighed again. The rate of burning was calculated according to the expression:
if the sample was burned totally, or using equation:
if the sample ceased burning, where:
Le – the length of burned fragment, measured as the difference 150 minus the length of unburned fragment (in mm). According to norms, if the burned fragment has the 125 mm length, the foam is considered as flammable.
tb, te– the time of propagation of flame measured at the distance between starting mark up to the end mark or as the time of flame cease.
The mass loss Δ m after burning was calculated from the formula:
where mo and m – mean the sample mass before and after burning, respectively.
Flammability of foams was evaluated for samples 100 × 100x10 mm in size using a cone microcalorimeter, a product of FTT Ltd. (United Kingdom), according to standard [50], by applying the heat flow 25 kW/m2 and the distance from ignition source 25 mm. During the tests, the time to ignite (TTI), total time of flaming (TTF), percentage mass loss (PML), heat release rate (HRR), effective heat of combustion (EHC) and total heat release (THR) were recorded.
3 Results and discussion
3.1 Synthesis of oligoetherols
The synthesis of HGCTP from HCCTP and GL in toluene was described in [24]. We have slightly modified the method using THF instead of toluene. TEA was used to bind HCl side product of condensation. The HGCTP (II) was isolated as a resin (Scheme 5) after removal of triethylammonium chloride and THF. We have noticed that low-temperature THF vacuum removal was necessary in order to avoid gelation of the product. Also, we have found that short reaction time or excess of GL (above 6 equivalents of GL per equivalent of HCCTP) resulted in gelation in presence of TEA according to the Scheme 6:

Gelation in presence of TEA
The HGCTP was reacted with EG and DEG at 70–80 °C in presence of TEA catalyst as shown in the Scheme 7:

Obtaining of oligoetherols
where R =—CH2CH2-, -CH2CH2-O-CH2CH2-, -CH2-CH(OH)-CH2- (III).
Elemental analyses of the products confirmed the composition of the oligoetherols III (Table 1). The IR spectra of obtained new oligoetherols are presented at Figs. 1a and 1b. The oxirane ring bands are absent in these spectra, indicating that oxirane ring opening took place according to the reaction of HGCTP with glycols. The valence and deformation O–H bands are observed at 3320–3360 cm−1 and 1032–1038 cm−1, respectively, the latter partially overlapped with ether group bands. The C-H valence bands and deformation bands are present at 2870–2940 cm−1 and 1455 cm−1, respectively. The cyclotriphosphazene ring bands are observed at: 1250 cm−1 (P = N) as well as P-O bands at 1010 and 1050 cm−1, confirming that oxygen linked cyclotriphosphazene ring is present at the structure of the products.

IR spectrum of oligoetherols HGCTP-EG (a), HGCTP-DEG (b) and HGCTP-GLYC (c)
The 1H NMR spectrum of HGCTP has characteristic multiplets of oxirane ring protons within 2.6–2.8 ppm, which are absent in the spectra of oligoetherols. Instead, the resonances at 3.5 ppm attributed to methylene and methine protons from oxyalkilene groups from EG (Fig. 2a) and those from disintegration of the oxirane ring are observed. For the oligoetherol obtained from HGCTP and DEG the methylene and methine proton resonances are occur in a wider range of the spectrum (Fig. 2b). The hydroxyl proton resonances at 4.5 and 7.2 ppm were identified by selective deuteration with D2O.

H-NMR spectrum of oligoetherols HGCTP-EG (a), HGCTP-DEG (b) and HGCTP-GLYC (c)
MALDI ToF spectra provided detailed information of the presence of low molecular weight side products in the isolated oligoetherols. Thus, the side products in the oligoetherol obtained from HGCTP and EG were oligomers of GL and products of its consecutive reaction with EG, as well as their dehydration products (Table 2, No. 3–5, 7, 8). Also, the products of reaction between TEA and alcohols were identified (Table 2, No. 6, 9). Products of partial reaction of HGCTP with EG equivalents were not detected except the mono-substituted HGCTP (Table 2, No. 17). Similar pattern of low molecular side products was found in the MALDI ToF spectrum of HGCTP-DEG oligoetherol.
The PUFs based on obtained oligoetherols and polymeric 4,4’-diphenylmethane diisocyanate (pMDI) were obtained at laboratory scale. The foaming compositions were optimized by choosing the amount of pMDI based on determined HN of the substrates. We aimed at rigid PUFs of improved thermal resistance and flame retardance. Preliminarily we have found that obtained PUFs mass loss upon 1 month exposure to 150 °C was about 22%, regardless the kind of oligoetherol. The obtained PUFs were more thermally resistant than classic PUFs, which showed 21% mass loss after 1 day exposure to 150 °C [51]. Horizontal flame retardant tests for obtained PUFs evidenced that PUFs obtained from DEG were flammable and released dark smoke upon flaming. The PUF obtained from EG was self-extinguishing; the PUF flame ceased after couple of seconds upon ignition.
With these results in hand, we focused on the PUFs obtained from HGCTP and EG. We have used another alcohol substrate, namely glycerol (GLYC), in order to obtain higher functionalized oligoetherol, which was supposed to give more crosslinked PUF. According to earlier studies, we assumed that increased crosslinking might result in obtaining the PUF of further reduced flammability [52] in comparison with thatof the PUF obtained from HGCTP and EG.
Elemental analysis of the HGCP-GLYC oligoetherol confirmed its assumed structure III (Table 1). The IR spectrum of HGCP-GLYC (Fig. 1c) was similar to those of other oligoetherols obtained previously. The strong stretching vibration OH band at 3200–3600 cm−1 was observed along with the sharp deformation band at 1030 cm−1. The band at 1252 cm−1 (P = N) revealed the cyclotriphosphazene ring remained untouched in HGCP-GLYC. The C-O and P-O-C bands at 1050 cm−1 overlapped partially with sharp O–H deformation band. In the 1H NMR of HGCP-GLYC, the methylene and methine proton resonances were found at 3.4 to 3.6 ppm, while hydroxyl proton resonances were identified at 7.5 and 4.0 ppm (Fig. 2c), as previously found for other oligoetherols synthesized here.
The fundamental physical properties of obtained oligoetherols, namely: density, viscosity, and surface tension were studied (Table 3). All the physical parameters fall inside the upper limits of oligoetherols suitable to obtain PUFs. Typical PUF-suitable polyetherols have a viscosity within 200—30,000 mPas [53]. From our previous experience, we have learned that suitable surface tension of PUF-suitable polyetherols should be within 30—50 mN/s. Low surface tension is preferred because it enable effective foaming process. Both obtained oligoetherols have relatively high surface tension, which imposes the use of relatively larger amount of surfactant in foaming composition. The obtained oligoetherols have large HN (Table 3), namely 642 and 702 mg KOH/g, which renders them promising substrates to obtain rigid PUFs. Thus, the PUFs were obtained in the reaction between the oligoetherols with pMDI and water, using TEA as catalyst and Silicon L-6900 surfactant.
3.2 Properties of polyurethane foams
To optimize the foaming compositions couple of pilot experiments were performed, and the optimized composition was established to obtain rigid PUF with small and uniform pores (Table 4). In optimized version, the molar ratio of isocyanate groups (from pMDI) to hydroxyl groups (–NCO/–OH) in foaming composition (the isocyanate coefficient) was 1. If larger amount of isocyanate was used the obtained PUFs were under-crosslinked, semi-rigid, and had sticky surface. The optimized water amount was 2 or 3% related to the mass of oligoetherol HGCTP-GLYC or HGCTP-EG, respectively. For such compositions, the rigid PUFs were obtained with regular pores. When 2% water was used for foaming with HGCTP-EG the resulting PUF had larger apparent density (ca 100 kg/m3) and low level of foaming. On the other hand, increased amount of water above 2% in foaming composition with oligoetherol HGCTP-GLYC, the obtained PUFs had too large pores. Generally, the uniform pores were obtained for compositions containing 3.9 or 4.7% surfactant. Optimal compositions were those with 6.5 or 5.8% catalyst related to the mass of oligoetherols: HGCTP-EG and HGCTP-GLYC, respectively. Lower amount of catalyst resulted in formation of under-crosslinked and semi-rigid PUFs. The optimized foaming composition obtained from HGCTP-EG had 15 s cream time, 30 s rise time and 12 s tack free time. The timing parameters were longer for compositions with HGCTP-GLYC, namely: 74, 155, and 64 s. This was attributed to presence of less reactive secondary hydroxyl groups in additions to the primary ones.
The physical properties of optimized PUFs were determined. The apparent density of the PUF obtained from HGCTP-EG was 37.7 kg/m3. If lower amount of water in foaming compositions with HGCTP-GLYC oligoetherol was used, the apparent density grew to 57.4 kg/m3 (Table 5). The PUFs were dimensionally stable at elevated temperatures. The foam obtained from HGCTP-EG oligoetherol shrinked 4.5% while the PUF obtained from HGCTP-GLYC oligoetherol showed even lower shrinkage, not exceeding 2.6%. We attribute this to higher functionality of this oligoetherol, which favors higher crosslinking of the PUF. The water uptake of PUF after 24 h exposure was low, below 6.3%, which suggested the pores of PUF were closed. This was also confirmed by microscopic imaging of the PUF with Microscope OPTA-TECH (Fig. 3). The oval pores had the length within 150–190 µm and width was within 90–120 µm for the PUF obtained from HGCTP-EG oligoetherol. The PUF obtained from HGCTP-GLYC oligoetherol had relatively larger pores, with 220–240 µm length and 180–200 µm width. Heat conductance coefficient for obtained PUFs is similar to that of typical rigid PUFs (0.0260 W/m·K) [52] and equals to 0.0270 W/m·K (Table 5).

Microscopic picture of foams obtained from oligoetherols HGCTP-EG (a) and HGCTP-GLYC (b)
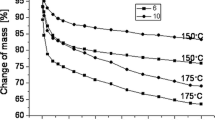
Thermal resistance of the PUFs was studied at 150 °C and 175 °C temperatures by monitoring mass loss and measuring mechanical properties before and after thermal exposure. A large mass loss was observed in the first day of thermal exposure of the studied PUFs (Fig. 4). Our preliminary results suggested that the PUFs were not resistant to thermal exposure at 200 °C. A 1-day exposure at 200 °C led to 22% mass loss. Long term exposure of PUFs at 150 °C was within 19.3–22.6%, while exposure to 175 °C for one month led to 25.5–29.6% mass loss. Generally, the thermal resistance of PUFs was higher for those, containing more contribution of cyclotriphosphazene subunit (obtained from HGCTP-EG oligoetherol), which is due to high thermal resistance of cyclotriphosphazene ring itself [32,33,34,35]. The PUF mass loss upon 1-month exposure at 150 °C was ca 20%. Simultaneously the PUF gained additional 80–100% compressive strength if compare with the not-annealed PUF (Table 5). This property seems useful for the PUF used as thermal isolation. The increase of compressive strength is due to additional crosslinking at elevated temperature. The compressive strength of the PUF annealed at 175 °C was still higher than that of non-annealed one, but slightly lower than that of annealed at 150 °C. It can be concluded that the PUF undergoes degradation in this condition (ca 25–30%), as evidenced by the greater weight loss (approx. 25–30%).

Change of mass of polyurethane foams after exposure in elevated temperature (reagents in synthesis of oligoetherol used to obtain polyurethane foams are shown in insert)
In order to monitor the structural changes accompanying annealing of PUFs, the IR spectra of PUFs exposed to heating at 150 and 175 °C were recorded (Fig. 5). In IR spectra of unheated foam were observed the band centered at 3400 cm−1 corresponding to stretching vibrations of N–H groups engaged in hydrogen bonding and also at 1600 cm−1 from N–H bending band, corresponding to stretching vibrations of N–H groups engaged in hydrogen bonding and also at 1600 cm−1 from N–H bending band. The presence of urethane bands and urea derivatives in PUFs was also found by amide band I at 1725 cm−1, characteristic for C = O group. The C-N and C = O are present in the region 1250–1000 cm−1, while C-H stretching vibration bands are present at 3000–2900 cm−1. The presence of cyclotriphosphazene ring in PUF was confirmed by the P = N band at 1223 cm−1, and bands within P-O-C at 1000–1050 cm−1. The band at 2276 cm−1 illustrated the presence of not–reacted isocyanate groups, while carbodiimide groups were identified by the band at 2136 cm−1. Upon annealing the carbodiimide and isocyanate group bands disappeared, presumably due to oxidation of these groups. The isocyanate group band disappearance can also be related to the thermally induced reaction between isocyanates and remaining hydroxyl groups, the reaction being responsible for additional crosslinking upon heating. Annealing was also accompanied by decrease of intensity of the band centered at 1725 cm−1, indicating the thermal decomposition of amide bonds. Long heating at 175 °C resulted in decrease of the cyclotriphosphazene ring P-O-C band at 1013 cm−1. Thermal exposure resulted in stepwise conversion of urethanes as well as allophanate and burette groups, which was illustrated by decrease of N–H valence bands and C = O bands.

IR spectrum of foam obtained from oligoetherol HGCTP-GLYC before exposure (green) and after exposure in temperature 150 °C (blue) and 175 °C (red)
Dynamic thermogravimetric analysis revealed that obtained PUFs have high thermal resistance. The 5% mass loss was observed at 209–212 °C temperature, while a temperature of maximum thermal decomposition was observed within 260–270 °C (Fig. 5 a, b; Table 5). This region corresponds to a decomposition of cyclotriphosphazene ring, which was determined to occur at 220 do 370 °C [25]. At this temperature also, the rigid fragments of PUF decompose and also an urethane bonds and fragments are converted into isocyanataes and alcohols. The isocyanates can further polymerize into carbodiimides and isocyanurates, which decompose at temperatures above 270 °C (second peak at ca 325 °C) to amines and carbon dioxide [53]. According to literature data [25], the P-O-C bonds linking the phosphorus of cyclotriphosphazene ring and oxygen of aliphatic chain dissociate at 250 °C. The flexible fragments of polyurethane (ether bonds) dissociate at higher temperatures, ca 450 °C. The 25% mass remained after heat. We noticed that no essential difference in initial temperature of pyrolysis and final mass loss of both PUFs, which seems reasonable, because both PUFs contain the same structural fragments derived from similar oligoetherols. The glass transition temperature (Tg) of obtained PUFs was determined by DSC. Two Tg were observed: at 75 and 102 °C, and confirmed that obtained PUFs are rigid. The higher Tg for PUF obtained from HGCTP-GLYC oligoetherol was related to denser crosslinking of the PUF imposed by higher functionality of the oligoetherol.
The flammability of PUFs was determined by horizontal flaming test and by oxygen index values (Table 6). The presence of phosphorus and nitrogen elements in obtained PUFs resulted in decreased flammability of the PUFs in comparison with PUFs obtained from typical commercial oligoetherols. This structural modification of PUFs resulted in a decrease of flaming rate to 0.9 mm/s in case of the PUF obtained from HGCTP-EG and to 0.5 mm/s in case of the PUF obtained from HGCTP-GLYC oligoetherol. For comparison, the PUF obtained from oligoetherol based on melamine and propylene oxide showed 6.2 mm/s flaming rate [54].
Moreover, the PUFs obtained here are self-extinguishing according to horizontal flaming test. The flame reached merely 24 mm or 35 mm from starting line. Their oxygen index was within 21.6–21.8% region, i.e. slightly above oxygen content in air. Thus the obtained PUFs can be considered as self-extinguishing [55]. The flame properties of the PUFs annealed previously for one month at 150 and 175 °C were also determined (Table 6). It has been found that that they do not ignite upon flame contact and their oxygen index increased to 32.8–38.4%. This clearly indicated that the PUFs have good properties to be applied as thermal isolators. The annealed PUFs have changed their composition (Table 7). The nitrogen percentage grew, while hydrogen percentage fallen down.
The relevant data on material flammability were obtained in calorimetric measurement (Table 8). The time to ignition (TTI) is the most reliable parameter enabling to determine thermal resistance of materials; the longer TTI, the longer is heating time to ignite and initiate a fire. We have found that the PUF obtained from HGCTP-EG oligoetherol ignites after 4 s, while that obtained from HGCTP-GLYC oligoetherol ignites, after 9 s, which is presumably due to compact structure of the latter, higher apparent density and higher crosslinking. These structural properties also result in relatively higher Tg (Table 5). Also, the TTF is longer for this PUF and equals 74 s. The HRR profiles vs time are shown at Fig. 6. The initial HRR waveforms are similar for both PUFs. HRR of obtained PUFs falls within typical range of flame retarded PUFs (which is 38–133 kW/m2) as was obtained by us previously and in other laboratories [56,57,58,59,60,61]. Small difference in elemental compositions of polyols (see Table 1) and PUFs obtained from them as well as similarity of structural fragments of both products (Table 4) may lead to little differences in HRR. In fact, more heat is released during combustion of the PUF obtained from HGCTP-GLYC oligoetherol (Table 5), which is understandable because the PUF has higher apparent density. Both PUFs reach the maximum HRR after 25–28 s, but time of heat release is longer for the denser PUF. Generally, the mass loss percent correlates with apparent density; the higher density the greater mass loss. Thus the PUF obtained from HGCTP-GLYC oligoetherol showed higher mass loss, above 80%, than that of PUF obtained from HGCTP-EG (70.8%). The effective heat of combustion (EHC) is similar for both PUFs, although the PUF with higher density was 2.6 MJ/kg larger (Table 8). It has been shown that decreased flammability of polymer containing incorporated cyclotriphosphazene is due to formation of porous foamed mass, which itself is diffusional barrier for gaseous products and additionally isolate the material from heat transfer and air accessibility [62]. The IR spectra of products of polymer combustion indicated the presence of phosphorus and carbon. Based on IR spectra, it was concluded that remnant carbonizate is composed of carbon networks with phosphorocarbon and phosphoroxide nodes [63] Fig. 7. El Gouri and coworkers [3] showed also that carbonizate had phosphorus and nitrogen nodes [24] (Scheme 8).

Thermogravimetric analysis of PUFs; mass change as a function of temperature (a), differential mass change as a function of temperature (b)

The course of changes in the HRR as a function of time during the combustion of polyurethane foams (reagents in synthesis of oligoetherol used to obtain polyurethane foams are shown in insert)

Decomposition of cyclotriphosphazene ring upon combustion
4 Summary and conclusions
-
1.
The reaction of hexachlorocyclotriphosphazene with six equivalents of glycidol and then with six equivalents of ethylene glycol or glycerol leads to oligoetherols based on cyclotriphosphazene.
-
2.
The oligoetherols with incorporated cyclotriphosphazene ring react with polymeric 4,4’-diphenylmethane diisocyanate and water to give rigid polyurethane foams.
-
3.
The polyurethane foams have similar properties as classic rigid polyurethane foams, except enhanced thermal resistance and diminished flammability.
-
4.
The polyurethane foams with incorporated cyclotriphosphazene ring can stand long lasting heating at 150 °C. After thermal exposure, they gain the compression strength of 80–100% in comparison with not heated foams.
-
5.
The flammability of obtained foams decreases considerably upon thermal treatment, which is promising properties to use them as heat isolating materials. The horizontal flaming tests and the values of oxygen index categorize them as self-extinguishing materials.
References
A. Kiliç, J. Qafqaz. Univ. 3, 133 (2000)
H. Özay, M. Yildirim, Ö. Özay, Turk. J. Chem. 39, 777 (2015)
J. Gruneich, P. Wisian-Neilson, Macromolecules 29, 5511 (1996)
H.R. Allcock, Phosphorus-nitrogen Compounds (Academic Press, New York, 1972)
D.W.J. Cruickshank, Acta Crystallogr. 17, 671 (1964)
H.R. Allcock, S. Kuharcik, K. Visscher, D. Ngo, J. Chem. Soc. Dalton Trans. 17, 2785 (1995)
R. Verma, Inorganic Chemistry. Silicones and Phosphazenes. Ed. University Department of Chemistry Magadh University, 2006.
J.R. Rabek, Contemporary knowledge of polymers - selected issues (PWN, Warsaw, 2008)
H.R. Allcock, R.L. Kugel, J. Am. Chem. Soc. 87, 4216 (1965)
H.R. Allcock, Ring-Opening Polymerization, in Phosphazene Chemistry. ed. by D.J. Brunelle (Hanser Publishers, Munich, 1993)
V. Chandrasekhar, M.G.R. Muralidhara, I.I. Selvaraj, Heterocycles 31, 2231 (1990)
R. Singler, N. Schneider, G. Hagnauer, Polym. Eng. Sci. 15, 321 (1975)
D. Dell, B. W. Fitzsimmons, and R.A. Shaw, J. Chem. Soc., 4070–4073 (1965).
JK. Dulesis, H. Rose, RA. Shaw. (1976). Naturforsch. 31b: 997
A. P. Carrol, and R. A. Shaw, J. Chem. Soc. A, 914–921 (1966).
N.L. Paddock, Q. Rev, Chem. Soc. 18, 168 (1964)
D. Kumar, Int. J. Adhes. Adhes. 18, 109 (1998)
E. Maaskant, H. Gojzewski, M.A. Hempenius, G.J. Vancso, N.E. Benes, Polym. Chem. 22, 3169 (2018)
I.S. Sirotin, Y.V. Bilichenko, K.A. Brigadnov, V.V. Kirev, O.V. Suraewa, R.S. Borisov, Russ. J. Appl. Chem. 86, 1903 (2013)
Z.P. Zhao, Q. Guo, X. Li, J.L. Sun, Z.J. Nie, Express Polym. Lett. 6, 308 (2012)
A.M. Caminade, A. Hameau, J.P. Majoral, Dalton Trans. 45, 1810 (2016)
R. Schwesinger, T.A. Dambacher, Z., Naturforsch. B 1233, 1229 (2006)
M.A. Abid, L. Wang, J. Wang, A. Xiao, Des Monomers Polym. 12, 357 (2009)
M. El Gouri, A. El Bachiri, S.E. Hegazi, M. Rafik, A. El Harfi, Polym. Degrad. Stab. 94, 2101 (2009)
M. El Gouri, A. El Bachiri, S.E. Hegazi, R. Ziraoui, M. Rafik, A. El Harfi, J. Mater. Environ. Sci. 2, 319 (2011)
H. Özay, Ö. Özay, Colloids Surf. A: Physicochem. Eng. Asp. 50, 99 (2014)
A.L. Baillargeon, K. Mequanint, Biomed. Res. Int. 2014, 1 (2014)
Y.W. Cho, J.-R. Lee, S.-C. Song, Bioconjugate Chem. 16, 1529 (2005)
Y. Fang, J. Miao, X. Yang, Y. Zhu, G. Wang, Chem. Eng. J. 385, 123830 (2020)
Y. Fang, X. Du, S. Yang, H. Wang, X. Cheng, Z. Du, Polym. Chem. 10, 4142 (2019)
J. Liu, Z. He, G. Wu, X. Zhang, C. Zhao, C. Lei, Chem. Eng. J. 390, 124620 (2020)
B. Zhao, W.J. Liang, J.S. Wang, F. Li, Y.Q. Liu, Polym. Degrad. Stab. 133, 162 (2016)
R. Yang, B. Wang, X. Han, B. Ma, J. Li, Polym. Degrad. Stab. 144, 62 (2017)
X. Zhou, S. Qiu, X. Mu, M. Zhou, W. Cai, L. Song, W. Xing, Y. Hu, Compos. B 202, 108397 (2020)
S. Qiu, Ch. Ma, X. Wang, X. Zhou, X. Feng, R.K.K. Yuen, Y. Hu, J. Hazard. Mater. 344, 839 (2018)
J. J. Pitts, Flame Retardancy of Polymeric Materials. Vol. 1, Ed. Marcel Dekker, New York 1973.
G. Janowska, W. Przygocki, A. Włochowicz, Flammability of polymers and polymer materials, WNT, Warsaw, 2007.
Ł Byczyński, M. Dutkiewicz, R. Januszewski, Prog. Org. Coat. 108, 51 (2017)
L. Qian, F. Feng, S. Tang, Polymer 55, 95 (2014)
Z. Brojer, Z. Hertz, P. Penczek, Epoxy Resins, WNT, Warsaw, 1972.
Polyethers for polyurethanes. Test methods. Determination of the hydroxyl number. Standards PN-93/C-89052.03. Ed. Polish Committee for Standardization.
T. Broniewski, A. Iwasiewicz, J. Kapko, and W. Płaczek, Test methods and evaluation of plastics pro-perties, WNT, Warsaw, 1967.
T. Dryński, Laboratory exercises in physics (PWN, Warsaw, 1967)
Cellular Plastics and Rubbers. Determination of apparent (bulk) Density, Polish (European) Standards PN-EN ISO 845–2000. Ed. Polish Committee for Standardization.
Cellular Plastics, Rigid. Determination of Water Absorption. Polish (European) Standards PN-EN ISO 2896–1986. Ed. Polish Committee for Standardization.
Cellular Plastics, Rigid. Test of dimensional Stability. Polish (European) Standards PN-EN ISO 2796–1986. Ed. Polish Committee for Standardization.
Cellular Plastics, Compression Test for rigid Materials. Polish (European) Standards PN-EN ISO 844–1978, Ed. Polish Committee for Standardization.
Plastics – Determination of burning behavior by oxygen index – Part 2. Ambient-temperature test. Polish (European) Standards PN-EN ISO 4589–2. 2006. Ed. Polish Committee for Standardization.
Flexible Cellular polymeric Materials – Laboratory Characteristics of small specimens Subject to a small Flame. Polish (European) Standards PN-EN ISO 3582–2002. Ed. Polish Committee for Standardization.
Fire Hazard Testing. Part 11–10: Test Flames. 50W Horizontal and Vertical Flame Test Method. Polish (European) Standards PN-EN 60695–11–10:2014–02. Ed. Polish Committee for Standardization.
J. Lubczak, E. Chmiel-Szukiewicz, J. Duliban, D. Głowacz-Czerwonka, R. Lubczak, B. Łuksiewicz, I. Zarzyka, A. Łodyga, P. Tyński, D. Minda-Data, M. Kozioł, Z. Majerczyk, Przemysl Chem. 10, 1690 (2014)
B. Czupryński, (2004) Questions of chemistry and technology of polyurethanes (The Publishing House of the Academy of Bydgoszcz, Bydgoszcz, 2004)
Z. Wirpsza, Poly. chem., technol. Appl., WNT, Warsaw, 1991.
J. Lubczak, R. Lubczak, J. Cell. Plast. 54, 561 (2018)
J.M. Cogen, T.S. Lin, R.E. Lyon, Fire Mater. 33, 33 (2009)
R. Lubczak, D. Szczęch, D. Broda, A. Szymańska, R. Wojnarowska-Nowak, M. Kus-Liśkiewicz, J. Lubczak, Polym. Test. 70, 403 (2018)
M. Borowicz, J. Paciorek-Sadowska, J. Lubczak, B. Czupryński, Polymers 11, 1 (2019)
J. Paciorek-Sadowska, M. Borowicz, E. Chmiel, J. Lubczak, Int. J. Mol. Sci. 22, 69 (2021)
E. Chmiel, J. Lubczak, Macromol. Res. 27, 543 (2019)
E. Chmiel, R. Oliwa, J. Lubczak, Polym. Plast. Technol. Eng. 58, 394 (2019)
W. Xi, L. Qian, L. Li, Polymers 11, 207 (2019)
M. El Gouri, S. Hegazi, M. Rafik, A. El Harfi, Ann. Chim. Sci. Mat. 35, 27 (2010)
J. Sun, Z. Yu, X. Wang, D. Wu, ACS Sustain. Chem. Eng. 2, 231 (2014)
Funding
This research did not receive any specific grant from funding agencies in the public, commercial sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Ethical approval
We confirm that neither the manuscript nor any parts of its content are currently under consideration or published in another journal. All authors have approved the manuscript and agree with its submission to Macromolecular Research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lubczak, J., Lubczak, R. Oligoetherols and polyurethane foams based on cyclotriphosphazene of reduced flammability. Macromol. Res. 31, 455–468 (2023). https://doi.org/10.1007/s13233-023-00121-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13233-023-00121-0