
Abstract
Diaporthe is an important plant pathogenic genus, which also occurs as endophytes and saprobes. Many Diaporthe species that are morphologically similar proved to be genetically distinct. The current understanding of Diaporthe taxonomy by applying morphological characters, host associations and multi-gene phylogeny are problematic leading to overestimation/underestimation of species numbers of this significant fungal pathogenic genus. Currently, there are no definite boundaries for the accepted species. Hence, the present study aims to re-structure the genus Diaporthe, based on single gene phylogenies (ITS, tef, tub, cal and his), multi-gene phylogeny justified by applying GCPSR (Genealogical Concordance Phylogenetic Species Recognition) methodology as well as the coalescence-based models (PTP—Poisson Tree Processes and mPTP—multi-rate Poisson Tree Processes). Considering all available type isolates of Diaporthe, the genus is divided into seven sections while boundaries for 13 species and 15 species-complexes are proposed. To support this re-assessment of the genus, 82 Diaporthe isolates obtained from woody hosts in Guizhou Province in China were investigated and revealed the presence of two novel species and 17 previously known species. Synonymies are specified for 31 species based on molecular data and morphological studies. Dividing Diaporthe into several specific sections based on phylogenetic analyses can avoid the construction of lengthy phylogenetic trees of the entire genus in future taxonomic studies. In other words, when one conducts research related to the genus, only species from the appropriate section need to be selected for phylogenetic analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diaporthe Nitschke (1870) encompasses important plant pathogenic, endophytic, and saprobic species with a diverse host range and a global distribution (Gomes et al. 2013; Hyde et al. 2014; Dissanayake et al. 2017b; Abeywickrama et al. 2022; Hilário and Gonçalves 2023). Giving priority to older name, Diaporthe Fuckel (1867) has been proposed recently, and the results are pending. The genus was typified with D. eres Nitschke (1870), and belongs to the taxonomic hierarchy of Diaporthaceae, Diaporthales in Sordariomycetes (Maharachchikumbura et al. 2016). Both generic names Diaporthe (sexual morph) and Phomopsis (asexual morph) were regularly used by mycologists and plant pathologists. According to the concept of ‘One fungus, one name’ Diaporthe, being the older generic name, has priority over Phomopsis (Rossman et al. 2015) and has been considered more auspicious in moving fowards with this pleomorphic genus (Gomes et al. 2013; Udayanga et al. 2014a, b; Rossman et al. 2015; Dissanayake et al. 2017b). Currently, Index Fungorum (www.indexfungorum.org) and MycoBank (www.mycobank.org) list more than 1200 taxa under Diaporthe, and almost 1000 species in Phomopsis (accessed in April 2024).
Morphological variation within Diaporthe species is inadequate in species identification and in defining novel species, and it is necessary to incorporate molecular data along with morphology, to describe species in the genus (Udayanga et al. 2014a, b; Gao et al. 2017; Guarnaccia and Crous 2017; Guo et al. 2020; Chaisiri et al. 2022; Monkai et al. 2023; Xiao et al. 2023; Aumentado and Balendres 2024). Multi-gene phylogenetic analyses of five gene markers: internal transcribed spacer region (ITS), translation elongation factor 1-alpha (tef), beta-tubulin (tub), calmodulin (cal) and histone H3 (his) gene regions have been promising tools for both identification of Diaporthe species and concluding evolutionary interactions among them (Udayanga et al. 2012a, b). Though multi-gene phylogenetic analyses are considered most effective for accurate reconstruction of species boundaries (Udayanga et al. 2012a; Gomes et al. 2013; Santos et al. 2017) it has made problematic issues by leading to overestimation or underestimation of species numbers in this significant fungal pathogenic group (Hilário et al. 2021a, b).
It is strongly believed that the splitting of this genus into numerous genera based on monophyletic grouping is not advisable as the genera share almost the same morphological characters in both sexual and asexual morphs with only minor variation in dimension of morphological features (Gao et al. 2017). When restructuring the genus, all available type sequence data which are available in public databases must be considered and more strains must be included for the analysis of each species. Hence, the main objective of the present project is to interpret the phylogenetic tree of the genus Diaporthe at the section level, and to define the boundaries of possible species/species-complexes in each section to avoid confusion in species identification. Hence, the resulting facts should serve as the perfect baseline for taxonomists, mycologists and plant pathologists who face continuous difficulties while dealing with this genus. This will not merely be significant in diversity perspectives, but also for when precise identification of plant pathogenic species is obligatory for quarantine and disease management.
Materials and methods
Sample collection, fungal isolation and morphological characterization
During the summer seasons (from 2017 to 2021), specimens were collected in field investigations of numerous unknown decomposing woody hosts to determine Diaporthe species in Karst region of Guizhou Province in China. Relevant data (location, date, etc.) were documented and samples were taken to the laboratory in either envelops or in ziploc plastic bags for fungal isolation and examination. The samples were brought to the laboratory under sterile conditions and stored in a refrigerator at 4 °C until the isolation was carried out.
Morphological observations of conidiomata or ascostromata were carried out using a Motic SMZ 168 series stereomicroscope and photographed using a Nikon E80i microscope camera system. Tarosoft Image Framework was used to measure morphological characters (Liu et al. 2010), and images were processed with Adobe Photoshop CS5. The isolation of single spore or conidia was carried out following (Senanayake et al. 2018). Germinated spores on water agar (WA) for 12–24 h were examined and then transferred to potato dextrose agar (PDA) media (OXOID CM0139) to give pure cultures. The cultures were incubated at 25 °C for two weeks, and colony characteristics and morphology of fungal structures were recorded. Colony color (Rayner 1970) was determined after 5–10 days of growth on PDA at 25 °C.
Herbarium specimens were deposited in Guizhou University Herbarium (GZAAS). Ex-type and other living cultures were deposited at the Guizhou University Culture Collection Center (GZCC) and in the China General Microbiological Culture Collection Center (CGMCC) in Beijing, China (Tables 2, 8, 10, 14, 16, 17).
DNA extraction and molecular based amplification
Using aerial mycelium of 7-day-old pure cultures grown on PDA at 25 °C, genomic DNA were extracted following the improved method of cetyltrimethyl ammonium bromide (CTAB) as described by Dissanayake et al. (2015). For identification of Diaporthe species ITS, tef, tub, cal and his gene regions were employed. The ITS region was amplified using universal primers ITS1 and ITS4 (White et al. 1990). The target region of the tef gene was amplified using primer pairs EF-728F and EF-986R (Carbone and Kohn 1999). A portion of the tub gene was amplified using the primers BT2a and BT2b (Glass and Donaldson 1995). The primer pair CAL228F and CAL737R were used to amplify the cal gene region (Carbone and Kohn 1999) while primers CYLH3F and H3-1b (Glass and Donaldson 1995) were used to amplify part of his gene. The PCR reactions were accomplished in a Bio Rad C1000 thermal cycler. The PCR mixture was composed of 0.3 μL of TaKaRa Ex-Taq DNA polymerase, 2.5 μL of 10 × Ex-Taq DNA polymerase buffer, 3.0 μL of dNTPs, 5–20 ng of genomic DNA, 1 μL of each primer and ddH2O up to 25 μL. Following the PCR amplification, products were visualized on 1% agarose gel under UV light using a Gel Doc™ XR Molecular Imager (BioRad, USA) following ethidium bromide staining. Sequence analysis was carried out by Sangon Biotech Co., Ltd (Shanghai, China).
Sequence alignment and phylogenetic analyses
All the type isolates and other representative strains (as per availability) of the genus Diaporthe, which were already published before January 2023 were obtained from GenBank based on previous literature. Single gene trees (ITS, tef, tub, cal and his) and a combined gene phylogenetic tree were constructed to experiment the possibility of robust sections and species/species-complexes of the genus. Sequences generated in previous studies were retrieved from GenBank and were aligned with the sequences of isolates obtained from this study, using MAFFT (Katoh and Toh 2010) (http://www.ebi.ac.uk/Tools/msa/mafft/) and were manually improved with BioEdit (Hall 2006) for maximum alignment.
Phylogenetic analyses of the combined sequence data were performed using, maximum likelihood (ML), maximum parsimony (MP) and Bayesian inference (BI) methods (Dissanayake et al. 2020b). Sequences generated in this study were deposited in GenBank (Tables 2, 8, 10, 14, 16, 17). Alignments and trees were deposited in TreeBASE (www.treebase.org, S6–S9).
Pairwise homoplasy index test and phylogenetic network analysis
For each species/species-complex, concatenated five loci trees were used to infer the existence of sexual recombination within the complex, using pairwise homoplasy index test (PHI) (Bruen 2005) executed in SplitsTree v.4.16.1 (www.splitstree.org) (Huson and Bryant 2006). Noteworthy recombination was reflected with a PHI index below 0.05 (Fw < 0.05). The affiliations among the closely associated taxa were envisioned by creating a phylogenetic linkage from the concatenated dataset of five loci, using the LogDet renovation and the NeighborNet process selections employed in SplitsTree v.4.16.1 (Huson and Bryant 2006).
Species delimitation based on coalescent methods
The inference of the Diaporthe species/species-complex boundaries were performed by coalescent-based methods, based on raxmlGUI2.0 (Edler et al. 2020) producing the Newick-format tree with five combined genes (ITS, tef, tub, cal and his).
Initially, the single Poisson Tree Processes model (PTP) (Zhang et al. 2013), was used. The ML tree produced by RAxML (newick format) was used for the PTP analysis with the following parameters: 1000,000 MCMC generations, thinning set to 100, burn-in at 100,000 generations and accompanied on the web server for PTP (http://species.h-its.org/ptp/). Subsequently, species delimitation using the multi-rate PTP (mPTP), which can lodge data sets encompassed of species with diverse levels of molecular diversity (Kapli et al. 2017), was carried out on the web server (http://mptp.h-its.org), with 1,000,000 MCMC generations -multi, mcmc_sample set to 1000, and minimum branch length (-minbr) which was calculated by mPTP was set to its specific number.
The specific species/species-complex boundaries were primarily identified incorporating coalescence-based models (PTP, mPTP) to the combined gene phylogeny. Based on these results, 28 phylogenetic analyses were conducted for the proposed species/species-complexes. The phylogenetic analyses of each species/species-complex contained almost all available strains of the species from GenBank as per availability. Putative species clusters resulted in PTP analysis are indicated in species/species complexes phylogenies using transitions between black-colored to red-colored branches. The complexes were named according to the oldest epithet or based on the ‘popularity’ of the specific species residing in each complex. Phylogenetic trees were rooted with one or few of the most closely related species, which reside out of the specific species/species-complex. These phylogenetic trees and the alignments of species/species-complexes were deposited in TreeBASE.
Results
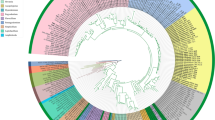
To evaluate the genus Diaporthe, single gene trees (ITS, tef, tub, cal and his) were constructed including all type strains (see supplementary materials Figs. S1–S5). Seven major sections (Section Betulicola, Section Crotalariae, Section Eres, Section Foeniculina, Section Psoraleae-pinnatae, Section Rudis and Section Sojae) were discovered, which mostly cluster separately in single gene trees (Fig. 1). The results were implemented and linked by the adoption of GCPSR based multiple gene genealogies and PHI test to assist the proposed sections (Fig. 2).

Section arrangement of the combined gene phylogeny and single gene phylogenies (ITS, tef, tub, cal and his). S1: Section Foeniculina, S2: Section Eres, S3: Section Sojae, S4: Section Rudis, S5: Section Betulicola, S6: Section Psoraleae-pinnatae and S7: Section Crotalariae

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the genus Diaporthe. Based on LogDet transformation and the NeighborNet algorithm inferred by SplitsTree, the genus was restructured into seven sections: Section Betulicola, Section Crotalariae, Section Eres, Section Foeniculina, Section Psoraleae-pinnatae, Section Rudis and Section Sojae. The scale bar represents the expected number of substitutions per nucleotide position
The combined gene tree (Fig. 3), which resulted from the ITS, tef, tub, cal and his alignments (including gaps) were determined to be approximately 695, 717, 1064, 664 and 706 base pair in size, respectively. The combined gene analysis of Diaporthe contained data for 749 isolates, including the outgroup taxon Diaporthella corylina (CBS 121124). The analyses consisted of 82 isolates from this study and sequences of 667 isolates originating from GenBank. Out of a total of 3846 characters in the MP analyses, 1186 were constant, and 541 were variable and parsimony uninformative. Ten most parsimonious trees resulted from the remaining 2136 parsimony-informative characters (CI = 0.384, RI = 0.804, RC = 0.309, HI = 0.616). In the ML analyses, the best scoring RAxML tree (Fig. 3) with a final likelihood value of -134,020.062780 is presented. The matrix had 3132 distinct alignment patterns, with 52.50% of undetermined characters or gaps. Estimated base frequencies were as follows: A = 0.217177, C = 0.324414, G = 0.237374, T = 0.221035; substitution rates AC = 1.069704, AG = 2.914692, AT = 1.152981, CG = 0.865015, CT = 4.116465, GT = 1.0; gamma distribution shape parameter alpha = 0.580991. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree (Fig. 3) is shown with bootstrap and posterior probabilities given for those well supported clades. Alignment and the tree were deposited in TreeBASE (www.treebase.org, study ID S30383). Taxonomic novelties were submitted to the Faces of Fungi database (www.facesoffungi.org) and Index Fungorum (www.indexfungorum.org).










Phylogram generated from maximum likelihood analysis of all type strains of Diaporthe species acquired from recent publications and GenBank, including the isolates obtained from Guizhou Province, based on combined internal transcribed spacer region (ITS), tef, tub, cal and his sequence data. Bootstrap values of ML, MP > 75% are shown near the nodes and branches in bold indicate BI probabilities > 0.95. Isolates obtained in this study are in red. Ex-type strains are in bold. The tree is rooted with Diaporthella corylina (CBS 121124). By applying GCPSR method and coalescence-based models to the multi-gene phylogeny, species/species complexes are proposed, and they are mentioned in colored rectangles
Concept of sections
In this manuscript, we propose seven sections in the genus Diaporthe (Section Betulicola, Section Crotalariae, Section Eres, Section Foeniculina, Section Psoraleae-pinnatae, Section Rudis and Section Sojae). These sections separately appeared in combined gene phylogeny as well as in several single gene trees (tub, cal and his; Fig. 1). We observed the species assigned to these sections always occurred together, forming clearly separated clades in combined phylogeny (Fig. 3) and in single gene phylogenies (Figs. S1–S5). This observation was proved by the adoption of PHI test, which assisted establishment of the proposed sections (Fig. 2).
Concept of species/species complexes
Within each section, we observed several species again clustered together and this was observed in combined gene phylogeny as well as in single gene phylogenies. By applying GCPSR method and coalescence-based models (PTP, mPTP) to these clusters, we could determine that some clusters formed species complexes (comprising several different species) while other ‘clusters’ appeared to be a single species. For these demarcations, we followed Hilário et al. (2021a, b), Norphanphoun et al. (2022) and Pereira et al. (2023).
Once the genus was restructured into seven sections (Section Betulicola, Section Crotalariae, Section Eres, Section Foeniculina, Section Psoraleae-pinnatae, Section Rudis and Section Sojae), each section was examined for possible species/species complexes by comparing the single gene phylogenies accompanying GCPSR principle and coalescence-based models. The difficulties in resolving closely related phylogenetic species are likely due to gene flow among species and recombination possibilities (Santos et al. 2011; Fan et al. 2018; Drenth et al. 2019).
Considering six phylogenetic trees (combined gene tree and single gene trees) of all available type strains of Diaporthe, the 28 species/species-complexes mentioned below are proposed within the genus. The complexes were named following Hilário et al. (2020, 2021a, b), Norphanphoun et al. (2022), Hongsanan et al. (2023), and Pereira et al. (2023) as well as based on the oldest epithet or the ‘popularity’ of the specific species residing in each complex. Synonymies are provided for 31 species. Species synonymized in the present study are mostly based on the type strains, while several strains which have no type sequences have been included as per their presence in previous publications.
Section Foeniculina
-
1.
Diaporthe anacardii
-
2.
Diaporthe arecae
-
3.
Diaporthe biconispora
-
4.
Diaporthe foeniculina
-
5.
Diaporthe hongkongensis
-
6.
Diaporthe oncostoma species complex—contains 10 species.
-
7.
Diaporthe pterocarpi
Section Eres
-
1.
Diaporthe citrichinensis species complex—contains 3 species.
-
2.
Diaporthe eres
-
3.
Diaporthe gardeniae species complex—contains 5 species.
-
4.
Diaporthe subclavata species complex—contains 2 species.
-
5.
Diaporthe virgiliae species complex—contains 5 species.
Section Sojae
-
1.
Diaporthe arctii species complex—contains 16 species.
-
2.
Diaporthe ganjae
-
3.
Diaporthe leucospermi species complex—contains 15 species.
-
4.
Diaporthe longicolla species complex—contains 9 species.
-
5.
Diaporthe schini species complex—contains 5 species.
-
6.
Diaporthe sclerotioides species complex—contains 3 species.
-
7.
Diaporthe siamensis species complex—contains 11 species.
-
8.
Diaporthe sojae
-
9.
Diaporthe tulliensis
Section Rudis
-
1.
Diaporthe amygdali
-
2.
Diaporthe pseudotsugae species complex—contains 5 species.
-
3.
Diaporthe rudis
Section Betulicola
-
1.
Diaporthe ampelina species complex—contains 6 species.
-
2.
Diaporthe betulicola species complex—contains 2 species.
Section Psoraleae-pinnatae
-
1.
Diaporthe psoraleae-pinnatae
Section crotalariae
-
1.
Diaporthe crotalariae species complex—contains 4 species)
Table 1 Proposed species/species-complexes and the arrangement of species within the single gene trees (ITS, tef, tub, cal and his)
Section Foeniculina
‘Section Foeniculina’ is composed of 38 type strains of Diaporthe (Table 2) and is separated from the other proposed sections by its arrangement of species within the combined phylogenetic analysis (Figs. 1–3). However, we observed several conflicts of the species arrangement in the multi-gene phylogeny as well as in the single gene trees (Figs. S1–S5).
In Section Foeniculina, three known species (D. arecae, D. lenispora and D. oncostoma) were isolated from various woody hosts in Guizhou Province, China. Additionally, synonymous names for eight Diaporthe species and seven species/species-complexes (D. anacardii, D. arecae, D. biconispora, D. foeniculina, D. hongkongensis, D. oncostoma and D. pterocarpi) are proposed (Figs. 3, 4).

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the species complexes within Section Foeniculina, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
Synonymies of Section Foeniculina
Diaporthe aseana Dissan., Tangthir. & K.D. Hyde, Fungal Diversity 80: 89 (2015)
= Diaporthe tectonigena Doilom, Dissan. & K.D. Hyde, Fungal Diversity 82: 164 (2016)
Host range: Tectona grandis (Doilom et al. 2016)
Known distribution: Thailand (Doilom et al. 2016; Hyde et al. 2016)
Notes: Hyde et al. (2016) described D. aseana from an unknown dead leaf in Thailand. Later, Doilom et al. (2016) introduced D. tectonigena which was associated with twig dieback of Tectona grandis in Thailand. These two species are morphologically indistinguishable with overlapping micromorphological characters of alpha conidia (5.0–8.3 × 2.5–3.5 μm vs 6–9 × 2–3 μm). A pairwise comparison of nucleotides showed that D. aseana and D. tectonigena are similar, since both species differ in 8 bp in ITS, 1 bp in tef, 2 bp in tub and 0 bp in cal. As D. aseana has no his sequences we were unable compare it with that of D. tectonigena.
Diaporthe chamaeropis (Cooke) R.R. Gomes, Glienke & Crous, Persoonia 31: 18 (2013)
= Diaporthe cytosporella (Penz. & Sacc.) Udayanga & Castl., Persoonia 32: 95 (2014).
Host range: Chamaerops humilis, Spartium junceum (Gomes et al. 2013), Citrus limonia (Udayanga et al. 2014a).
Known distribution: Croatia, Greece (Gomes et al. 2013), Italy (Udayanga et al. 2014a)
Notes: Gomes et al. (2013) described D. chamaeropis as a comb. nov. from a dead branch of Spartium junceum in Croatia. Later, Udayanga et al. (2014a) introduced D. cytosporella from Citrus limonia in Italy. In the combined gene phylogenetic tree, these two species clustered together (Fig. 3). They are morphologically indistinguishable with overlapping micromorphological characters of alpha conidia (6–9 × 2.0–2.5 μm vs 7–10 × 2–3 μm). Though beta conidia were observed in D. chamaeropis, they were not seen in D. cytosporella. A pairwise comparison of nucleotides showed that D. chamaeropis and D. cytosporella are similar differing in 2 bp in ITS, 1 bp in tef, 6 bp in tub and 0 bp in cal. Neither species has his sequences available.
Species/species-complexes residing in Section Foeniculina
1. Diaporthe anacardii
1.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates in this clade contained six species, designated here as D. anacardii. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 5). The five loci combined alignment comprised 2151 characters (532 characters from ITS, 314 from tef, 386 from tub, 465 from cal, and 454 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 16 ingroup taxa (all 17 taxa were retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. anacardii’ is provided in Table S6. As the ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe anacardii’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PP) greater than 0.95 are indicated as thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host where it was isolated. Synonymies are given within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. isoberliniae (CPC 22549). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), tef and tub provided the most informative sequences for D. anacardii, followed by cal and his loci (Table 1). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of taxa in D. anacardii (Fig. S1).
1.2 Species delimitation based on the GCPSR principle
To assess the boundaries of species residing in the D. anacardii clade, the GCPSR principle was applied. To disclose concordant branches, individual gene trees were compared. The results revealed several conflicts between individual phylogenies, where some species do not clade within the complex while some additional species claded within the complex. For example, D. phillipsii does not clade within the complex in the ITS gene tree (Fig. S1). Diaporthe incompleta (CGMCC3.18288) is unusually claded within the complex in tub single gene tree (Fig. S3). According to cal and his single gene trees (Figs. S4, S5), D. macadamiae and D. nebulae had no cal and his sequences.
However, individual gene trees are concordant and have the same topology for the well-delimited outgroup taxon D. isoberliniae denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the D. anacardii complex and it is herein regarded as a single species, demarcating its species boundaries.
1.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences detected significant recombination within the D. anacardii complex (Φw = 0) and the network relationships are shown in Fig. 6. Moreover, based on the comparable remoteness of species and the arrangement of the phylogenetic network, all six species in the D. anacardii complex should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the strains belonging to ‘Diaporthe anacardii’ and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
1.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and showed the D. anacardii complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a specific species instead of diverse taxa. Hence, each species presently recognized in the D. anacardii complex should be accepted as a single species (Figs. 5, 6).
1.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. anacardii, which was earlier considered to be six species but herein represents a single species.
Diaporthe anacardii (Early & Punith.) R.R. Gomes, Glienke & Crous, Persoonia 31: 15 (2013)
= Diaporthe macadamiae Y.P. Tan, Akinsanmi & R.G. Shivas, Pl. Path. 69: 916 (2020)
= Diaporthe nebulae Lesuthu, Mostert, Spies, Moyo & Halleen, Pl. Dis. 103: 813 (2019)
= Diaporthe phillipsii S. Hilário, L. Santos & A. Alves, Mycologia 112: 301 (2020)
= Diaporthe portugallica Guarnaccia, Phytopath. Mediterr. 27: 316 (2018)
= Diaporthe velutina Y.H. Gao & L. Cai, IMA Fungus 8: 178 (2017)
Host range: Anacardium occidentale (Gomes et al. 2013), Camellia sinensis (Guarnaccia and Crous 2018), Macadamia sp. (Wrona et al. 2020), Neolitsea sp. (Gao et al. 2017), Vaccinium corymbosum (Hilário et al. 2020), Vitis viniferae (Lesuthu et al. 2019).
Known distribution: China (Gao et al. 2017), East Africa (Gomes et al. 2013), Portugal (Guarnaccia and Crous 2018; Hilário et al. 2020), South Africa (Wrona et al. 2020; Lesuthu et al. 2019).
Description: Sexual morph not reported. See Gomes et al. (2013) for illustrations and descriptions of asexual morph.
Notes: Gomes et al. (2013) epitypified D. anacardii (Phomopsis anacardii) which was described from Anacardium occidentale in Kenya, and has also been reported from Nigeria, Guinea and Cuba (Early and Punithalingam 1972). Later, D. velutina was introduced from diseased leaves of Neolitsea sp. in China (Gao et al. 2017) and D. portugallica was introduced from leaf lesions of Camellia sinensis in Portugal (Guarnaccia and Crous 2018). Lesuthu et al. (2019) introduced D. nebulae from wood of Vitis viniferae from South Africa while D. phillipsii was introduced from twig blight and dieback of Vaccinium corymbosum in Portugal by Hilário et al. (2020). Later, Wrona et al. (2020) described D. macadamiae from husk rot of Macadamia sp. in South Africa. In the phylogenetic analysis of Wrona et al. (2020), strains of D. anacardii were not included. Here we show that D. anacardii, D. macadamiae, D. nebulae, D. phillipsii, D. portugallica and D. velutina cluster in a well-supported clade and are closely related (Figs. 5, 6) supported by the GCPSR principle and coalescent methods of PTP and mPTP. These species are also morphologically indistinguishable (Table 3). Synonymous names of D. anacardii are provided.
1.6. Morphology of the strains belonging to ‘Diaporthe anacardii'. See Table 3.
2. Diaporthe arecae
Norphanphoun et al. (2022) included 45 species in D. arecae species complex. Pereira et al. (2023) noted that a group of species including D. acuta, D. anhuiensis, D. annellsiae, D. arengae, D. arecae, D. averrhoae, D. bounty, D. camelliae-oleiferae, D. ceratozamiae, D. cercidis, D. chamaeropicola, D. chrysalidocarpi, D. delonicis, D. drenthii, D. endocitricola, D. fraxini-angustifoliae, D. fulvicolor, D. gossiae, D. guangxiensis, D. hongheensis, D. howardiae, D. huangshanensis, D. hunanensis, D. krabiensis, D. limonicola, D. liquidambaris, D. litchiicola, D. loropetali, D. meliae, D. melitensis, D. millettiae, D. musigena, D. nelumbonis, D. norfolkensis, D. oculi, D. osmanthi, D. pandanicola, D. pascoei, D. pescicola, D. phyllanthicola, D. podocarpi-macrophylli, D. pseudomangiferae, D. pseudooculi, D. pseudophoenicicola, D. pterocarpicola, D. schimae, D. searlei, D. sennae, D. spinosa, D. taiwanensis, D. taoicola, D. viciae and D. viniferae, occupy a monophyletic clade in Diaporthe phylogenies. By employing the phylogenetic analysis, GCPSR and coalescence-based models they determined that the D. arecae species complex constitutes a single species. We observed the same results as Pereira et al. (2023) and verify D. arecae is a single species. Additionally, we observed the strains of D. caricae-papayae, D. euginiae, D. persiae and D. phoenicicola also clustered in this species clade.
3. Diaporthe biconispora
3.1. Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates residing in this clade contain five species, designated here as ‘D. biconispora’. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 7). The five loci combined alignment comprised 2209 characters (543 characters from ITS, 372 from tef, 387 from tub, 435 from cal, and 472 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 16 ingroup taxa (17 taxa were retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. biconispora’ is provided in Table S6. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe biconispora’ based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host from where they were isolated. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. undulata (CGMCC3.18293). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), all single gene trees (ITS, tef, tub, cal and his) displayed the most informative sequences for D. biconispora (Table 1).
3.2 Species delimitation based on the GCPSR principle
To assess boundaries of the species residing in the D. biconispora complex, the GCPSR principle was applied. To disclose concordant branches, the individual gene trees were compared. Almost all single gene trees were phylogenetically similar, although an additional species (D. megabiguttulata) is claded in the tef gene tree (Fig. S2).
Individual gene trees are concordant and have the same topology for the well-delimited outgroup taxon D. undulata denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the D. biconispora complex and it is herein regarded as a single species, demarcating its species boundaries.
3.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences detected significant recombination within the D. biconispora complex (Φw = 0.014) and the network relationships are shown in Fig. 8. Moreover, based on the comparable remoteness of species and the arrangement of the phylogenetic network, all five species in this complex should be regarded as one single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the strains belonging to ‘Diaporthe biconispora’ and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
3.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. biconispora complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as one species instead of diverse taxa. Hence, D. biconispora complex should be accepted as a single species (Figs. 7, 8).
3.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. biconispora, which was earlier known as five different species but herein represent a single species.
Diaporthe biconispora F. Huang, K.D. Hyde & Hong Y. Li, Fungal Biol 119: 338 (2015)
= Diaporthe longiconidialis M. Luo, W. Guo, Manawas., M.P. Zhao, K.D. Hyde & C.P. You, Journal of Fungi 8:14 (2022)
= Diaporthe pometiae S.T. Huang, J.W. Xia, W.X. Sun & X.G. Zhang, MycoKeys 78: 70 (2021)
= Diaporthe salsuginosa Vrijmoed, K.D. Hyde & E.B.G. Jones, Mycol. Res. 98: 699 (1994)
= Diaporthe tersa (Sacc.) Udayanga & Castl., IMA Fungus 7: 291 (2016)
Host range: Avicennia marina (Vrijmoed et al. 1994), Citrus grandis (Huang et al. 2015), Morinda officinalis (Luo et al. 2022), Pometia pinnata (Huang et al. 2021), deep-sea sediments (Rossman et al. 2016)
Known distribution: China (Vrijmoed et al. 1994; Huang et al. 2015, 2021; Rossman et al. 2016; Luo et al. 2022)
Description: Sexual morph not reported. See Huang et al. (2015) for illustrations and descriptions of asexual morph.
Notes: All five species reported in this complex have been described from China. Vrijmoed et al. (1994) identified D. salsuginosa from Avicennia marina while D. tersa was identified from deep-sea sediments by Rossman et al. (2016). Diaporthe pometiae was introduced by Huang et al. (2021) from diseased leaves of Pometia pinnata, and D. longiconidialis was identified from a healthy root of Morinda officinalis (Luo et al. 2022). Here we show that D. biconispora, D. longiconidialis, D. pometiae, D. salsuginosa, and D. tersa cluster in a well-supported clade and are closely related supported by the GCPSR principle and coalescent methods of PTP and mPTP (Figs. 7, 8) and are morphologically indistinguishable (Table 4). Synonymous names of D. biconispora are provided.
Norphanphoun et al. (2022) introduced a species complex named ‘Diaporthe biconispora’, and it contained two species (D. biconispora and D. salsuginosa). Hence, we delimit the boundaries of D. biconispora integrating single gene trees, multi-gene tree, GCPSR and coalescence-based models (PTP, mPTP).
3.6. Morphology of the strains belonging to ‘Diaporthe rudis’
4.Diaporthe foeniculina species-complex
4.1 Phylogenetic analyses and informative characters
Hilário et al. (2021c) synonymized D. baccae and D. ravennica under D. foeniculina. In this study, the multi-gene phylogenetic tree (Fig. 3) showed that the isolates belonging to ‘D. foeniculina’ cluster in one clade containing seven species. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 9). The five loci combined alignment comprises 2127 characters (534 characters from ITS, 308 from tef, 389 from tub, 448 from cal, and 448 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 23 ingroup taxa (24 taxa were retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of ‘D. foeniculina’ are provided in Table S6. As MP, ML and BI analyses resulted in trees that were topologically similar, only ML tree is shown.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe foeniculina’ based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host from where they were isolated. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. forlicesenica (MFLUCC 17-1015). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
No single gene tree possessed 100% similar topologies as additional taxa were observed within tef, tub gene trees (Figs. S1–S5).
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), tub displayed the most informative sequence for D. foeniculina (Table 1). ITS presented the lowest percentage, as D. baccae, D. ravennica, D. rhusicola and D. zaobaisu were located outside the clade, indicating that this locus is unreliable for the delimitation of taxa in D. foeniculina (Fig. S1). In the tef single gene tree, only D. zaobaisu was located outside the clade. Diaporthe nigra, D. ravennica, D. rhusicola and D. rumicicola had no cal and his sequences. Diaporthe canthii is unusually claded in ITS single gene tree while D. cytosporella is unusually claded in tub single gene tree.
4.2 Species delimitation based on the GCPSR principle
According to the GCPSR principle, the results revealed conflicts between individual phylogenies, where some species do not clade within the complex or some additional species are claded within the complex. For example, in the ITS phylogeny, species in the D. foeniculina complex formed two distinct clades within the tree (Fig. S1). Moreover, D. chamaeropis, D. cytosporella and D. parvae are unusually claded within the complex in tef single gene tree (Fig. S2) while D. cytosporella (FAU 461) is unusually claded within the complex in tub single gene tree (Fig. S3). By analyzing the individual gene trees (Figs. S1–S5), and given their position within the complex, we suggest that all the species mentioned above should be assigned to ‘D. foeniculina’. Moreover, a lack of high bootstrap and posterior probability values on both individual and combined trees in several branches were observed, revealing poor phylogenetic support among the taxa. However, combined gene tree (Fig. 9) is composed of a well-delimited out-group taxon (D. forlicesenica), indicating these clades represent different species. Thus, by implementing the GCPSR principle, based on the comparison of more than one gene genealogy to identify phylogenetic concordance, we verified that D. foeniculina is a single species rather than a species complex.
4.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of the concatenated multi-locus sequences ITS, tef, tub, cal and his revealed significant recombination within the D. foeniculina complex (Φw = 0.015). The network relationships in D. foeniculina are shown in Fig. 10. Additionally, based on the relative distance of species and structure of the phylogenetic network, all seven species in ‘D. foeniculina’ should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe foeniculina species complex and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
4.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. foeniculina complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a single species instead of diverse taxa. Hence, D. foeniculina complex should be accepted as a single species (Figs. 9, 10).
4.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. foeniculina, which was earlier known as seven different species but herein represent a single species. Hilário et al. (2021c) synonymized D. baccae and D. ravennica under D. foeniculina. In the present study we include four additional species (D. nigra, D. rhusicola, D. rumicicola and D. zaobaisu) under this synonymy. Diaporthe nigra always clustered within the taxa of D. foeniculina and this was observed in combined gene tree and in all the single gene trees (Figs. 3, S1–S5).
Diaporthe foeniculina (Sacc.) Udayanga & Castl., Persoonia 32: 95 (2014)
= Diaporthe baccae L. Lombard, Polizzi & Crous, Phytopath. Mediterr. 53: 93 (2014)
= Diaporthe nigra Brahmanage & K.D. Hyde, Fungal Diversity 100: 185 (2020)
= Diaporthe ravennica Thambug., Camporesi & K.D. Hyde, Fungal Diversity 82: 245 (2016)
= Diaporthe rhusicola Crous, Persoonia 26: 135 (2011)
= Diaporthe rumicicola Manawasinghe, Camporesi & K.D. Hyde, Fungal Diversity: 96: 139 (2019)
= Diaporthe zaobaisu Y.S. Guo & G.P. Wang, Persoonia 45: 156 (2020)
Host range: See Hilário et al. (2021c) for the hosts for D. foeniculina. Ballota nigra (Hyde et al. 2020), Juglans regia (Chen et al. 2014), Pyrus bretschneideri cv. Zaobaisu (Guo et al. 2020), Rhus pendulina (Crous et al. 2011), Rumex sp. (Hyde et al. 2019), Tamarix sp. (Thambugala et al. 2016), Vaccinium corymbosum (Lombard et al. 2014)
Known distribution: See Hilário et al. (2021c) for the distribution of D. foeniculina. China (Guo et al. 2020), Italy (Lombard et al. 2014; Thambugala et al. 2016; Hyde et al. 2019, 2020), South Africa (Crous et al. 2011), USA (Chen et al. 2014)
Description: Sexual morph and asexual morph have been reported. See Phillips (2003) and Udayanga et al. (2014a) for illustrations and descriptions.
Notes: Hilário et al. (2021c) synonymized D. baccae and D. ravennica under D. foeniculina, but their analysis did not include D. nigra, D. rhusicola, D. rumicicola and D. zaobaisu. Here we show that D. nigra, D. rhusicola, D. rumicicola and D. zaobaisu are phylogenetically closely associated with this species complex. A pairwise comparison showed that both D. nigra and foeniculina are phylogenetically 100% similar in ITS, tef and tub. As D. nigra had no cal and his sequence data we were unable to compare its nucleotide variations. A pairwise comparison showed that D. rhusicola and D foeniculina are phylogenetically similar since both species differ in 8 bp in ITS. Since D. rhusicola had no tef, tub, cal and his sequence data we were unable to compare its nucleotide variations. A pairwise comparison showed that D. zaobaisu and the ex-type of D. foeniculina (CBS 11553) are phylogenetically distinct but D. zaobaisu shows an affinity to D. ravennica since both species differ in 2 bp in ITS. Unusually, for D. zaobaisu we observed an insertion of 18 base pairs (773 to 790) in the tef gene region, which is not seen for other strains in the complex. Diaporthe foeniculina and D. zaobaisu differ in 2 bp in tub and 6 bp in his. As D. zaobaisu has no cal sequence data we were unable to compare its nucleotide variations. However, we observed D. zaobaisu always clusters within the D. foeniculina complex in combined gene tree (Fig. 9) as well as in ITS, tef, tub, his single gene trees (Figs. S1–S5). This group of seven species cluster together in our combined gene phylogenetic tree (Fig. 9) as well as in most single gene trees (Figs. S1–S5). This was supported by the GCPSR principle and coalescent methods of PTP and mPTP. For comparison, a synopsis of conidiomata, conidiogenous cells and conidia characteristics are provided for all these species in Table 5. Synonymous names of D. foeniculina are provided.
4.6 Morphology of the strains belonging to ‘Diaporthe foeniculina’. See Table 5.’
5. Diaporthe hongkongensis
5.1 Phylogenetic analyses and informative characters
The preliminary multi-gene phylogenetic tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this clade contains eight species, which is designated here as ‘D. hongkongensis’. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 11). The five loci combined alignment comprises 2176 characters (539 characters from ITS, 332 from tef, 385 from tub, 468 from cal, and 452 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 28 ingroup taxa (29 taxa were retrieved from GenBank). Alignment details of MP, ML and BI analyses are provided in Table S6. Resulting MP, ML and BI trees were topologically similar and only ML tree is shown here.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe hongkongensis’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. xishuangbanica (CGMCC3.18282). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
Considering the single gene trees, tef and tub gene trees possessed 100% identical topology followed by cal and his (Figs. S2–S5, Table 1). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of taxa in D. hongkongensis (Fig. S1).
5.2 Species delimitation based on the GCPSR principle
To assess the species boundaries in the D. hongkongensis complex, the GCPSR principle was applied. Our results revealed conflicts between individual phylogenies, where some species do not clade within the complex or some species are unusually claded within the complex. It is also evident that isolates of the same species cluster in different clades in the combined gene tree. For instance, most of the isolates of D. eucalyptorum, D. hongkongensis and D. lithocarpus cluster in various places in the combined phylogeny (Fig. 11) showing their phylogenetic distinction. In the ITS phylogeny, we observed the taxa belonging to this complex are claded in two distinct clades (Fig. S1), while they group together in the tef gene tree (Fig. S2). Diaporthe conorum, D. tectonigena, D. xishuangbanica and D. zhaoqingensis are unusually claded in ITS single gene tree. Moreover, there was a lack of high bootstrap and posterior probability values in several branches of both individual and combined trees, revealing poor phylogenetic support among the species. However, combined gene tree (Fig. 11) is composed of a well-delimited outgroup species (D. xishusngbanica), indicating these clades represent different species.
Thus, by implementing the GCPSR principle, based on the comparison of more than one gene genealogy to identify phylogenetic concordance, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles D. hongkongensis which constitutes a single species rather than a species complex.
5.3 Pairwise homoplasy test and phylogenetic networks
Application of the PHI test to the concatenated multi-locus sequences ITS, tef, tub, cal and his revealed significant recombination within the D. hongkongensis complex (Φw = 0.0046). Additionally, based on the relative distance of species and structure of the phylogenetic network (Fig. 12), all eight species in the D. hongkongensis complex should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe hongkongensis species complex and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
5.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. hongkongensis complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a single species instead of diverse taxa. Hence, D. hongkongensis complex should be accepted as a single species (Figs. 11, 12).
5.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. hongkongensis, which was earlier known as eight different species but herein represents a single species.
Diaporthe hongkongensis R.R. Gomes, Glienke & Crous, Persoonia 31: 23 (2013)
= Diaporthe australiana R.G. Shivas, Akinsanmi & Y.P. Tan, Pl. Path. 69: 916 (2020)
= Diaporthe eucalyptorum Crous & R.G. Shivas, Persoonia 28: 153 (2015)
= Diaporthe eucommiae (F.X. Chao & P.K. Chi) Y.H. Gao & L. Cai, IMA Fungus 8: 183 (2017)
= Diaporthe lagerstroemiae (C.Q. Chang, Xiang & P.K. Chi) Y.H. Gao & L. Cai, IMA Fungus 8: 183 (2017)
= Diaporthe lithocarpus Y.H. Gao, W. Sun & L. Cai, Mycol. Progr. 13: 115 (2014)
= Diaporthe rhodomyrti C.M. Tian & Qin Yang, MycoKeys 91: 41 (2022)
= Diaporthe salinicola Dayarathne, Mycosphere 11: 91 (2020)
Host range: Camelia sinensis (Gao et al. 2016), Citrus sp. (Huang et al. 2015), Dichroa febrifuga (Gomes et al. 2013), Eucalyptus sp. (Crous et al. 2012), Eucommia ulmoides (Jiang et al. 2018), Lagerstroemia indica (Gao et al. 2017), Lithocarpus glabra (Gao et al. 2014), Rhodomyrtus tomentosa (Cao et al. 2022), Vitis vinifera (Dissanayake et al. 2015), Macadamia sp. (Wrona et al. 2020), mangrove wood (Dayarathne et al. 2020).
Known distribution: Australia (Crous et al. 2012; Gomes et al. 2013; Wrona et al. 2020), China (Gao et al. 2014, 2016, 2017; Dissanayake et al. 2015; Huang et al. 2015; Jiang et al. 2018; Cao et al. 2022), Thailand (Dayarathne et al. 2020).
Notes: Micromorphology of the asexual morph of all taxa belonging to D. hongkongensis (Table 6) match the original description of D. hongkongensis reported by Gomes et al. (2013). Culture characteristics are similar to those reported by Gomes et al. (2013): colonies spreading on PDA in a radial pattern with white, aerial, cottony mycelium, sometimes with brown aerial mycelium at the center, becoming grey at edges of plate, and reverse white to ivory color in concentric zones, becoming brownish to black with age. Conidiomata, conidiophores, conidiogenous cells and alpha conidia with shape and dimensions that correlate with that reported by those authors (globose to subglobose, dark brown to black conidiomata; hyaline, smooth, densely aggregated conidiophores; phialidic, hyaline, cylindrical conidiogenous cells; hyaline, aseptate, fusiform, guttulate, 5–8 × 2–3 μm alpha conidia). Diaporthe lithocarpus was introduced by Gao et al. (2014) from Lithocarpus glabra in China while D. eucalyptorum was introduced by Crous et al. (2012) from Eucalyptus sp. in Australia. Gao et al. (2017) introduced D. lagerstroemiae from Lagerstroemia indica in China while Jiang et al. (2018) introduced D. eucommiae from Eucommia ulmoides in China. Diaporthe australiana was introduced from Macadamia sp. in Australia (Wrona et al. 2020) while D. salinicola was introduced from mangrove wood in Thailand (Dayarathne et al. 2020). Dayarathne et al. (2020) reported the sexual morph of D. salinicola; there are no sexual morph reports for the other species. Diaporthe rhodomyrti was introduced from Rhodomyrtus tomentosa in China (Cao et al. 2022). All eight species in this group claded within the strains of D. hongkongensis in our combined gene phylogenetic tree (Fig. 11) as well as mostly in single gene trees (Figs. S1–S5). This was supported by the GCPSR principle and coalescent methods of PTP and mPTP. A comparative synopsis of conidiomata, conidiogenous cells and conidia morphology is provided in Table 6. Synonymous names of D. hongkongensis are provided.
5.6 Morphology of the strains belonging to ‘Diaporthe hongkongensis’. See Table 6.
6. Diaporthe sclerotioides species complex
6.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this clade contains 10 species, designated here as ‘D. oncostoma species complex’. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 13). The five loci combined alignment comprises 2537 characters (585 characters from ITS, 342 from tef, 704 from tub, 454 from cal, and 452 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 31 ingroup taxa (one from this study and 31 taxa retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. oncostoma species complex’ is provided in Table S6. All ML, MP and BI analyses resulted in trees that were topologically similar, and only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe oncostoma species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. stictica (CBS 370.54). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), tef displayed the most informative sequences for D. oncostoma species complex, followed by tub, cal and his loci (Table 1). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of taxa in D. oncostoma species complex (Fig. S1).
6.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. oncostoma species complex, the GCPSR principle was applied. Individual gene trees are concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. stictica) denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. oncostoma species complex’ by demarcating its species boundaries.
6.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant evidence for recombination within the D. oncostoma species complex (Φw = 0.99) and the network relationships are shown in Fig. 14. Moreover, based on the comparable remoteness of species and the arrangement of the phylogenetic network, all 10 species in the D. oncostoma species complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe oncostoma species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
6.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. oncostoma species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed the boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, D. oncostoma species complex should be accepted as different species (Figs. 13, 14).
6.5 Taxonomy.
Species residing in Diaporthe oncostoma species complex
Diaporthe oncostoma (Duby) Fuckel, Jb. nassau. Ver. Naturk. 23–24: 205 (1870)
Diaporthe camelliae-sinensis S.T. Huang, J.W. Xia, X.G. Zhang & Z. Li, MycoKeys 77: 71 (2021)
Diaporthe canthii Crous, Persoonia 28: 159 (2012)
Diaporthe cissampeli Crous & Roets, Persoonia 36: 361 (2016)
Diaporthe hickoriae Wehm., Monogr. Gen. Diaporthe Nitschke & Segreg., Univ. Mich. Stud., Sci. Ser. 9: 149 (1933)
Diaporthe maytenicola Crous, Persoonia 31: 259 (2013)
Diaporthe melastomatis S.T. Huang, J.W. Xia, X.G. Zhang & Z. Li, MycoKeys 77: 83 (2021)
Diaporthe parapterocarpi Crous, Persoonia 32: 229 (2014)
Diaporthe saccarata (J.C. Kang, L. Mostert & Crous) Crous, Persoonia 31: 32 (2013)
Diaporthe vangueriae Crous, Persoonia 32: 227 (2014)
Synonymies in Diaporthe oncostoma species complex
Diaporthe vangueriae Crous, Persoonia 32: 227 (2014)
= Diaporthe macintoshii R.G. Shivas, S.M. Thomps. & Y.P. Tan, Persoonia 35: 45 (2015)
Hosts: Rapistrum rugosum (Thompson et al. 2015), Vangueria infausta (Crous et al. 2014)
Known distribution: Australia (Thompson et al. 2015), Zambia (Crous et al. 2014)
Notes: Crous et al. (2014) introduced D. vangueriae from Vangueria infausta in Zambia. Later, Thompson et al. (2015) described D. macintoshii from Rapistrum rugosum in Australia. Diaporthe vangueriae has not produced alpha conidia while D. macintoshii produces both alpha and beta conidia. According to the morphological descriptions, the beta conidia of both species are morphologically overlapping. However, Thompson et al. (2015) did not include D. vangueriae in their phylogenetic analysis. A pairwise comparison showed that D. vangueriae and D. macintoshii are similar, differring by only 1 bp in tub. The ITS gene region of both species was 100% identical. The tef sequence was unavailable for D. vangueriae and neither species had his sequence data.
Norphanphoun et al. (2022) introduced a species complex named ‘Diaporthe oncostoma’, but in this study, the boundaries of D. biconispora was determined integrating single gene trees, multi-gene tree, GCPSR and coalescence-based models (PTP, mPTP).
7. Diaporthe pterocarpi
7.1 Phylogenetic analyses and informative characters
The multi-gene phylogenetic tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this clade contains four species, which is designated here as ‘D. pterocarpi’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 15). The five loci combined alignment comprises 2448 characters (531 characters from ITS, 308 from tef, 388 from tub, 462 from cal, and 451 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 13 ingroup taxa (14 taxa were retrieved from GenBank). Alignment details of MP, ML and BI analyses are provided in Table S6. Resulting MP, ML and BI trees were topologically similar and only ML tree is shown here. Considering the single gene trees, ITS, tef, tub, cal and his gene trees possessed 100% same topology to the combined multi-gene tree (Figs. S1–S5, Table 1).

Maximum likelihood (ML) tree of the species complex of Diaporthe pterocarpi and related species, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. isoberliniae (CPC 22549). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
7.2 Species delimitation based on the GCPSR principle
To assess the species boundaries in the D. pterocarpi complex, the GCPSR principle was applied. Our results did not reveal any conflict between individual phylogenies (Figs. 15, S1–S5). Moreover, a lack of high bootstrap and posterior probability values in several branches of individual gene trees were observed, revealing poor phylogenetic support among the species. The combined gene tree (Fig. 15) is composed of one well-delimited outgroup species (D. isoberliniae), indicating these clades represent different species.
Thus, by implementing the GCPSR principle, based on the comparison of more than one gene genealogy to identify phylogenetic concordance, we verified that D. pterocarpi is a single species rather than a species complex.
7.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences detected significant recombination within the D. pterocarpi species complex (Φw = 7.6 × 10–8), and the network relationship is shown in Fig. 16. Additionally, based on the relative distance of species and structure of the phylogenetic network, all four species in the D. pterocarpi complex should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe pterocarpi species complex and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
7.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. pterocarpi complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a single species instead of diverse taxa. Hence, D. pterocarpi complex should be accepted as a single species (Figs. 15, 16).
7.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. pterocarpi, which was earlier known as four species but herein represent a single species.
Diaporthe pterocarpi (S. Hughes) Udayanga, Xing Z. Liu & K.D. Hyde, Cryptog. Mycol. 33: 305 (2012)
= Diaporthe inconspicua R.R. Gomes, Glienke & Crous, Persoonia 31: 23 (2013)
= Diaporthe lutescens S.T. Huang, J.W. Xia, X.G. Zhang & Z. Li, MycoKeys 77: 81 (2021)
= Diaporthe pseudoinconspicua T.G.L. Oliveira, J.D.P. Bezerra, A.R. Machado, Souza-Motta & O.M.C. Magalh., Persoonia 40: 275 (2018)
Host range: Aloe vera, Jatropha curcas, Ougeinia dalbergioides, Pterocarpus santalinoides, P. angolensis, P. erinaceus, P. indicus, P. violaceus (Udayanga et al. 2012b), Chrysalidocarpus lutescens (Sun et al. 2021), Maytenus ilicifolia (Gomes et al. 2013), Poincianella pyramidalis (Crous et al. 2018)
Known distribution: Brazil, Ghana, India, Sierra Leone, Thailand, Togo, Zambia (Udayanga et al. 2012b), Brazil (Gomes et al. 2013; Crous et al. 2018), China (Sun et al. 2021)
Description: Sexual morph not reported. See Udayanga et al. (2012b) for illustrations and descriptions of asexual morph.
Notes: An ex-epitype of D. pterocarpi was designated by Udayanga et al. (2012b) from Pterocarpus indicus in Thailand. Later, Gomes et al. (2013) introduced D. inconspicua from Maytenus ilicifolia in Brazil. Diaporthe pseudoinconspicua was introduced from Poincianella pyramidalis in Brazil by Crous et al. (2018) while D. lutescens was introduced from Chrysalidocarpus lutescens in China (Sun et al. 2021). All four species in this group claded within the strains of D. pterocarpi in our combined gene phylogenetic tree (Fig. 15) as well as mostly in single gene trees (Figs. S1–S5). This was supported by the GCPSR principle and coalescent methods of PTP and mPTP. A morphological synopsis of conidiomata, conidiogenous cells and conidia characteristics are provided in Table 7. Synonymous names of D. pterocarpi are provided.
7.6 Morphology of the strains belonging to ‘Diaporthe pterocarpi’. See Table 7.
Section Eres
‘Section Eres’ is composed of 18 type strains in the genus Diaporthe (Table 8) and is separated from the other sections by its species arrangement within the phylogenetic analysis (Figs. 1, 2, 3). However, we observed several conflicts of the clade arrangement in the single gene trees (Figs. S1–S5).
In Section Eres, five known species (D. apiculata, D. citrichinensis, D. conica, D. eres and D. irregularis) were isolated from various woody hosts in Guizhou Province. Additionally, synonymous names for four Diaporthe species and five species/species-complexes (D. citrichinensis, D. eres, D. gardeniae, D. subclavata and D. virgiliae) are proposed (Figs. 3, 17).

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the species complexes within Section Eres, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
Species complexes residing in Section Eres
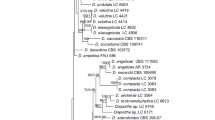
1. Diaporthe citrichinensis
1.1 Phylogenetic analyses and informative characters
The combined gene tree and the individual gene trees (Figs. S1-S5) showed that the isolates residing in this species complex cluster in a clade containing three species, designated here as ‘D. citrichinensis species complex’ (Fig. 3). Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 18). The five loci combined alignment comprises 2165 characters (518 characters from ITS, 328 from tef, 397 from tub, 474 from cal, and 448 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 25 ingroup taxa (four from this study and 23 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. citrichinensis species complex are provided in Table S7. As ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe citrichinensis species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. sennicola (CFCC 51634, CFCC 51635). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
Considering the single gene trees, ITS, tef, cal and his possessed 100% similar topology (Figs. S1-S5) to the combined gene tree (Fig. 3). Only the tub single gene tree is not consistent and retained in two specific groups (Fig. S3). Diaporthe acerigena and D. fraxinicola do not clade within these two groups of tub single gene tree.
1.2 Species delimitation based on the GCPSR principle
The GCPSR principle was applied to assess the boundaries of the ‘D. citrichinensis species complex’. Our results revealed less conflicts between individual phylogenies (Figs. S1–S5), as all the single gene trees possess 100% similarity to the combined gene phylogeny, except the tub single gene tree. The combined gene tree is accordant with a well-delimited outgroup species (D. sennicola). Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. citrichinensis species complex’ by demarcating its species boundaries.
1.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant recombination within the D. citrichinensis species complex (Φw = 0.8), and the network relationships are shown in Fig. 19. Additionally, based on the relative distance of species and structure of the phylogenetic network, all three species in the D. citrichinensis species complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the ‘Diaporthe citrichinensis species complex’ based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
1.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. citrichinensis species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, D. citrichinensis species complex should be accepted as different species (Figs. 18, 19).
1.5 Taxonomy
Species residing in Diaporthe citrichinensis species complex
Diaporthe citrichinensis F. Huang, K.D. Hyde & H.Y. Li, Fungal Divers 61: 247 (2013)
Diaporthe conica C.M. Tian & Qin Yang, MycoKeys 39: 130 (2018)
Diaporthe oraccinii Y.H. Gao, F. Liu & L. Cai, Syst. Biodiv. 14: 111 (2016)
Interestingly, all species in this complex were introduced from China, and most are from Shaanxi Province (Gao et al. 2016; Fan et al. 2018; Yang et al. 2018, 2020, 2021).
Synonymies in Diaporthe citrichinensis species complex
Diaporthe citrichinensis F. Huang, K.D. Hyde & H.Y. Li, Fungal Divers 61: 247 (2013)
= Diaporthe acerigena C.M. Tian & Q. Yang, MycoKeys 39: 118 (2018)
= Diaporthe albosinensis C.M. Tian & Q. Yang, MycoKeys 67: 9 (2020)
= Diaporthe coryli C.M. Tian & Q. Yang, MycoKeys 67: 11 (2020)
= Diaporthe fraxinicola C.M. Tian & Qin Yang, MycoKeys 39: 133 (2018)
= Diaporthe tibetensis C.M. Tian, Qin Yang & X.L. Fan, Mycol. Progr. 17: 847 (2018)
= Diaporthe ukurunduensis C.M. Tian & Qin Yang, MycoKeys 39: 137 (2018)
Host range: Acer sp., A. ukurunduense, Alangium chinense, Betula albosinensis, Camellia sinensis, Corylus mandshurica, Fraxinus chinensis, Juglandis regia, (Gao et al. 2016; Fan et al. 2018; Yang et al. 2018, 2020, 2021).
Known distribution: China (Shaanxi, Zhejiang, Jiangxi, Tibet Autonomous Region) (Gao et al. 2016; Fan et al. 2018; Yang et al. 2018, 2020, 2021).
Notes: All the taxa synonymized under ‘D. citrichinensis’ are reported from China (Gao et al. 2016; Fan et al. 2018; Yang et al. 2018, 2020, 2021).
Diaporthe oraccinii Y.H. Gao, F. Liu & L. Cai, Syst. Biodiv. 14: 111 (2016)
= Diaporthe ganzhouensis C.M. Tian & Q. Yang, MycoKeys 77: 53 (2021)
Host range: Camellia sinensis (Gao et al. 2016), dead wood (Yang et al. 2021)
Known distribution: China (Gao et al. 2016; Yang et al. 2021)
Notes: Diaporthe oraccinii was introduced by Gao et al. (2016) from healthy leaves of Camellia sinensis, in China while D. ganzhouensis was introduced by Yang et al. (2021) from unknown dead wood in China. In the multigene phylogenetic analysis, these two species clustered together and thus D. ganzhouensis is synonymized under D. oraccinii.
2. Diaporthe eres
Diaporthe eres is the type species of the genus and was originally described by Nitschke (1870), from Ulmus sp. in Germany (Udayanga et al. 2014b). Based on morphological characters, several synonyms under D. eres were listed by Wehmeyer (1933) and a few of them were accepted by several authors based on molecular data (Gomes et al. 2013; Udayanga et al. 2014b). Later, there were attempts to define the species limits of D. eres and closely related species based on multi-locus analyses (Guarnaccia et al. 2018; Yang et al. 2018; Guo et al. 2020) as well as employing the GCPSR principle coupled with haplotype network analysis and population genetic diversity (Chaisiri et al. 2021). Hilário et al. (2021b), using GCPSR and the coalescent-based model Poisson Tree Processes (PTPs) evaluated the presence of recombination within the D. eres complex. They identified D. eres complex as a single species, suggesting that D. eres constitutes a population rather than different lineages. Hence, D. alleghaniensis, D. alnea, D. betulae, D. betulina, D. bicincta, D. biguttusis, D. brevicancria, D. camptothecicola, D. celastrina, D. celeris, D. chensiensis, D. cotoneastri, D. ellipicola, D. fukushii, D. helicis, D. henanensis, D. longicicola, D. lonicerae, D. mahothocarpus, D. maritima, D. momicola, D. neilliae, D. nobilis, D. padina, D. phragmitis, D. pulla, D. rosicola, D. vaccinii, D. vacuae, Phomopsis oblonga and Phomopsis velata were recognized as synonyms of D. eres.
We obtained the same results as Hilário et al. (2021b) and mention our observations here as confirmation to their study (Figs. 20, 21). The five loci combined alignment comprises 2243 characters (543 characters from ITS, 348 from tef, 397 from tub, 474 from cal, and 481 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 73 ingroup taxa (22 from this study and 52 taxa retrieved from GenBank).

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe eres’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. shennongjiaensis (CNUCC201905). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the strains belonging to ‘Diaporthe eres’ and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
Several other species (Diaporthe dejiangensis, D. eucommiigena, D. magnoliicola and D. tongrensis), also reside in ‘Diaporthe eres’, but were not included by Hilário et al. (2021b). A morphological synopsis of conidiomata, conidiophores, conidiogenous cells and conidia characteristics are provided in Table 9.
3. Diaporthe gardeniae
3.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates in this species complex cluster in a clade containing five species, designated here as the ‘D. gardeniae species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 22). The five loci combined alignment comprises 2181 characters (512 characters from ITS, 330 from tef, 402 from tub, 486 from cal, and 451 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 15 ingroup taxa (17 taxa were retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. gardeniae species complex are provided in Table S7. All ML, MP and BI analyses resulted in trees that were topologically similar, thus only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe gardeniae species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. irregularis (CGMCC3.20092, GZCC 19–0344). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of tef displayed the most informative sequences for the D. gardeniae species complex, followed by tub, cal and his (Figs. S2–S5, Table 1). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of the D. gardeniae species (Fig. S1).
3.2 Species delimitation based on the GCPSR principle
To assess the species boundaries in the D. gardeniae species complex, the GCPSR principle was applied. Our results revealed conflicts between individual phylogenies, where some species do not clade within the complex or some additional species are claded within the complex. Moreover, a lack of high bootstrap and posterior probability values on both individual and combined trees in several branches were observed, revealing poor phylogenetic support among the species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. gardeniae species complex’ by demarcating its species boundaries.
3.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find significant evidence for recombination within the D. gardeniae species complex (Φw = 0.972), and the network relationships are shown in Fig. 23. Additionally, based on the relative distance of species and structure of the phylogenetic network, all five species in the D. gardeniae complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe gardeniae species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
3.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. gardeniae species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, demonstrating that the complex should be regarded as diverse taxa. Hence, D. gardeniae species complex should be accepted as different species (Figs. 22, 23).
3.5 Taxonomy
Species residing in Diaporthe gardeniae species complex
Diaporthe gardeniae (Buddin & Wakf.) R.R. Gomes, Glienke & Crous, Persoonia 31: 22 (2013)
Diaporthe apiculata Y.H. Gao, F. Liu & L. Cai, Syst. Biodiv. 14: 106 (2016)
Diaporthe azadirachtae Udayanga & Castl., IMA Fungus 7: 291 (2016)
Diaporthe charlesworthii R.G. Shivas, S.M. Thomps. & Y.P. Tan, Persoonia 35: 43 (2015)
Diaporthe sennicola C.M. Tian & Qin Yang, Phytotaxa 302: 150 (2017)
4. Diaporthe subclavata species Complex
4.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this clade contains two species, designated here as ‘D. subclavata species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 24). The five loci combined alignment comprises 2255 characters (527 characters from ITS, 326 from tef, 477 from tub, 442 from cal, and 483 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxa and 13 ingroup taxa (14 taxa were retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. subclavata species complex’ is provided in Table S7. All ML, MP and BI analyses resulted in trees that were topologically similar, thus only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe subclavata species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. shennongjiaensis (CNUCC 201906). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), ITS and tef displayed the most informative sequences for D. subclavata species complex, followed by tub, cal and his loci (Table 1).
4.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. subclavata species complex, the GCPSR principle was applied. Individual gene trees are concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. shennongjiaensis) denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. subclavata species complex’ by demarcating its species boundaries.
4.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant evidence for recombination within the D. subclavata species complex (Φw = 0.65) and the network relationships are shown in Fig. 25. Moreover, based on the comparable remoteness of species and the arrangement of the phylogenetic network, two species in the D. subclavata species complex should be regarded as distinct species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe subclavata species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
4.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. subclavata species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, D. subclavata species complex should be accepted as different species (Figs. 24, 25).
4.5 Taxonomy
Species reside in Diaporthe subclavata species complex
Diaporthe subclavata F. Huang, K.D. Hyde & Hong Y. Li, Fungal Biology 119: 343 (2015).
Diaporthe collariana R.H. Perera & K.D. Hyde, Studies in Fungi 3: 145 (2018).
Synonymies in Diaporthe subclavata species complex
Diaporthe subclavata F. Huang, K.D. Hyde & Hong Y. Li, Fungal Biology 119: 343 (2015).
= Diaporthe heliconiae S.T. Huang, J.W. Xia, X.G. Zhang & Z. Li, MycoKeys 77: 76 (2021).
Hosts: Citrus unshiu (Huang et al. 2015), Heliconia metallica (Sun et al. 2021).
Known distribution: China (Huang et al. 2015; Sun et al. 2021).
Notes: Diaporthe subclavata was introduced from a leaf of Citrus unshiu with citrus scab in China (Huang et al. 2015), while D. heliconiae was identified from a symptomatic petiole of Heliconia metallica in China (Sun et al. 2021). Although Sun et al. (2021) stated that D. heliconiae can be distinguished from its phylogenetically closely related species D. subclavata in having smaller alpha conidia, we observed these two species are morphologically and phylogenetically indistinguishable. Hence, here we synonymized D. heliconiae under D. subclavata.
Diaporthe collariana R.H. Perera & K.D. Hyde, Studies in Fungi 3: 145 (2018)
= Diaporthe litchii S.T. Huang, J.W. Xia, X.G. Zhang & Z. Li, MycoKeys 77: 80 (2021)
Hosts: Litchi chinensis (Sun et al. 2021), Magnolia champaca (Perera et al. 2018)
Known distribution: China (Sun et al. 2021), Thailand (Perera et al. 2018)
Notes: Diaporthe collariana was introduced on Magnolia champaca in Thailand (Perera et al. 2018). Later, Sun et al. (2021) introduced D. litchi on infected leaves of Litchi chinensis in China (Sun et al. 2021). According to the phylogenetic analysis, we observed both species cluster together and we herein synonymize D. litchi under D. collariana.
5. Diaporthe virgiliae species complex
5.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates used in this species complex cluster in a clade containing five species, designated here as the D. virgiliae species complex. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 26). The five loci combined alignment comprises 2114 characters (508 characters from ITS, 325 from tef, 397 from tub, 466 from cal, and 418 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 11 ingroup taxa (12 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. virgiliae species complex are provided in Table S7. All ML, MP and BI analyses resulted in trees that were topologically similar, thus only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe virgiliae species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. shennongjiaensis (CNUCC201905). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of tub displayed the most informative sequences for the D. virgiliae species complex, followed by tef, his and cal (Figs. S2–S5, Table 1). ITS presented the lowest percentage, indicating that this locus is unreliable for delimitation of the D. virgiliae species complex (Fig. S1).
5.2 Species delimitation based on the GCPSR principle
The GCPSR principle was applied to assess the species boundaries in the D. virgiliae species complex. Our results revealed less conflict between individual phylogenies. It is also evident that isolates of the complex cluster in different clades in the same individual gene tree. For instance, isolates of D. virgiliae are phylogenetically distant in the ITS gene tree (Fig. S1) as it formed two clades. Moreover, a lack of high bootstrap and posterior probability values on both individual and combined gene trees in several branches were observed, revealing poor phylogenetic support among the species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. virgiliae species complex’ by demarcating its species boundaries.
5.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find significant recombination within the D. virgiliae species complex (Φw = 0.338), and the network relationships are shown in Fig. 27. Additionally, based on the relative distance of species and structure of the phylogenetic network, all species in the D. virgiliae complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe virgiliae species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
5.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. virgiliae species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. virgiliae species complex should be accepted as a different species (Figs. 26, 27).
5.5 Taxonomy
Species residing in Diaporthe virgiliae species complex
Diaporthe grandiflori S.T. Huang, J.W. Xia, X.G. Zhang & Z. Li, MycoKeys 77: 75 (2021)
Diaporthe heterophyllae Guarnaccia & Crous, Stud. Mycol. 92: 59 (2018)
Diaporthe penetriteum Y.H. Gao, F. Liu & L. Cai, Syst. Biodiv. 14: 113 (2016)
Diaporthe virgiliae Maching., Dreyer & Roets, Pl. Path. 64: 1153 (2015)
Diaporthe zaofenghuang X.H. Wang & G.P. Wang, MycoKeys 80: 84 (2021)
Section Sojae
‘Section Sojae’ is composed of 84 type strains in the genus Diaporthe (Table 10) and is separated from the other proposed sections by its species arrangement within the phylogenetic analysis (Figs. 1, 2, 3). However, we observed several conflicts of the clade arrangement in single gene trees (Figs. S1–S5). In the ITS phylogeny (Fig. S1), the strains of D. brasiliensis, D. caatingaensis and D. paranensis claded outside Section Sojae. In the tef single gene tree (Fig. S2) an unusual clade arrangement of Section Sojae was observed. The strains of D. brasiliensis and D. caatingaensis claded outside Section Sojae in cal phylogeny (Fig. S4). We did not observe any conflict of Section Sojae, within the tub and his single gene trees (Figs. S3, S5).
In Section Sojae, we provide descriptions and illustrations of one novel species (D. submersa), and six known species (D. cinnamomi, D. discoidispora, D. guttulata, D. minusculata, D. passiflorae and D. sojae) isolated from various woody hosts in Guizhou Province. Additionally, synonymous names for 14 Diaporthe species and nine species/species-complexes (D. arctii, D. ganjae, D. leucospermi, D. longicolla, D. schini, D. sclerotioides, D. siamensis, D. sojae and D. tulliensis) are proposed (Figs. 3, 28).

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the species complexes of Section Sojae, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
Diaporthe submersa Y.Y. Chen, A.J. Dissanayake & Jian K. Liu sp. nov.
Index Fungorum number: IF559781; Facesoffungi number: FoF12617, Fig. 29

Diaporthe submersa (GZAAS 22-0008, holotype). a, b Ascomata on host surface. c Section of ascomata. d–g Asci. h Ascospores. i, j 10-days-old culture on PDA from above and below. Scale bars: c, d = 100 μm; e–g = 50 μm; h = 15 μm
Etymology: Refers to the submerged ascomata in the host.
Holotype: GZAAS 22-0008.
Saprobic on decaying wood. Sexual morph Ascomata 225–350 μm diam., black, globose to subglobose, clustered and scattered on dead twigs, submerged in host tissue, protruding through substrata. Asci (55–)63–70(–75) × (9–)10–18(–19) μm, 8-spored, unitunicate, sessile, elongate to clavate. Ascospores (11–)12–14(–15) × (3–)3.5–4(–5) μm, hyaline, 2-celled, often tetra-guttulate, with larger guttules at centre and smaller at ends, elongated to clavate. Asexual morph Not observed.
Culture characteristics: Colonies covering 9 cm diam. PDA Petri dishes after 10 d at 25 °C, with moderate aerial mycelium. Surface ochreous and reverse pale luteous.
Material examined: China, Guizhou Province, Chishui City, saprobic on decaying woody host, May 2019, Y.Y. Chen (GZAAS 19-1848, holotype); ex-type culture CGMCC3.24297 = GZCC 19–0129; ibid, Guiyang District, saprobic on decaying woody host, March 2020, Y.Y. Chen (GZAAS 22-0008), living culture GZCC 22-0007.
Notes: The phylogenetic results showed that two isolates of Diaporthe submersa clustered close to D. caryae forming a distinct lineage (Fig. 3) with maximum support (ML/MP/BI = 100/100/1.0). Diaporthe submersa can be distinguished from D. caryae in the concatenated alignment differing by 3/512 bp in ITS, 8/413 bp in tef, 12/453 bp in tub, 11/506 bp in cal and 12/515 bp in his. Morphologically, D. submersa cannot be compared with D. caryae as it does not have an asexual morph (Yang et al. 2018).
Synonymies in Section Sojae
Diaporthe amaranthophila (Inácio, Dianese & Carlos) Rossman & Udayanga, IMA Fung 6: 150 (2015)
= Diaporthe sinensis X.X. Feng & C.L. Zhang, Phytotaxa 425: 264 (2019)
Host range: Amaranthus tricolor (Rosskopf et al. 2000; Feng et al. 2019)
Known distribution: China (Feng et al. 2019), Japan (Rosskopf et al. 2000)
Notes: Rosskopf et al. (2000) introduced Phomopsis amaranthicola from Amaranthus tricolor from Japan. Later, Rossman et al. (2015) named this species as Diaporthe amaranthophila comb. nov. Feng et al. (2019) introduced D. sinensis from Amaranthus sp. in China. In our multigene phylogenetic tree both species clustered in the same clade (Fig. 3). A pairwise comparison of nucleotides showed that D. amaranthophila and D. sinensis are similar, since both species differ in 4 bp in his and 0 bp in ITS, tef, tub. As D. sinensis had no cal sequences we were unable compare it with that of D. amaranthophila.
Diaporthe raonikayaporum R.R. Gomes, Glienke & Crous, Persoonia 31: 31 (2013)
= Diaporthe neoraonikayaporum Doilom, Dissan. & K.D. Hyde, Fungal Diversity 54: (2016)
Host range: Spondias mombin (Gomes et al. 2013), Tectona grandis (Doilom et al. 2016)
Known distribution: Brazil (Gomes et al. 2013), Thailand (Doilom et al. 2016)
Notes: Gomes et al. (2013) introduced D. raonikayaporum from a leaf of Spondias mombin in Brazil. Later, D. neoraonikayaporum was introduced by Doilom et al. (2016) on dieback lesion of Tectona grandis branches in Thailand. These two species are morphologically indistinguishable with overlapping micromorphological characters of alpha conidia (6–8 × 2–3 μm vs 5–8 × 2–3 μm). A pairwise comparison of nucleotides showed that D. raonikayaporum and D. neoraonikayaporum are similar, differing in 7 bp in ITS, 4 bp in tef, 3 bp in tub and 3 bp in cal. As D. neoraonikayaporum has no his sequences we were unable to compare it with that of D. raonikayaporum.
Diaporthe passiflorae Crous & L. Lombard, Persoonia 28: 149 (2012)
= Diaporthe malorum L. Santos & A. Alves, Mycosphere 8: 494 (2017)
= Diaporthe eucommiigena S.Y. Wang, Yong Wang bis & Y. Li, Journal of Fungi 8: 1301 (2022)
Host range: Eucommia ulmoides (Wang et al. 2022), Malus domestica (Santos et al. 2017), Passiflora edulis (Crous et al. 2012)
Known distribution: China (Wang et al. 2022), Portugal (Santos et al. 2017), South America (Crous et al. 2012)
Notes: Diaporthe passiflorae was introduced by Crous et al. (2012) on fruits of Passiflora edulis in South America. Diaporthe malorum was introduced by Santos et al. (2017) from Malus domestica fruit with rot symptoms in Portugal. Diaporthe eucommiigena was introduced by Wang et al. (2022) on dead wood of Eucommia ulmoides in China. These three species are morphologically indistinguishable with overlapping micromorphological characters of alpha conidia (5–8 × 2–3.5 μm vs 5–7.5 × 1.5–3.2 μm vs 5.5–8 × 1.5–3 μm). A pairwise comparison of nucleotides showed that D. passiflorae and D. malorum are similar, differing in 3 bp in ITS and 10 bp in his. As D. passiflorae had no tef, tub and cal sequences we were unable compare it with that of D. malorum. A pairwise comparison of D. malorum and D. eucommiigena are similar since both species differ in 10 bp in ITS. As D. eucommiigena has no tef and tub sequences while D. malorum has no his sequences we were unable compare them. Neither species has cal sequences.
Species complexes residing in Section Sojae
1. Diaporthe arctii species complex
1.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates used in this species complex cluster in a clade containing 16 species, designated here as the ‘D. arctii species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 30). The five loci combined alignment comprises 2721 characters (579 characters from ITS, 383 from tef, 771 from tub, 484 from cal, and 504 from his, including alignment gaps), while the analyses included one well-delimited outgroup taxon and 70 ingroup taxa (one from this study and 70 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. arctii species complex are provided in Table S8. All ML, MP and BI analyses resulted in trees that were topologically similar, thus only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe arctii species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. arezzoensis (MFLU 19–2880). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of tef and cal displayed the most informative sequences for the D. arctii species complex, followed by tub and his loci (Figs. S2–S5). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of the D. arctii species complex (Fig. S1).
1.2 Species delimitation based on the GCPSR principle
To assess the species boundaries of the species residing in the D. arctii complex, the GCPSR principle was applied. Our results revealed conflicts between individual phylogenies, where some species do not clade within the complex or some additional species are claded within the complex. For example, D. acericola and D. schoeni did not clade within the complex of tub gene tree and two specific groups of the complex were observed in ITS single gene tree. To assess the boundary of the D. arctii species complex, the GCPSR principle was applied. Individual gene trees are concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. arezzoensis) denoting no conflicts among the individual gene trees (except the ITS single gene tree) and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. arctii species complex’ by demarcating its species boundaries.
1.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find significant recombination within the D. arctii species complex (Φw = 0.79), and the network relationships are shown in Fig. 31. Additionally, based on the relative distance of species and structure of the phylogenetic network, all 16 species in the D. arctii complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe arctii species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
1.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. arctii species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. arctii species complex should be accepted as a distinct species (Figs. 30, 31).
1.5 Taxonomy.
Species residing in Diaporthe arctii species complex
Diaporthe arctii (Lasch) Nitschke, Pyrenomyc. Germ. 2: 268 (1870)
Diaporthe angelicae (Berk.) Wehm., Monogr. & Segreg., Stud., Sci. Ser. 9: 204 (1933)
Diaporthe cichorii Dissan., Camporesi & K.D. Hyde, Mycosphere 8: 864 (2017)
Diaporthe cucurbitae (McKeen) Udayanga & Castl., Fungal Biology 119: 395 (2014)
Diaporthe cuppatea (E. Jansen, Lampr. & Crous) Udayang, Crous & Hyde, Fungal Divers 56: 166 (2012)
Diaporthe gulyae R.G. Shivas, S.M. Thomps. & A.J. Young, Persoonia 27: 85 (2011)
Diaporthe guttulata Y.Y Chen, Dissanayake & Jian K. Liu, Journal of Fungi 6: 19 (2020)
Diaporthe lusitanicae A.J.L. Phillips & J.M. Santos, Fungal Diversity 34: 118 (2009)
Diaporthe monetii Gomzhina, Persoonia 114: 569 (2022)
Diaporthe neoarctii R.R. Gomes, Glienke & Crous, Persoonia 31: 25 (2013)
Diaporthe novem J.M. Santos, Vrandečić & A.J.L. Phillips, Persoonia 27: 14 (2011)
Diaporthe phaseolorum (Cooke & Ellis) Sacc., Syll. fung. (Abellini) 1: 692 (1882)
Diaporthe schoeni Dissan., Camporesi & K.D. Hyde, Mycosphere 8: 870 (2017)
Diaporthe stewartii A.L. Harrison, Mycologia 27: 525 (1935)
Diaporthe subordinaria (Desm.) R.R. Gomes, Glienke & Crous, Persoonia 31: 34 (2013)
Diaporthe vangoghii Gomzhina, Persoonia 114: 565 (2022)
Synonymies in Diaporthe arctii species complex
Diaporthe novem J.M. Santos, Vrandečić & A.J.L. Phillips, Persoonia 27: 14 (2011)
= Diaporthe pseudolongicolla K. Petrović, L. Riccioni & M. Vidić, Ratarstvo i povrtarstvo 55: 106 (2018)
Notes: See Petrović et al. (2018) for the synonymy.
Diaporthe schoeni Dissan., Camporesi & K.D. Hyde, Mycosphere 8: 870 (2017)
= Diaporthe acericola Dissan., Camporesi & K.D. Hyde, Mycosphere 8: 864 (2017)
Host range: Acer negundo, Schoenus nigricans (Dissanayake et al. 2017a)
Known distribution: Italy (Dissanayake et al. 2017a)
Notes: Both D. acericola and D. schoeni were introduced by Dissanayake et al. (2017a) and D. acericola forms a sister clade to D. schoeni. As the two species differed by 62 nucleotides in the concatenated alignment, Dissanayake et al. (2017a) introduced them as two different novel species. However, we observed that D. acericola cannot be differentiated from D. schoeni by their morphology and they always cluster close in the phylogenetic analysis (Figs. 30, 31). Hence, we synonymize D. acericola under D. schoeni.
2. Diaporthe ganjae
2.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the species in this complex cluster in a clade containing five species, designated here as ‘D. ganjae’. Based on these details, another phylogenetic tree including more isolates of this species was constructed (Fig. 32). The five loci combined alignment comprises 2151 characters (528 characters from ITS, 344 from tef, 405 from tub, 410 from cal, and 464 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 11 ingroup taxa (13 taxa retrieved from GenBank). Alignment details and results of MP, ML and BI analyses of D. ganjae are provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe ganjae’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. sclerotioides (CBS 296.67, CBS 710.76). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, all single gene phylogenetic analyses (ITS, tef, tub, cal and his) displayed 100% similarity to the combined gene phylogeny for the D. ganjae species complex (Figs. 3, S1–S5, Table 1).
2.2 Species delimitation based on the GCPSR principle
To assess the species boundaries of the species residing in the D. ganjae complex, the GCPSR principle was applied. To disclose concordant branches, the individual gene trees were compared. The results revealed concordance in the individual phylogenies (Figs. S1–S5, Table 1). Diaporthe compacta had no cal sequences (Fig. S4), while D. elonis composed only ITS sequences.
However, individual gene trees are concordant and have the same topology for the well-delimited outgroup taxon D. sclerotioides denoting no conflicts among the individual gene trees. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the D. ganjae complex and it is herein regarded as a single species, demarcating its species boundaries.
2.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences detected significant recombination within the D. ganjae complex (Φw = 0.033) and the network relationships are shown in Fig. 33. Additionally, based on the relative distance of species and structure of the phylogenetic network, all five species in the D. ganjae complex should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe ganjae species complex and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
2.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. ganjae complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a single species instead of diverse taxa. Hence, each taxon presently recognized in the D. ganjae complex should be accepted as a single species.
2.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are present to delimit the species boundaries of D. ganjae, which was earlier known as five species but herein represent a single species.
Diaporthe ganjae (McPartl.) R.R. Gomes, Glienke & Crous, Persoonia 31: 22 (2013)
= Diaporthe compacta Y.H. Gao, F. Liu & L. Cai, Syst. Biodiv. 14: 110 (2016)
= Diaporthe elonis H. Liang
= Diaporthe manihotia Punith., Kavaka 3: 29 (1976) [1975]
= Diaporthe sambucusii C.M. Tian & Qin Yang, Phytotaxa 336: 164 (2018)
Host range: Camellia sinensis (Gao et al. 2016), Cannabis sativa (Gomes et al. 2013), Manihot utilissima (Gomes et al. 2013), Sambucus williamsii (Yang et al. 2018)
Known distribution: China (Gao et al. 2016; Yang et al. 2018), Rwanda (Gomes et al. 2013), USA (Gomes et al. 2013)
Description: See Gomes et al. (2013) for illustrations and descriptions of asexual morph and Abeywickrama et al. (2020) for sexual morph.
Notes: Diaporthe ganjae was introduced by Gomes et al. (2013) from a dead leaf of Cannabis sativa in USA. Later, D. manihotia was introduced from Manihot utilissima in Rwanda (Gomes et al. 2013), D. compacta was introduced from Camellia sinensis in China (Gao et al. 2016), D. elonis was identified from Prunella vulgaris in China, while D. sambucusii was introduced from Sambucus williamsii in China (Yang et al. 2018). Here we show that D. compacta, D. elonis, D. ganjae, D. manihotia and D. sambucusii cluster in a well-supported clade and are closely related (Figs. 32, 33) supported by the GCPSR principle and coalescent methods of PTP and mPTP, the above species are morphologically indistinguishable (Table 11). Synonymous names of D. ganjae are provided.
2.6 Morphology of the strains belonging to ‘Diaporthe ganjae. See Table 11.
3. Diaporthe leucospermispecies complex
3.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this species complex clusters in a clade containing 15 species, designated here as the ‘D. leucospermi species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 34). The five loci combined alignment comprises 2187 characters (532 characters from ITS, 331 from tef, 403 from tub, 482 from cal, and 439 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 37 ingroup taxa (two from this study and 37 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. leucospermi species complex are provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe leucospermi species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. Synonymies are mentioned within brackets. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. rosiphthora (COAD 2913, COAD 2914). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, all single gene phylogenetic analyses (ITS, tef, tub, cal, his) displayed 100% similarity to the D. leucospermi species complex (Table 1). Though cal and his single gene trees formed a similar clade arrangement as of the combined gene phylogeny, sequences were unavailable in GenBank for most of the species.
3.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. leucospermi species complex, the GCPSR principle was applied. Though D. rosiphthora is unusually claded in ITS single gene tree, individual gene trees are concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. rosiphthora) denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. leucospermi species complex’ by demarcating its species boundaries.
3.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant evidence for recombination within the D. leucospermi species complex (Φw = 0.35) and the network relationships are shown in Fig. 35. Additionally, based on the relative distance of species and structure of the phylogenetic network, all 15 species in the D. leucospermi species complex should be regarded as distinct species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe leucospermi species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
3.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. leucospermi species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed the boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. leucospermi species complex should be accepted as different species (Figs. 34, 35).
3.5 Taxonomy
Species residing in Diaporthe leucospermi species complex
Diaporthe leucospermi Crous & Summerell, Persoonia 27: 32 (2011)
Diaporthe acaciarum Crous & M.J. Wingf., Persoonia 33: 243 (2014)
Diaporthe beilharziae R.G. Shivas, Jacq. Edw & Y.P. Tan, Fungal Divers 61: 254 (2013)
Diaporthe caryae C.M. Tian & Qin Yang, MycoKeys 39: 124 (2018)
Diaporthe chimonanthi (Chang, Xiang, Chi) Y.H. Gao, L. Cai, IMA Fungus 8: 183 (2017)
Diaporthe infecunda R.R. Gomes, Glienke & Crous, Persoonia 31: 24 (2013)
Diaporthe juglandigena S.Y. Wang, Yong Wang, Y. Li, Journal of Fungi 8: 1301 (2022)
Diaporthe machili S.T. Huang, J.W. Xia, W.X. Sun, X.G. Zhang, Mycokeys 78: 63 (2021) Diaporthe middletonii Shivas, L. Morin, S.M. Thomps., Tan, Persoonia 35: 45 (2015)
Diaporthe myracrodruonis A.P.S.L. Pádua, T.G.L. Oliveira, Souza-Motta, X.L. Fan & J.D.P. Bezerra, Acta Bot. Brasil. 33: 171 (2019)
Diaporthe orixae Q.T. Lu & Z. Zhang, Phytotaxa 544: 45 (2022)
Diaporthe pachirae Milagres, Belisário, Pinho, Furtado, Trop Plant Pathol 43: 460 (2018)
Diaporthe sackstonii R.G. Shivas, S.M. Thomps. & Y.P. Tan, Persoonia 35: 46 (2015)
Diaporthe serafiniae R.G. Shivas, S.M. Thomps. & Y.P. Tan, Persoonia 35: 46 (2015)
Diaporthe submersa Y.Y. Chen, A.J. Dissanayake & J. K. Liu (this study)
Synonymies in Diaporthe leucospermi species complex
Diaporthe leucospermi Crous & Summerell, Persoonia 27: 32 (2011)
= Diaporthe pyracanthae L. Santos & A. Alves, Mycosphere 8: 493 (2017)
= Diaporthe rossmaniae S. Hilário, I. Amaral, L. Santos & A. Alves, Mycologia 112: 302 (2020)
Hilário et al. (2021c) synonymized D. pyracanthae and D. rossmaniae under D. leucospermi and we obtained the same results (Figs. 34, 35). Hence, we follow and accept the synonymies of D. leucospermi.
Diaporthe chimonanthi (C.Q. Chang, M.M. Xiang & P.K. Ch) Y.H. Gao & L. Cai, IMA Fungus 8: 183 (2017)
= Diaporthe michelina Y.H. Gao & L. Cai IMA Fungus 8: 184 (2017)
Hosts: Chimonanthus praecox, Michelia alba (Gao et al. 2017)
Known distribution: China (Gao et al. 2017)
Notes: Diaporthe chimonanthi and D. michelina were introduced as new combinations by Gao et al. (2017) on living branches of Chimonanthus praecox and on living branches of Michelia alba from China, respectively. However, according to the multi-gene phylogenetic analysis, we observed these two species clustering together.
4. Diaporthe longicolla species complex
4.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates in this species complex clustered in a clade containing nine species, designated here as the ‘D. longicolla species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 36). The five loci combined alignment comprises 2235 characters (535 characters from ITS, 325 from tef, 415 from tub, 487 from cal, and 473 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 35 ingroup taxa (37 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. longicolla species complex are provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe longicolla species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. convolvuli (CBS 124654, FAU 649). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of tef, cal and his displayed most informative sequences followed by ITS and tub for the D. longicolla species complex (Figs. S1–S5).
4.2 Species delimitation based on the GCPSR principle
To assess the boundaries of the species residing in D. longicolla species complex, the GCPSR principle was applied. Our results revealed conflicts between individual phylogenies, where some species do not clade within the complex. For example, D. thunbergiicola does not clade within the complex in the ITS gene tree (Fig. S1) whereas D. guangdongensis and D. melonis are unusually claded in ITS single gene tree. Thus, by implementing the GCPSR principle, based on the comparison of more than one gene genealogy to identify phylogenetic concordance, we verified that D. longicolla is a species complex.
4.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find significant recombination within the D. longicolla species complex (Φw = 0.61) and the network relationships are shown in Fig. 37. Additionally, based on the relative distance of species and structure of the phylogenetic network, all nine species in the D. longicolla species complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe longicolla species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
4.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. longicolla species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. longicolla species complex should be accepted as a different species (Figs. 36, 37).
4.5 Taxonomy
Species residing in Diaporthe longicolla species complex
Diaporthe longicolla (Hobbs) Santos, Vrandečić & A.J.L. Phillips, Persoonia 27: 13 (2011)
Diaporthe breyniae Y. Marín & C. Lamb., MycoKeys 90: 96 (2022)
Diaporthe morindendophytica M. Luo, W. Guo, Manawas., M.P. Zhao, K.D. Hyde & C.P. You, J of Fungi 8: 806 (2022)
Diaporthe rosae Samarak. & K.D. Hyde, Fungal Diversity 89: 185 (2018)
Diaporthe tectonendophytica Doilom, Dissan. & K.D. Hyde, Fungal Diversity: 54: (2016)
Diaporthe thunbergiicola Udayanga & K.D. Hyde, Fungal Diversity: (2015)
Diaporthe ueckerae Udayanga & Castl. Fungal Biology 119: 401 (2014)
Diaporthe unshiuensis F. Huang, K.D. Hyde & Hong Y. Li, Fungal Biology 119: 344 (2015)
Diaporthe vochysiae S.A. Noriler, R.R. Gomes & C. Glienke, Fitoterapia 138: 273 (2018)
Synonymies in Diaporthe longicolla species complex
Diaporthe thunbergiicola Udayanga & K.D. Hyde, Fungal Diversity: (2015)
= Diaporthe absenteum Gao, Liu, Cai, Systematics and Biodiversity 14: 107 (2016)
= Diaporthe durionigena L.D. Thao, L.T. Hien, N.V. Liem, H.M. Thanh & T.N. Khanh, Fungal Systematics and Evolution 7: 288 (2021)
= Diaporthe passifloricola Crous & M.J. Wingf., Persoonia 36: 395 (2016)
Hosts: Camellia sinensis (Gao et al. 2016), Durio zibethinus (Crous et al. 2020), Passiflora foetida (Crous et al. 2016), Thunbergia laurifolia (Liu et al. 2015)
Known distribution: China (Gao et al. 2016), Malaysia (Crous et al. 2016), Thailand (Liu et al. 2015), Vietnam (Crous et al. 2020)
Notes: Diaporthe thunbergiicola was described from Thunbergia laurifolia from Thailand (Liu et al. 2015). Later, D. absenteum was introduced from Camellia sinensis in China (Gao et al. 2016), while D. passifloricola was introduced from Passiflora foetida in Malaysia. Diaporthe durionigena was introduced from Durio zibethinus in Malaysia (Crous et al. 2016). These authors have shown that despite the similarities of conidial dimensions between these four species, they differ in several nucleotide positions. However, we observed that these four species are phylogenetically indistinguishable (Fig. 36).
Diaporthe ueckerae Udayanga & Castl. Fungal Biology 119: 401 (2014)
= Diaporthe miriciae R.G. Shivas, S.M. Thomps. & Y.P. Tan, Persoonia 35: 46 (2015)
Hosts: Cucumis melo (Udayanga et al. 2015), Glycine max (Thompson et al. 2015)
Known distribution: Australia (Thompson et al. 2015), USA (Udayanga et al. 2015)
Notes: Udayanga et al. (2015) introduced D. ueckerae based on isolates from Cucumis melo in Oklahoma, USA while D. miriciae was recorded from Glycine max by Thompson et al. (2015). According to the multi-gene phylogenetic analysis these two species were phylogenetically indistinguishable.
Diaporthe unshiuensis F. Huang, K.D. Hyde, Hong Y. Li, Fungal Biology 119: 344 (2015)
= Diaporthe megabiguttulata M. Luo, W. Guo, Manawas., M.P. Zhao, K.D. Hyde & C.P. You, Journal of Fungi 8: 806 (2022)
Hosts: Citrus unshiu (Huang et al. 2015), Morinda officinalis (Luo et al. 2022)
Known distribution: China (Huang et al. 2015; Luo et al. 2022)
Notes: Diaporthe unshiuensis was introduced from Citrus unshiu in China (Huang et al. 2015), while D. megabiguttulata was isolated from a healthy stem of Morinda officinalis in China (Luo et al. 2022). These authors have shown that despite the similarities of conidial dimensions between these two species, they differ in several nucleotide positions. However, we observed that these two species are phylogenetically indistinguishable (Fig. 36).
5. Diaporthe schini species complex
5.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this species complex contains five species, designated here as the ‘D. schini species complex’. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 38). The five loci combined alignment comprises 2283 characters (540 characters from ITS, 348 from tef, 402 from tub, 505 from cal, and 488 from his, including alignment gaps), while the analyses included three well-delimited outgroup taxa and 11 ingroup taxa (14 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. schini species complex are provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe schini species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 60% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. hordei (CBS 481.92). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of tef, tub and cal displayed the most informative sequences followed by his for the D. schini species complex (Figs. S2–S5). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of the taxa in D. schini species complex (Fig. S1).
5.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. schini species complex, the GCPSR principle was applied. Individual gene trees are concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. hordei) denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. schini species complex’ by demarcating its species boundaries.
5.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find significant recombination within the D. schini species complex (Φw = 0.393) and the network relationships are shown in Fig. 39. Additionally, based on the relative distance of species and structure of the phylogenetic network, all five species in the D. schini complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe schini species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
5.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. schini species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed the boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. schini species complex should be accepted as different species (Figs. 38, 39).
5.5 Taxonomy
Species residing in Diaporthe schini species complex
Diaporthe schini R.R. Gomes, Glienke & Crous, Persoonia 31: 32 (2013)
Diaporthe racemosae Guarnaccia & Crous, Stud. Mycol. 92: 64 (2018)
Diaporthe rosiphthora Pereira,Ferreira & Barreto, Crop Protection 139: 6 (2020)
Diaporthe tecomae Sacc. & P. Syd., Syll. fung. (Abellini) 14: 550 (1899)
Diaporthe terebinthifolii R.R. Gomes, Glienke & Crous, Persoonia 31: 35 (2013)
6.Diaporthe sclerotioides
6.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this clade contains three species, designated here as ‘D. sclerotioides species complex’. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 40). The five loci combined alignment comprises 2740 characters (613 characters from ITS, 369 from tef, 815 from tub, 470 from cal, and 473 from his, including alignment gaps), while the analyses included five well-delimited outgroup taxa and 15 ingroup taxa (20 taxa retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. sclerotioides species complex’ is provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe sclerotioides species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. oxe (CBS 133186). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), tef displayed the most informative sequences for D. sclerotioides species complex (Table 1).
6.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. sclerotioides species complex, the GCPSR principle was applied. Individual gene trees are concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. oxe) denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. sclerotioides species complex’ by demarcating its species boundaries.
6.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant evidence for recombination within the D. sclerotioides species complex (Φw = 0.985) and the network relationships are shown in Fig. 41. Moreover, based on the comparable remoteness of species and the arrangement of the phylogenetic network, all three species in the D. sclerotioides species complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe sclerotioides species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
6.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. sclerotioides species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed the boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. sclerotioides species complex should be accepted as different species (Figs. 40, 41).
6.5 Taxonomy
Species residing in Diaporthe sclerotioides species complex
Diaporthe sclerotioides (Kest) Udayanga, Crous & K.D. Hyde, Fungal Divers 56: 166 (2012)
Diaporthe cyatheae Fu, Hsieh, Chen, Chang, Huang, Ju, Mycologia 105:861 (2013)
Diaporthe longispora (Wehm.) R.R. Gomes, Glienke & Crous, Persoonia 31: 24 (2013)
Synonymies in Diaporthe sclerotioides species complex
Diaporthe sclerotioides (Kest) Udayanga, Crous & K.D. Hyde, Fungal Divers 56: 166 (2012)
= Diaporthe columnaris (D.F. Farr & Castl.) Udayanga & Castl., IMA Fungus 7: 291 (2016).
Hosts: Cucumis sativus (Udayanga et al. 2012b), Vaccinium vitis-idaea (Rossman et al. 2016)
Known distribution: Netherlands (Udayanga et al. 2012b), USA (Rossman et al. 2016)
Notes: The new combination Diaporthe sclerotioides was introduced for a species on roots of Cucumis sativus in Netherlands (Udayanga et al. 2012b). Later, Rossman et al. (2016) introduced D. columnaris from Vaccinium vitis-idaea in USA. According to the phylogenetic analysis and PTP results D. columnaris is synonymized under D. sclerotioides (Figs. 40, 41).
7. Diaporthe siamensis species complex
7.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates in this species complex cluster in a clade containing 11 species, designated here as the ‘D. siamensis species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 42). The five loci combined alignment comprises 2221 characters (530 characters from ITS, 340 from tef, 408 from tub, 480 from cal, and 463 from his, including alignment gaps), while the analyses included three well-delimited outgroup taxa and 38 ingroup taxa (three from this study and 38 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. siamensis species complex are provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the Diaporthe siamensis species complex, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. tarchonanthi (CPC 37479). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, all single gene trees (ITS, tef, tub, cal, his) displayed the most informative sequences for the D. siamensis species complex (Figs. S1–S5). However, most of the species had no sequences in cal and his gene regions (Table 1).
7.2 Species delimitation based on the GCPSR principle
Our results revealed several conflicts between individual phylogenies, where some species do not clade within the complex or some additional species are claded within the complex. For example, D. ueckerae (D. thunbergiicola MFLUCC 12-0033) is unusually claded within the complex in ITS single gene tree (Fig. S1). Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. siamensis species complex’ by demarcating its species boundaries.
7.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find significant recombination within the D. siamensis species complex (Φw = 0.99) and the network relationships are shown in Fig. 43. Additionally, based on the relative distance of species and structure of the phylogenetic network, all 11 species in the D. siamensis complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe siamensis species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
7.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. siamensis species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed the boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. siamensis species complex should be accepted as a different species (Figs. 42, 43).
7.5 Taxonomy
Species residing in Diaporthe siamensis species complex
Diaporthe siamensis Udayanga, Xing Z. Liu & K.D. Hyde, Crypto. Mycol. 33: 298 (2012)
Diaporthe biguttulata F. Huang, K.D. Hyde & Hong Y. Li, Fungal Biol. 119: 338 (2015)
Diaporthe chiangraiensis (Senan & Hyde) Norph., (2022)
Diaporthe cinnamomi C.M. Tian & Qin Yang, MycoKeys 39: 127 (2018)
Diaporthe citriasiana F. Huang, K.D. Hyde & Hong Y. Li, Fungal Divers 61: 246 (2013)
Diaporthe discoidispora F. Huang, K.D. Hyde & Hong Y. Li, Fungal Biol 119: 341 (2015)
Diaporthe eleutherrhenae Song, Landrein (2022)
Diaporthe fici-septicae Tennakoon, C.H. Kuo & K.D. Hyde, Fungal Diversity: (2021)
Diaporthe kyushuensis Kajitani & Kanem., Mycoscience 41:111 (2000)
Diaporthe quercicola Qin Yang, MycoKeys 91: 40 (2022)
Diaporthe yunnanensis Y.H. Gao & L. Cai, IMA Fungus 8: 180 (2017)
8. Diaporthe sojae
The ‘D. sojae complex’ was comprehensively re-assessed by Udayanga et al. (2015), with special reference to the re-definition and epitypification of this prevalent pathogenic species. A phylogenetic tree inferred with a selection of ex-type isolates available in the genus was used by Udayanga et al. (2015) to illustrate the limits of the D. sojae complex. They defined the D. sojae complex which was originally described from soybean, cucurbits, sunflower, and other herbaceous field crops. However, with the introduction of more novel species within this complex during the past few years, we observed an expansion and a complexity within the taxa, which were earlier assigned to D. sojae complex. Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. sojae, which was earlier known as 10 different species but herein represents a single species.
8.1 Phylogenetic analyses and informative characters
The multi-gene phylogenetic tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates residing in this clade comprise 11 species, designated here as ‘D. sojae’. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 44). The five loci combined alignment comprises 2227 characters (539 characters from ITS, 318 from tef, 420 from tub, 477 from cal, and 473 from his, including alignment gaps), while the analyses included 10 well-delimited outgroup taxa and 50 ingroup taxa (one from this study and 59 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. sojae are provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe sojae’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. tectoendophyta (MFLUCC 13-0471). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), tef and tub displayed the most informative sequences for D. sojae, followed by cal and his loci (Figs. S2–S5) (Table 1). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of the taxa in D. sojae (Fig. S1).
8.2 Species delimitation based on the GCPSR principle
To assess the species boundaries in ‘D. sojae’, the GCPSR principle was applied. Our results revealed less conflict between individual phylogenies, where some species do not clade within the complex and some additional species are claded within the complex. For example, in the ITS phylogeny, the species were scattered in three distinct clades within the tree (Fig. S1). Moreover, D. phaseolorum (CBS 116019) was unusually claded within the complex in all single gene trees (Figs. S1–S5). Both in cal and his single gene trees, many taxa had no available sequences in GenBank (Figs. S4, S5). Moreover, a lack of high bootstrap and posterior probability values on both individual and combined trees in several branches were observed, revealing poor phylogenetic support among the species. The combined gene tree (Fig. 44) is composed of six well-delimited outgroup species (D. convolvuli, D. ovalispora, D. tectoendophytica, D. thunbergiicola D. ueckerae and D. unshiuensis), indicating these clades represent different species. Thus, by implementing the GCPSR principle, based on the comparison of more than one gene genealogy to identify phylogenetic concordance, we verified that the D. sojae is a single species rather than a species complex.
8.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of the concatenated multi-locus sequences ITS, tef, tub, cal and his revealed significant recombination within the D. sojae species complex (Φw = 0.047). The network relationships in the D. sojae species complex are shown in Fig. 45. Additionally, based on the relative distance of species and structure of the phylogenetic network, all species in the D. sojae complex should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe sojae species complex and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
8.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. sojae as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a single species instead of diverse taxa. Hence, each taxon presently recognized in the D. sojae complex should be accepted as a single species.
8.5 Taxonomy
Diaporthe sojae Lehman, Ann. Mo. bot. Gdn 10: 128 (1923)
= Diaporthe actinidiae N.F. Sommer & Beraha, Mycologia 67: 650 (1975)
= Diaporthe camptothecae (C.Q. Chang, Z.D. Jiang, P. Chi) Y.H. Gao & L. Cai, IMA Fungus 8: 183 (2017)
= Diaporthe chromolaenae Mapook & K.D. Hyde, Fungal Diversity 101: 130 (2020)
= Diaporthe endophytica R.R. Gomes, Glienke & Crous, Persoonia 31: 20 (2013)
= Diaporthe fructicola Minosh., T. Ono & Hirooka, Persoonia 42: 409 (2019)
= Diaporthe heterostemmatis S.T. Huang, J.W. Xia & Z. Li, MycoKeys 77: 78 (2021)
= Diaporthe kochmanii R.G. Shivas, S.M. Thomps. & A.J. Young, Pers 27: 86 (2011)
= Diaporthe kongii R.G. Shivas, S.M. Thomps. & A.J. Young, Persoonia 27: 86 (2011)
= Diaporthe masirevicii R.G. Shivas, L. Morin & Y.P. Tan, Persoonia 35: 45 (2015)
= Diaporthe subellipicola S.K. Huang & K.D. Hyde, Mycosphere 9: 381 (2018)
Host range: See Udayanga et al. (2015). Chromolaena odorata (Mapook et al. 2020), Chrysanthemoides monilifera (Thompson et al. 2011), Helianthus annuus (Thompson et al. 2011), Heterostemma grandiflorum (Sun et al. 2021), Passiflora edulis (Minoshima et al. 2020), Schinus terebinthifolius (Gomes et al. 2013).
Known distribution: See Udayanga et al. (2015). Australia (Thompson et al. 2011, 2015), Brazil (Gomes et al. 2013), China (Sun et al. 2021), Japan (Minoshima et al. 2020), Thailand (Mapook et al. 2020).
Notes: Though Udayanga et al. (2015) assessed the D. sojae complex, due to continuous introduction of novel species we observed an expansion of this complex. Hence, we re-structured the complex according to the clade arrangement of multigene combined phylogenies (Figs. 3, 44) and single gene trees (Figs. S1–S5) supported by the GCPSR principle and coalescent methods of PTP and mPTP. To compare the micromorphological characteristics of all these species, a synopsis of conidiomata, conidiogenous cells and conidia characteristics are provided in Table 12. Synonymous names of D. sojae are provided and we propose D. sojae as a single species.
Norphanphoun et al. (2022) introduced a species complex named ‘Diaporthe sojae’, having the same name as of this study, but it contains 92 species. Hence, here we signify the boundaries of D. sojae integrating single gene trees, multi-gene tree, GCPSR and coalescence-based models (PTP, mPTP).
8.6. Morphology of the strains belonging to ‘Diaporthe sojae’. See Table 12
9. Diaporthe tulliensis
9.1 Phylogenetic analyses and informative characters
The multi-gene phylogenetic tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this clade contains seven species, designated here as ‘D. tulliensis’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 46). The five loci combined alignment comprises 2191 characters (521 characters from ITS, 352 from tef, 406 from tub, 462 from cal, and 450 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 39 ingroup taxa (41 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. tulliensis species complex are provided in Table S8. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades. Except tub single gene tree, other single gene trees did not possess 100% similar topology as of the combined gene tree.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe tulliensis’ based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. ambigua (CBS 114015, CBS 117167). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
9.2 Species delimitation based on the GCPSR principle
To assess the species boundaries in the D. tulliensis complex, the GCPSR principle was applied. Our results revealed conflicts between individual phylogenies, where some species do not clade within the complex. For example, in the ITS phylogeny, D. celtidis (NCYU 19–0357, NCYU 19–0358) claded outside the complex (Fig. S1). Both in cal and his single gene trees, many taxa had no available sequences in GenBank (Figs. S4, S5). Moreover, a lack of high bootstrap and posterior probability values on both individual and combined trees in several branches were observed, revealing poor phylogenetic support among the species. However, combined gene tree (Fig. 46) is composed of two well-delimited species (D. ambigua and D. sclerotioides), indicating these clades represent different species. Thus, by implementing the GCPSR principle, based on the comparison of more than one gene genealogy to identify phylogenetic concordance, we verified that D. tulliensis is a single species rather than a species complex.
9.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of the concatenated multi-locus sequences (ITS, tef, tub, cal and his) revealed significant recombination within the D. tulliensis complex (Φw = 0). The network relationships in the D. tulliensis species complex are shown in Fig. 47. Additionally, based on the relative distance of species and structure of the phylogenetic network, all species in the D. tulliensis complex should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of ‘Diaporthe tulliensis’ and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
9.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. tulliensis complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing the complex should be regarded as a single species instead of diverse taxa. Hence, each taxon presently recognized in the D. tulliensis complex should be accepted as a single species.
9.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. tulliensis, which was earlier thought to comprise seven different species but herein represent a single species.
Diaporthe tulliensis R.G. Shivas, Vawdrey & Y.P. Tan, Persoonia 35: 301 (2015)
= Diaporthe alangii C.M. Tian & Qin Yang, MycoKeys 39: 118 (2018)
= Diaporthe celtidis Tennakoon, C.H. Kuo & K.D. Hyde, Fungal Diversity: 108: 124 (2021)
= Diaporthe glabrae (C.Q. Chang, Z.D. Jiang & P.K. Chi) Y.H. Gao & L. Cai, IMA Fungus 8: 183 (2017)
= Diaporthe hubeiensis Dissanayake, X.H. Li & K.D. Hyde, Frontiers in Microbiology 10: 20 (2019)
= Diaporthe morindae M. Luo, W. Guo, Zhao, Manawas., K.D. Hyde, C.P. You, J of Fungi 8: 806 (2022)
= Diaporthe tectonae Doilom, Dissan. & K.D. Hyde, Fungal Diversity 82: 100 (2016)
Host range: Alangium kurzii (Yang et al. 2018), Bougainvillea glabra (Gao et al. 2017), Celtis formosana (Tennakoon et al. 2021), Morinda officinalis (Luo et al. 2022), Tectona grandis (Doilom et al. 2016), Theobroma cacao (Crous et al. 2015), Vitis vinifera (Manawasinghe et al. 2019).
Known distribution: Australia (Crous et al. 2015), China (Gao et al. 2017; Yang et al. 2018; Manawasinghe et al. 2019; Tennakoon et al. 2021; Luo et al. 2022), Thailand (Doilom et al. 2016).
Notes: Crous et al. (2015) introduced D. tulliensis from Theobroma cacao in Australia. Six species which were introduced later; D. tectonae (Doilom et al. 2016), D. glabrae (Gao et al. 2017), D. alangii (Yang et al. 2018), D. hubeiensis (Manawasinghe et al. 2019), D. celtidis (Tennakoon et al. 2021) and D. morindae (Luo et al. 2022) were claded within the strains of D. tulliensis in our combined gene phylogenetic tree (Fig. 46) as well as mostly in single gene trees (Figs. S1–S5). This was supported by the GCPSR principle and coalescent methods of PTP and mPTP. To compare and study the micromorphological characteristics of these species a morphological synopsis of conidiomata, conidiophores, conidiogenous cells and conidia characteristics are provided in Table 13. Synonymous names of D. tulliensis are provided.
9.6. Morphology of the strains belonging to ‘Diaporthe tulliensis’. See Table 13.
Section Rudis
‘Section Rudis’ is composed of 18 type strains of Diaporthe species (Table 14) and is separated from the other proposed sections by its species arrangement within the phylogenetic analysis (Figs. 1–3). However, we observed several conflicts of the clade arrangement in single gene trees (Figs. S1–S5). In the ITS single gene tree, the strains of D. australafricana (D. asheicola), D. obtusifoliae and D. pseudotsugae claded outside Section Rudis (Fig. S1). In the tef phylogeny (Fig. S2), Section Crotalariae is unusually claded within Section Rudis. In the tub single gene (Fig. S3) tree, the strains of D. schoeni which belong to Section Sojae, are unusually claded within Section Rudis. The strains of D. silvicola claded outside Section Rudis in cal single gene tree (Fig. S4). There were no conflicts in his phylogeny in Section Rudis, but an unusual clade arrangement was observed (Fig. S5).
In Section Rudis, we provide descriptions and illustrations of one novel species D. breviconidiophora and one known species (D. guizhouensis) isolated from various woody hosts in Guizhou Province. Additionally, synonymous names for one Diaporthe species and three species/species-complexes (D. amygdali, D. pseudotsugae and D. rudis) are proposed (Figs. 3, 48).

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the species complexes of Section Rudis, based on the LogDet transformation and NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
Diaporthe breviconidiophora Y.Y. Chen, A.J. Dissan. & Jian K. Liu sp. nov.
Index Fungorum number: IF559780; Facesoffungi number: FoF12616, Fig. 49

Diaporthe breviconidiophora (GZAAS 22-0027, holotype). a, b Conidiomata on host surface. c Section of conidioma. d Alpha conidia. e Alpha conidia stained in methylene blue. f Beta conidia attached to conidiophores. g Beta conidia. h, i 10 days-old-culture on PDA from above and from reverse. Scale bars: c = 100 μm; d–g = 20 μm
Etymology: Refers to the short conidiophores.
Holotype: GZAAS 22-0027.
Saprobic on decaying wood. Sexual morph Not observed. Asexual morph Conidiomata 200–350 μm diam., globose to irregular, erumpent at maturity. Peridium 60–150 μm diam., parenchymatous, consisting of 3–4 layers of medium brown textura angularis. Conidiophores 20–45 × 2–2.4 μm, cylindrical, hyaline, smooth, branched, ampulliform, straight to sinuous. Conidiogenous cells 0.5–1 μm, phialidic, cylindrical, terminal, with slight tapering towards apex. Paraphyses 20–40 × 1–2 μm, abundant among conidiophores. Alpha conidia (6–)7–8(–9) × 2–3.5 μm (\(\overline{\text{x}}\) = 7.5 × 3, n = 30), aseptate, hyaline, smooth, ovate to ellipsoidal, biguttulate, base subtruncate. Beta conidia (17–)19–21(–24) × (1–)1.5–2(–2) μm, aseptate, hyaline, smooth, fusiform or hooked, base subtruncate.
Culture characteristics: On PDA, colonies covering entire 9 cm diam. Petri dishes after 10 d at 25 °C, producing abundant white aerial mycelium. Reverse white, turning to grey in centre.
Material examined: China, Guizhou Province, Xingyi City, saprobic on decaying woody host, June 2020, Y.Y. Chen (GZAAS 22-0027, holotype); ex-type culture CGMCC3.24298 = GZCC 22-0031; ibid, DuShan County, saprobic on decaying woody host, May 2020, Y.Y. Chen (GZAAS 22-0050), living culture GZCC 22-0049.
Notes: The phylogenetic results showed that two isolates of Diaporthe breviconidiophora clustered close to D. pustulata and D. silvicola forming a distinct lineage (Fig. 3) with maximum support (ML/MP/BI = 100/100/1.0). Diaporthe breviconidiophora can be distinguished from D. pustulata in the concatenated alignment by 13/512 bp in ITS, 8/413 bp in tef, 14/453 bp in tub, 12/506 bp in cal and 5/515 bp in his and from D. silvicola in the concatenated alignment by 11/512 bp in ITS, 6/413 bp in tef, 9/453 bp in tub.
Species/ species-complexes residing in Section Rudis
1. Diaporthe amygdali
Diaporthe amygdali (Delacr.) Udayanga, Crous, K.D. Hyde, Fungal Divers 56: 166 (2012).
= Diaporthe chongqingensis Y.S. Guo & G.P. Wang, Persoonia 45: 146 (2020)
= Diaporthe fusicola Y.H. Gao & L. Cai, Fungal Biology 119: 304 (2015)
= Diaporthe garethjonesii Dissan., Tangthir. & K.D. Hyde, Fungal Diversity 80: 171 (2015)
= Diaporthe kadsurae C.M. Tian & Qin Yang, MycoKeys 39: 135 (2018)
= Diaporthe mediterranea Leon, Rodríguez-Reina & J. Armengol, Agronomy 10: 17 (2020)
= Diaporthe ovoicicola Y.H. Gao & L. Cai, Fungal Biology 119: 302 (2015)
= Diaporthe sterilis L. Lombard, Polizzi & Crous, Phytopath. Mediterr. 53: 94 (2014)
= Diaporthe ternstroemiae Y.H. Gao, W. Sun & L. Cai, Fungal Biology 119: 306 (2015)
Hilário et al. (2021a) noted that a group of species including D. amygdali, D. garethjonesii, D. sterilis, D. kadsurae, D. ternstroemia, D. ovoicicola, D. fusicola, D. chongqingensis and D. mediterranea, occupy a monophyletic clade in Diaporthe phylogenies. By employing the phylogenetic analysis, GCPSR and coalescence-based models they determined that the D. amygdali species complex constitutes a single species. We observed the same results as Hilário et al. (2021a), and verify D. amygdali is a single species rather than a species complex (Figs. 50, 51). The five loci combined alignment comprises 2179 characters (539 characters from ITS, 329 from tef, 400 from tub, 457 from cal, and 454 from his, including alignment gaps), while the analyses included six well-delimited outgroup taxa and 35 ingroup taxa (41 taxa retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. amygdali’ is provided in Table S9. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe amygdali’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. spartinicola (CPC 24951). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of ‘Diaporthe amygdali’ and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
According to the clade arrangement provided by the ML analyses, all single gene phylogenetic analyses (ITS, tef, tub, cal and his) displayed the most informative sequences for the D. amygdali species complex. Many taxa of the complex have no his sequences (Fig. S5).
All nine species in this group were claded within the strains of D. amygdali in our combined gene phylogenetic tree (Fig. 50) as well as mostly in single gene trees (Figs. S1–S5). This was supported by the GCPSR principle and coalescent methods of PTP and mPTP.
2. Diaporthe pseudotsugae species complex
2.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that this clade contains five species, designated here as ‘D. pseudotsugae species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 52). The five loci combined alignment comprises 2261 characters (563 characters from ITS, 332 from tef, 491 from tub, 428 from cal, and 447 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 13 ingroup taxa (two from this study and 13 taxa retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. pseudotsugae species complex’ is provided in Table S9. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the ‘Diaporthe pseudotsugae species complex’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. nothofagi (CBS 370.54). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), tef displayed the most informative sequences for D. pseudotsugae species complex (Table 1).
2.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. pseudotsugae species complex, the GCPSR principle was applied. Individual gene trees are mostly concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. nothofagi) denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. pseudotsugae species complex’ by demarcating its species boundaries.
2.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant evidence for recombination within the D. pseudotsugae species complex (Φw = 0.351) and the network relationships are shown in Fig. 53. Moreover, based on the comparable remoteness of species and the arrangement of the phylogenetic network, all five species in the D. pseudotsugae species complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe pseudotsugae species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
2.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. pseudotsugae species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed the boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. pseudotsugae species complex should be accepted as different species (Figs. 52, 53).
2.5 Taxonomy
Species residing in Diaporthe pseudotsugae species complex
Diaporthe pseudotsugae Dissan., Camporesi & K.D. Hyde, Mycosphere 8: 869 (2017)
Diaporthe araucanorum M. Zapata, M.A. Palma, M.J. Anninat, & E. Piontelli, Int. J. Syst. Evol. Micro.: 6 (2020)
Diaporthe beckhausii Nitschke, Pyrenomyc. Germ. 2: 295 (1870)
Diaporthe foikelawen M. Zapata, M.A. Palma, M.J. Anninat & E. Piontelli, Int. J. Syst. Evol. Microb.: 7 (2020)
Diaporthe guizhouensis Y.Y. Chen, A.J. Dissan. & Jian K. Liu, Fungal Divers 114: 387 (2022)
3. Diaporthe rudis
3.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates used in this species complex cluster in a clade containing nine species, designated here as ‘D. rudis’. Based on these details, another phylogenetic tree including more isolates of this complex was constructed (Fig. 54). The five loci combined alignment comprises 2099 characters (520 characters from ITS, 296 from tef, 401 from tub, 440 from cal, and 442 from his, including alignment gaps), while the analyses included seven well-delimited outgroup taxa and 31 ingroup taxa (38 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. rudis species complex are provided in Table S9. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe rudis’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. ocoteae (CPC 26217). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of tef possessed 100% similar topology displaying the most informative sequences for the D. rudis species complex (Fig. S2). Several other taxa have been claded within the tub single gene tree (Fig. S3). Many taxa of the complex were absent in both cal and his gene trees (Figs. S4, S5). ITS presented the lowest percentage as two species claded outside the complex, indicating that this locus is less reliable for the delimitation of ‘D. rudis’ (Fig. S1).
3.2 Species delimitation based on the GCPSR principle
To assess boundaries of the species residing in the D. rudis complex, the GCPSR principle was applied. To disclose concordant branches, the individual gene trees were compared. The results revealed several conflicts between individual phylogenies, where some species do not clade within the complex or some additional species are claded within the complex. For example, D. asheicola and D. perniciosa do not clade within the complex in the ITS gene tree (Fig. S1) while D. acericola, D. pseudotsugae and D. schoeni are unusually claded within the group in tub single gene tree (Fig. S3).
However, individual gene trees are mostly concordant and have the same topology for the well-delimited outgroup taxa D. crousii, D. nothofagi, D. cassines and D. ocoteae denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the D. rudis complex and it is herein regarded as a single species, demarcating its species boundaries.
3.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of the concatenated multi-locus sequences ITS, tef, tub, cal and his detected significant recombination within D. rudis (Φw = 0.0199). The network relationships in the D. rudis group are shown in Fig. 55. Additionally, based on the relative distance of species and structure of the phylogenetic network, all nine species in the D. rudis complex should be regarded as a single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of Diaporthe rudis and other well- delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
3.4 Species delimitation based on coalescent methods.
PTP and mPTP analyses provided consistent outcomes and accepted the D. rudis complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a single species instead of diverse taxa. Hence, each taxon presently recognized in the D. rudis complex should be accepted as a single species.
3.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. rudis, which was earlier known as nine different species but herein represents a single species.
Diaporthe rudis (Fr.) Nitschke, Pyrenomyc. Germ. 2: 282 (1870)
= Diaporthe asheicola L. Lombard & Crous, Phytopath. Mediterr. 53: 93 (2014)
= Diaporthe australafricana Crous & van Niekerk, Australas. Pl. Path. 34: 33 (2005)
= Diaporthe cynaroidis Marinc., M.J. Wingf. & Crous, CBS Diversity Ser. 7: 39 (2008)
= Diaporthe italiana Chethana, Camporesi & K.D. Hyde, Fungal Diversity 96: 1 (2019)
= Diaporthe patagonica M. Zapata, M.A. Palma, M.J. Anninat & E. Piontelli, Int. J. Syst. Evol. Microbiol. 70: 9 (2020)
= Diaporthe perniciosa Marchal & É.J. Marchal, Bull. Soc. R. Bot. Belg. 54: 117 (192)
= Diaporthe salicicola R.G. Shivas, Jacq. Edwards & Y.P. Tan, Fungal Diversity 61: 258 (2013)
= Diaporthe subcylindrospora S.K. Huang & K.D. Hyde, Mycosphere 9: 381 (2018)
Host range: See Udayanga et al. (2014a). Corylus sp. (Guerrero and Perez 2013), Protea cynaroides (Marincowitz et al. 2008), Prunus dulcis and Salix sp. (Tan et al. 2013; Lawrence et al. 2015), Vaccinium sp. (Latorre et al. 2012; Elfar et al. 2013; Lombard et al. 2014), Vitis vinifera (van Niekerk et al. 2005).
Known distribution: See Udayanga et al. (2014a). Chile (Latorre et al. 2012; Elfar et al. 2013; Guerrero and Perez 2013; Lombard et al. 2014), South Africa (van Niekerk et al. 2005), USA (Lawrence et al. 2015).
Notes: Diaporthe rudis was fully described and illustrated with a review of all synonyms based on molecular data and morphological studies by Udayanga et al. (2014a) and an epitype was designated isolated from Laburnum anagyroides in Austria. van Niekerk et al. (2005) introduced D. australafricana from Vitis vinifera in South Africa while D. cynaroidis was introduced by Marincowitz et al. (2008) from Protea cynaroides in South Africa. Later, Tan et al. (2013) described D. salicicola from Salix purpurea in Australia while Lombard et al. (2014) described D. asheicola from Vaccinium ashei in Chile. In their phylogenetic analysis, Lombard et al. (2014) observed D. asheicola claded close to D. australafricana but based on few base pair differences, they introduced D. asheicola as a different species. Hyde et al. (2018) defined D. subcylindrospora from Salix sp. in China while Hyde et al. (2019) introduced D. italiana from Morus alba in Italy and in their phylogenetic analysis, they also noted D. italiana was phylogenetically closely related to D. rudis. Zapata et al. (2020) introduced D. patagonica from Aristotelia chilensis in China. However, in our phylogeny (Fig. 54), we observed all of the above-mentioned species cannot be distinguished phylogenetically from D. rudis. This was supported by the GCPSR principle and coalescent methods of PTP and mPTP. To compare and study the micromorphological characteristics of all these species a synopsis of conidiomata, conidiogenous cells and conidia characteristics are provided in Table 15. Synonymous names of D. rudis are provided.
Norphanphoun et al. (2022) introduced a species complex named ‘Diaporthe rudis’, having the same name as ours, but it contains sixteen species. In this study, the boundary of D. rudis was determined integrating single gene trees, multi-gene tree, GCPSR and coalescence-based models (PTP, mPTP).
3.6. Morphology of the strains belonging to ‘Diaporthe rudis’. See Table 15.
Section Betulicola
‘Section Betulicola’ is composed of 10 type strains of Diaporthe (Table 16) and is separated from the other proposed sections by its species arrangement within the combined phylogenetic analysis (Figs. 1, 2, 3). However, we observed several conflicts of the clade arrangement in single gene trees (Figs. S1–S5). In the ITS single gene tree, three different groups of the species belong to Section Betulicola (Fig. S1). In the tub single gene tree (Fig. S3), the strains of D. hispaniae and D. hungariae are claded outside Section Betulicola. There are no conflicts in tef, cal and his phylogenies in Section Betulicola (Figs. S2, S4, S5).
In Section Betulicola, one known species (D. minima) from woody hosts in Guizhou Province was identified. Additionally, synonymous names for two Diaporthe species and two species-complexes (D. ampelina and D. betulicola) are proposed (Figs. 3, 56).

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the species complexes of Section Betulicola, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
Synonymies in Section Betulicola
Diaporthe rostrata C.M. Tian, X.L. Fan & K.D. Hyde, Mycol. Progr. 14: 4 (2015)
= Diaporthe juglandicola Qin Yang, Mycosphere 8: 821 (2017)
Host range: Juglans mandshurica (Fan et al. 2015; Yang et al. 2017)
Known distribution: China (Fan et al. 2015; Yang et al. 2017.
Notes: Diaporthe rostrata was introduced from Juglans mandshurica in China by Fan et al. (2015). Later, Yang et al. (2017) described D. juglandicola from the same host and from the same country. Both sexual and asexual morph were observed in D. rostrata as well as in D. juglandicola (Fan et al. 2015; Yang et al. 2017). A pairwise comparison showed that D. rostrata and D. juglandicola are phylogenetically similar, since the species differ in 1 bp in tef, 3 bp cal and 5 bp in his. Both species were 100% similar in ITS and tub. By comparing morphological details and phylogenetic analysis we synonymize D. juglandicola under D. rostrata.
Species-complexes residing in Section Betulicola
1. Diaporthe ampelina species-complex
1.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates in this complex cluster in a clade containing six species, designated here as the ‘D. ampelina species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 57). The five loci combined alignment comprises 2203 characters (550 characters from ITS, 319 from tef, 396 from tub, 462 from cal, and 476 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 27 ingroup taxa (29 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. ampelina species complex are provided in Table S9. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the ‘Diaporthe ampelina species complex’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. schisandrae (CFCC 51988, CFCC 51989). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of tef, cal and his displayed the most informative sequences for the D. ampelina species complex, followed by ITS and tub loci (Figs. S2–S5). Moreover, D. hispaniae and D. hungariae do not clade within the complex in the tub gene tree (Fig. S3).
1.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. ampelina species complex, the GCPSR principle was applied. Most of the individual gene trees have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. schisandrae) denoting less conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. ampelina species complex’ by demarcating its species boundaries.
1.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant recombination within the D. ampelina species complex (Φw = 0.16) and the network relationships are shown in Fig. 58. Additionally, based on the relative distance of species and structure of the phylogenetic network, all six species in the D. ampelina complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe ampelina species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
1.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. ampelina species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. ampelina species complex should be accepted as a different species (Figs. 57, 58).
1.5 Taxonomy
Species residing in Diaporthe ampelina species complex
Diaporthe ampelina (Berk. & M.A. Curtis) R.R. Gomes, Glienke & Crous, Persoonia 31: 14 (2013)
Diaporthe fibrosa (Pers.) Fuckel, Jb. nassau. Ver. Naturk. 23–24: 204 (1870)
Diaporthe hispaniae Guarnaccia, Armengol & Crous, Persoonia 40: 148 (2018)
Diaporthe hungariae Guarnaccia, Armengol & K.Z. Váczy, Persoonia 40: 149 (2018)
Diaporthe impulsa (Cooke & Peck) Sacc., Syll. fung. (Abellini) 1: 618 (1882)
Diaporthe toxicodendri Y. Ando, Masuya & Tabata, Mycosphere 8: 1161 (2017)
2. Diaporthe betulicola species-complex
2.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates residing in this clade comprise two species, designated here as ‘D. betulicola species complex’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 59). The five loci combined alignment comprises 2447 characters (474 characters from ITS, 349 from tef, 701 from tub, 449 from cal, and 474 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 13 ingroup taxa (15 taxa retrieved from GenBank). Alignment details of ML, MP, and BI analyses of ‘D. betulicola species complex’ is provided in Table S9. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the ‘Diaporthe betulicola species complex’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. schisandrae (CFCC 51988, CFCC 5198). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), tef, cal and his displayed the most informative sequences for D. betulicola species complex, followed by tub (Table 1). ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of the taxa in D. betulicola species complex (Fig. S1). Diaporthe decorticans and D. woolworthii are unusually claded in his single gene tree.
2.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. betulicola species complex, the GCPSR principle was applied. Individual gene trees are mostly concordant (except ITS single gene tree) and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. schisandrae) denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. betulicola species complex’ by demarcating its species boundaries.
2.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find statistically significant evidence for recombination within the D. betulicola species complex (Φw = 0.23) and the network relationships are shown in Fig. 60. Moreover, based on the comparable remoteness of species and the arrangement of the phylogenetic network, the two species in the D. betulicola species complex should be regarded as separate.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe betulicola species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
2.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. betulicola species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. betulicola species complex should be accepted as a different species (Figs. 59, 60).
2.5 Taxonomy
Species residing in Diaporthe betulicola species complex
Diaporthe betulicola C.M. Tian & Z. Du, Phytotaxa 269: 96 (2016)
Diaporthe bohemiae Guarnaccia, Eichmeier & Crous, Persoonia 40: 146 (2018)
Synonymies in Diaporthe betulicola species complex
Diaporthe betulicola C.M. Tian & Z. Du, Phytotaxa 269: 96 (2016)
= Diaporthe huairouensis Y.K. Bai & X.L. Fan, Plant Pathology 71: 1980 (2022)
Hosts: Betula albosinensis (Du et al. 2016), Corylus heterophylla (Bai et al. 2022)
Known distribution: China (Du et al. 2016; Bai et al. 2022)
Notes: Du et al. (2016) introduced D. betulicola from Betula albosinensis from China. In the phylogenetic analysis we observed D. huairouensis cluster with D. betulicola (Figs. 3, 59). Hence, we synonymize D. huairouensis under D. betulicola.
Section Psoraleae-pinnatae
The entire Section Psoraleae-pinnatae has been identified as a single species (Table 17) and named as Diaporthe psoraleae-pinnatae. There are no conflicts in any of the single gene trees (ITS, tef, tub, cal and his) for Section Psoraleae-pinnatae (Figs. S1–S5). This complex was named as D. varians by Norphanphoun et al. (2022), however, there are no available type sequences for D. varians.
In Section Psoraleae-pinnatae, we identified one known species (D. psoraleae-pinnatae) isolated from various woody hosts in Guizhou Province. Additionally, synonymous name for one Diaporthe species and details of a single species group, D. psoraleae-pinnatae is provided (Fig. 3).
Species residing in Section Psoraleae-pinnatae
1. Diaporthe psoraleae-pinnatae
1.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates used in this species complex cluster in a clade containing nine species, designated here as ‘D. psoraleae-pinnatae’. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 61). The five loci combined alignment comprises 2265 characters (540 characters from ITS, 338 from tef, 404 from tub, 492 from cal, and 491 from his, including alignment gaps), while the analyses included three well-delimited outgroup taxa and 20 ingroup taxa (five from this study and 18 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. psoraleae-pinnatae species complex are provided in Table S9. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the strains belonging to ‘Diaporthe psoraleae-pinnatae’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 75% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. The isolates from this study are indicated in green. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. crotalariae (CBS 162.33). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the ML analyses, single gene phylogenetic analyses of ITS, tef, cal and his displayed the most informative sequences for D. psoraleae-pinnatae complex, followed by tub locus (Figs. S1–S5, Table 1).
1.2 Species delimitation based on the GCPSR principle
To assess the boundaries of the species residing in the D. psoraleae-pinnatae complex complex, the GCPSR principle was applied. To disclose concordant branches, the individual gene trees were compared. The results revealed less conflicts between individual phylogenies. We observed D. incompleta does not clade within the complex in the tub gene tree (Fig. S3). Moreover, sequences are unavailable for some species in cal and his single gene trees.
However, individual gene trees are concordant and have the same topology for the well-delimited outgroup taxon D. crotalariae denoting no conflicts among the individual gene trees and revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the D. psoraleae-pinnatae complex and it is herein regarded as a single species, demarcating its species boundaries.
1.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences detected significant recombination within D. psoraleae-pinnatae group (Φw = 0) and the network relationships are shown in Fig. 62. Additionally, based on the relative distance of species and structure of the phylogenetic network, all nine species in the D. psoraleae-pinnatae complex should be regarded as one single species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of Diaporthe psoraleae-pinnatae and other well-delimited species, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
1.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided consistent outcomes and accepted the D. psoraleae-pinnatae complex as a single species. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in a single clade only, showing that the complex should be regarded as a single species instead of diverse taxa. Hence, each taxon presently recognized in the D. psoraleae-pinnatae complex should be accepted as a single species.
1.5 Taxonomy
Combined gene phylogeny based on the GCPSR method, coalescent methods (PTP, mPTP), and micromorphological comparisons are presented to delimit the species boundaries of D. psoraleae-pinnatae, which was earlier known as 10 different species but herein represents a single species.
Diaporthe psoraleae-pinnatae Crous & M.J. Wingf., Persoonia 31: 205 (2013)
= Diaporthe aquatica D.M. Hu, L. Cai & K.D. Hyde, Mycologia 104: 1481 (2012)
= Diaporthe bauhiniae C.M. Tian & Q. Yang, MycoKeys 77: 51 (2021)
= Diaporthe ellipsospora Ya Ya Chen, A.J. Dissanayake & Jian K. Liu, Journal of Fungi 6: 17 (2020)
= Diaporthe incompleta Y.H. Gao & L. Cai, IMA Fungus 8: 175 (2017)
= Diaporthe jinxiu X.H. Wang & G.P. Wang, MycoKeys 80: 83 (2021)
= Diaporthe rhoina Feltgen, Vorstud Pilzfl. Luxemb., Nachtr. III: 145 (1903)
= Diaporthe shaanxiensis C.M. Tian & Q. Yang, MycoKeys 67: 13 (2020)
= Diaporthe varians (Curr.) Sacc., Syll. fung. (Abellini) 1: 614 (1882)
Host range: Bauhinia purpurea (Yang et al. 2021), Camellia sinensis (Gao et al. 2017), Liana sp. (Yang et al. 2021), Prunus persica (Wang et al. 2021), Psoralea pinnata (Crous et al. 2013).
Known distribution: China (Hu et al. 2012; Gao et al. 2017; Dissanayake et al. 2020a; Wang et al. 2021; Yang et al. 2021), South Africa (Crous et al. 2013).
Notes: Except for D. rhoina and D. varians (neither species has reported type material) all other species have been introduced from China. Although D. aquatica, which was introduced from an aquatic habitat in China, is the oldest species in this group, we did not name this group by that name as it has only ITS sequences. All nine species in this group claded within the strains of D. psoraleae-pinnatae in our combined gene phylogenetic tree (Fig. 61) as well as mostly in single gene trees (Figs. S1–S5). This was supported by the GCPSR principle and coalescent methods of PTP and mPTP. Synonymous names of D. psoraleae-pinnatae are provided. To compare the micromorphological characteristics of all these species a synopsis of conidiomata, conidiophores, conidiogenous cells and conidia characteristics are provided in Table 18.
1.6 Morphology of the strains belonging to ‘Diaporthe psoraleae-pinnataeSee Table 18’
Section Crotalariae
‘Section Crotalariae’ is composed of five species of Diaporthe (Table 19) and is separated from the other proposed sections by its species arrangement within the combined phylogenetic analysis (Figs. 1–3).
In Section Crotalariae, synonymous name for one Diaporthe species and one species complex named D. crotalariae is proposed (Fig. 3).
Species complex residing in Section Crotalariae
1. Diaporthe crotalariae species complex
1.1 Phylogenetic analyses and informative characters
The combined gene tree (Fig. 3) and the individual gene trees (Figs. S1–S5) showed that the isolates used in this species complex cluster in a clade containing four species, designated here as the D. crotalariae species complex. Based on these details, another phylogenetic tree including more isolates of this species complex was constructed (Fig. 63). The five loci combined alignment comprises 2247 characters (533 characters from ITS, 334 from tef, 409 from tub, 480 from cal, and 491 from his, including alignment gaps), while the analyses included two well-delimited outgroup taxa and 18 ingroup taxa (20 taxa retrieved from GenBank). Alignment details and the results of MP, ML and BI analyses of D. crotalariae species complex are provided in Table S9. As all ML, MP and BI analyses resulted in trees that were topologically similar, only ML tree is shown with bootstrap and posterior probabilities given for those well supported clades.

Maximum likelihood (ML) tree of the ‘Diaporthe crotalariae species complex’, based on ITS, tef, tub, cal and his loci. ML and MP bootstrap values greater than 70% are shown at the nodes and posterior probabilities (PPs) greater than 0.95 are indicated in thickened branches. Ex-type strains are in bold. Species names are followed by the country and the host. The scale bar represents the expected number of nucleotide substitutions per site. The tree is rooted with D. caulivora (CBS 127268, CBS 178.55). Putative species clusters resulted in PTP analysis are indicated using transitions between black-colored to red-colored branches
According to the clade arrangement provided by the single gene phylogenetic trees (Figs. S1–S5), ITS displayed the most informative sequences for D. crotalariae species complex (Table 1).
1.2 Species delimitation based on the GCPSR principle
To assess the boundary of the D. crotalariae species complex, the GCPSR principle was applied. Our results revealed conflicts between individual phylogenies, where some species do not clade within the complex. For example, D. marina claded outside the species complex in the ITS gene tree (Fig. S1), while D. carpini claded outside the species complex in tef, tub, cal and his gene trees (Figs. S2–S5, Table 1).
Individual gene trees are mostly concordant and have the same topology for the well-delimited ingroup taxa and the outgroup taxon (D. caulivora) revealing these clades represent different species. Hence, comparing one gene genealogy to accomplish GCPSR principle, we confirmed that the node delineating the conversion from concordant branches to incongruence resembles the ‘D. crotalariae species complex’ by demarcating its species boundaries.
1.3 Pairwise homoplasy test and phylogenetic networks
The PHI test of ITS, tef, tub, cal and his sequences did not find significant recombination within the D. crotalariae species complex (Φw = 0.74) and the network relationships are shown in Fig. 64. Additionally, based on the relative distance of species and structure of the phylogenetic network, all four species in the D. crotalariae complex should be regarded as different species.

Phylogenetic network from concatenated data (ITS, tef, tub, cal and his) representing the structure of the Diaporthe crotalariae species complex, based on LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position
1.4 Species delimitation based on coalescent methods
PTP and mPTP analyses provided different results for the D. crotalariae species complex. In the analyses, the shift from blue-colored to red-colored branches (in PTP, data not shown), displayed the boundaries of the species and the shift from brown-colored to red-colored (in mPTP, data not shown), proved that all species were encompassed in the species complex, showing that the complex should be regarded as diverse taxa. Hence, each taxon presently recognized in the D. crotalariae species complex should be accepted as a different species (Figs. 63, 64).
1.5 Taxonomy
Species residing in Diaporthe crotalariae species complex
Diaporthe crotalariae G.F. Weber, Phytopathology 23: 602 (1933).
Diaporthe aspalathi E. Jansen, Castl. & Crous, Stud. Mycol. 55: 71 (2006).
Diaporthe carpini (Pers.) Fuckel, Jb. nassau. Ver. Naturk. 23–24: 205 (1870).
Diaporthe marina Dayar., Mycosphere 11: 99 (2020).
Synonymies in Diaporthe crotalariae species complex
Diaporthe aspalathi E. Jansen, Castl. & Crous, Stud. Mycol. 55: 71 (2006).
= Diaporthe meridionalis Sacc., Michelia 1: 387 (1878).
= Diaporthe woodii Punith., Mycol. Pap. 136: 51 (1974).
Hosts: Aspalathus linearis (van Rensburg et al. 2006), Glycine max (Gomes et al. 2013), Lupinus sp. (Williamson et al. 1994).
Known distribution: Australia (Williamson et al. 1994), South Africa (van Rensburg et al. 2006).
Notes: Diaporthe aspalathi was introduced from Aspalathus linearis in South Africa (van Rensburg et al. 2006). According to our phylogenetic analysis both D. meridionalis and D. woodii cluster close to D. aspalathi (Figs. 3, 63). This was proved by both GCPSR principle and coalescent methods.
Two species: D. litoricola and D. pseudoalnea did not clade with any of the sections. Hence, these two species are regarded as singletons.
Discussion
Combination of morphological characteristics, together with multigene sequence data, have been frequently used to delineate species in the genus Diaporthe (Gomes et al. 2013; Gao et al. 2017; Guo et al. 2020). Mycologists have referred to the clades with higher bootstrap support in single or multilocus phylogenies as species (Stewart et al. 2014), ignoring the contrary of single gene trees, which is an important aspect influencing the species status (Taylor et al. 2000). Besides, it is a challenge to depend on the number of base pair differences when it comes to distinguishing two species in this genus, thus nucleotide variation is not a significant factor to demarcate boundaries. This is crucial in the genus Diaporthe since it has been mistakenly assumed that substantial intraspecific variability serves to distinguish species, leading to an overestimation of species diversity (Gao et al. 2017). In this context, our study aimed to clearly delimit the boundaries of species/species-complexes noticeably visible within the multigene tree as well as in single gene trees by applying GCPSR methodology and coalescent methods (PTP, mPTP).
The GCPSR principle was applied to each proposed species/species-complex of this study. One of the key tenets of the GCPSR is that when comparing gene genealogies, the change from concordance to incongruent establishes the boundary of species (Taylor et al. 2000). This has been used increasingly to identify cryptic speciation in Cladosporium herbarum complex (Schubert et al. 2007), species complexes in Colletotrichum (Damm et al. 2009), speciation in Didymellaceae (Aveskamp et al. 2010), Fusarium (Summerell et al. 2010; Crous et al. 2021), and Phyllosticta (Wulandari et al. 2009). The GCPSR concept was used in this work to comprehend the boundaries of species in each complex. Phylogenetic discordance was seen in the tree topologies following analysis of the individual gene genealogies, which revealed incongruent points and contradictory branches.
For assessing and categorizing species diversity, molecular species delimitation has been recently established as a standard procedure. Large-scale surveys benefit greatly from the use of barcoding techniques since they provide quick species detection and biodiversity estimations. As well as reviewing the concepts and methodologies integrated to define species, Maharachchikumbura et al. (2021) presented a case study applying various species demarcation methods to recognize species boundaries in the cryptic genus Phyllosticta in Botryosphaeriales. They realized that a variety of DNA-based methods may over-split the taxa while in other methods they fall into a single species. Hence, Maharachchikumbura et al. (2021) suggested to resolve these problems by using multiple loci and coalescence-based methods. Moreover, they comprehended integrative approaches which are decisive for demarcating species boundaries while numerous examples for species delimitation were provided. The most popular option among them is a distance-based technique since it scales properly for huge datasets, although it can be sensitive to similarity threshold values and neglect evolutionary relationships. A phylogeny-aware methodology that is not reliant on such criteria is the recently introduced "Poisson Tree Processes" (PTP) method (Kapli et al. 2017). The theoretical and technological problems with PTP are addressed by the enhanced approach known as multi-rate PTP (mPTP). It considers various degrees of intraspecific genetic variety resulting from variations in either each species' evolutionary history or genetic sample. According to empirical data, mPTP regularly produces more precise delimitations regarding the taxonomy, making it preferable to PTP and other distance-based approaches (i.e., recognize more taxonomic species, conclude species numbers closer to the taxonomy). When compared to PTP, it achieves a speedup of at least five orders of magnitude, making it possible to delimitate species in massive (meta-) barcoding data. Additionally, for millions of steps, Markov chain Monte Carlo sampling offers a thorough assessment of the inferred delimitation in just a few seconds, regardless of tree size (Kapli et al. 2017).
Morphological traits are also incorporated to determine species (Liu et al. 2016), but even this is not consistently conspicuous because ‘species’ identified by these traits frequently include several taxa (Harrington and Rizzo 1999; Taylor et al. 2000). For many years, the morphological species conceptualization predominated in Diaporthe, where all known fungi were identified by their morphological characteristics (Rehner and Uecker 1994). Due to the presence of preserved morphological characteristics in this genus, this technique is no longer appropriate (Mostert et al. 2001; Udayanga et al. 2012a).
Various new taxa and new host records of Diaporthe have been described in recent years, and the quantity of species recognized through molecular data has risen rapidly (Dissanayake et al. 2017b; Gao et al. 2017; Guarnaccia and Crous 2017; Boonmee et al. 2021; de Silva et al. 2022; Jayawardena et al. 2022; Norphanphoun et al. 2022; Hongsanan et al. 2023; Hyde et al. 2023; Zhang et al. 2023; Zhou et al. 2023). Researchers working with this specific plant pathogenic genus face many taxonomic problems due to no precise boundaries for the species or species complexes. Although Hilário et al. (2021a, b) resolved the species boundaries of ‘D. amygdali’ and ‘D. eres’ while Pereira et al. (2023) resolved the species boundaries of ‘D. arecae’, there is still no appropriate study to resolve the problems in the whole genus Diaporthe, in a scientific manner. Norphanphoun et al. (2022) presented the species-group concept of Diaporthe, based on results of multi-gene phylogeny and single gene tree analysis and introduced 13 species complexes, of which five complexes (D. arecae, D. biconispora, D. oncostoma, D. rudis and D. sojae) have the same names as used in the current study.
Hence, our research aimed to split the genus Diaporthe into several strong sections and to respective species or species-complexes within each section, based on thorough studies of single gene trees (ITS, tef, tub, cal and his) and multi-gene phylogeny by including all available type strains of Diaporthe. Most importantly, the results were justified by applying the GCPSR methodology as well as the coalescence-based models to the whole genus, sections, and complexes. The tef, tub, cal and his single gene phylogenies seems to overlap with multi-gene phylogeny (Fig. 1). However, we observed numerous Diaporthe species lack tub, cal and his sequence data. In tef phylogeny, we observed ‘Section Rudis’ has interfered with ‘Section Crotalariae’. Considering this information, we do not suggest any single gene phylogeny to symbolize the best for this genus. Hence, due to the inconsistencies in single gene phylogenies, we highly recommend following the combined gene phylogeny for sections. In Fig. 2 we have provided additional support of phylogenetic network from concatenated data of ITS, tef, tub, cal and his to prove the existence of these sections.
To support this re-assessment of the genus, the genetic diversity of Diaporthe isolates obtained from woody hosts in South-Western China were investigated and analyzed. As a result, 82 isolates revealed two novel species (D. breviconidiophora and D. submersa) and 17 known species (D. apiculata, D. arecae, D. cinnamomi, D. citrichinensis, D. conica, D. discoidispora, D. eres, D. guizhouensis, D. guttulata, D. irregularis, D. lenispora, D. minima, D. minusculata, D. oncostoma, D. passiflorae, D. psoraleae-pinnatae and D. sojae).
Most of the recognized species/species-complexes in this study were proposed based on additional strains (we tried to represent more strains where the species distribution is world-wide), which might be the decisive factor for assessment of species boundaries, as advocated by Gao et al. (2016). The complexes were named according to the oldest epithet or based on the popularity of the species residing in each complex. We noted that several strains of the same species were distributed throughout different clades in individual phylograms, which probably indicates its intraspecific variability, horizontal gene transfer, etc. It has therefore been proposed that many strains, ideally from different locations and hosts, should always be included to delimitate species or to describe novel taxa in the genus Diaporthe (Gao et al. 2016; Liu et al. 2016). Assembling the genus into several precise sections will avoid the need for continuous construction of unnecessary, lengthy phylogenetic trees, as very recent publications have reported about 300 type species in Diaporthe. Hence, forthcoming studies related to this genus could be directed in a formal/strict way by choosing only the appropriate ‘Diaporthe section’/’Diaporthe species or species-complex’, which would perfectly fit with the particular study. Hence, we suggest that future studies follow the recent modifications to this genus.
References
Abeywickrama PD, Wanasinghe DN, Karunarathna SC, Jayawardena RS et al (2020) A new host report of Diaporthe manihotia (Diaporthales, Ascomycota) from Camellia sp. in Yunnan Province. China Asian Journal of Mycology 3:472–487
Abeywickrama PD, Camporesi E, Jayawardena RS, Hyde KD et al (2022) Novel and surprising host associations of Diaporthe (Diaporthaceae, Diaporthales) species from Italy. Chiang Mai J Sci 49:223–247
Aveskamp MM, de Gruyter J, Woudenberg JHC, Verkley GJM, Crous PW (2010) Highlights of the Didymellaceae: a polyphasic approach to characterise Phoma and related pleosporalean genera. Stud Mycol 65:1–60
Aumentado HD, Balendres MA (2024) Novel species and new records of Diaporthe causing eggplant leaf and fruit blight in the Philippines. Mycol Prog 23:23
Bai Y, Pan M, Gao H, Lin L et al (2022) Studies of Diaporthe species causing hazelnut canker disease in Beijing, China, with two new species described. Plant Pathol 71:1980–1991
Boonmee S, Wanasinghe DN, Calabon MS, Huanraluek N et al (2021) Fungal diversity notes 1387–1511: taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers 111:1–335
Bruen TC (2005) A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681
Cao L, Luo D, Lin W, Yang Q, Deng X (2022) Four new species of Diaporthe (Diaporthaceae, Diaporthales) from forest plants in China. MycoKeys 91:25–47
Carbone I, Kohn LM (1999) A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91:553–556
Chaisiri C, Liu X, Lin Y, Fu Y et al (2021) Phylogenetic and haplotype network analyses of Diaporthe eres species in China based on sequences of multiple loci. Biology 10:179
Chaisiri C, Liu X, Lin Y, Luo C (2022) Diaporthe citri: a fungal pathogen causing melanose disease. Plants 11:1600
Chen SF, Morgan DP, Michailides TJ (2014) Botryosphaeriaceae and Diaporthaceae associated with panicle and shoot blight of pistachio in California, USA. Fungal Divers 67:157–179
Crous PW, Groenewald JZ, Shivas RG, Edwards J et al (2011) Fungal Planet description sheets: 69–91. Persoonia 26:108–156
Crous PW, Summerell BW, Shivas RG, Burgess TI et al (2012) Fungal Planet description sheets, 107–127. Persoonia 28:138–182
Crous PW, Wingfield MJ, Guarro J, Cheewangkoon R et al (2013) Fungal Planet description sheets, 154–213. Persoonia 31:188–296
Crous PW, Shivas RG, Quaedvlieg W, van der Bank M et al (2014) Fungal planet description sheets: 214–280. Persoonia 32:184–306
Crous PW, Wingfield MJ, Le Roux JJ, Richardson DM et al (2015) Fungal Planet description sheets: 371–399. Persoonia 35:264–327
Crous PW, Wingfield MJ, Richardson DM, Roux JJL et al (2016) Fungal planet description sheets: 400–468. Persoonia 36:316–458
Crous PW, Wingfield MJ, Burgess TI, Hardy GESt et al (2018) Fungal Planet description sheets: 716–784. Persoonia 40:240–393
Crous PW, Wingfield MJ, Schumacher RK, Akulov A et al (2020) New and interesting fungi. 3. Fungal Syst Evol 6:157–231
Crous PW, Lombard L, Sandoval-Denis M, Hyde KD (2021) Fusarium: more than a node or a foot cell. Stud Mycol 98:100–116
Damm U, Woudenberg J, Cannon P, Crous P (2009) Colletotrichum species with curved conidia from herbaceous hosts. Fungal Divers 39:45–87
de Silva NI, Hyde KD, Lumyong S, Phillips AJL et al (2022) Morphology, phylogeny, host association and geography of fungi associated with plants of Annonaceae, Apocynaceae and Magnoliaceae. Mycosphere 13:955–1076
Dayarathne MC, Jones EBG, Maharachchikumbura SSN, Devadatha B et al (2020) Morpho-molecular characterization of microfungi associated with marine based habitats. Mycosphere 11:1–188
Dissanayake AJ, Liu M, Zhang W, Chen Z et al (2015) Morphological and molecular characterisation of Diaporthe species associated with grapevine trunk disease in China. Fungal Biol 119:283–294
Dissanayake AJ, Camporesi E, Hyde KD, Wei Z et al (2017a) Molecular phylogenetic analysis reveals seven new Diaporthe species from Italy. Mycosphere 8:853–877
Dissanayake AJ, Phillips AJL, Hyde KD, Yan JY, Li XH (2017b) The current status of species in Diaporthe. Mycosphere 8:1106–1156
Dissanayake AJ, Chen YY, Liu JK (2020a) Unravelling Diaporthe species associated with woody hosts from Karst formations (Guizhou) in China. J Fungi 6:251
Dissanayake AJ, Bhunjun CS, Maharachchikumbura SSN, Liu JK (2020b) Applied aspects of methods to infer phylogenetic relationships amongst fungi. Mycosphere 11:2653–2677
Doilom M, Dissanayake AJ, Wanasinghe DN, Boonmee S et al (2016) Microfungi on Tectona grandis (teak) in northern Thailand. Fungal Divers 82:107–182
Drenth A, McTaggart AR, Wingfield BD (2019) Fungal clones win the battle, but recombination wins the war. IMA Fungus 10:1–6
Du Z, Fan XL, Hyde KD, Yang Q et al (2016) Phylogeny and morphology reveal two new species of Diaporthe from Betula spp. in China. Phytotaxa 269:090–102
Early MP, Punithalingam E (1972) Phomopsis anacardii sp. nov. on Anacardium occidentale. Transactions of British Mycological Society 59:345–347
Edler D, Klein J, Antonelli A, Silvestro D (2020) raxmlGUI 2.0: a graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol Evol 12:373–377
Elfar K, Torres R, Diaz GA, Latorre BA (2013) Characterization of Diaporthe australafricana and Diaporthe spp. associated with stem canker of blueberry in Chile. Plant Dis 97:1042–1050
Fan XL, Hyde KD, Udayanga D, Wu XY, Tian CM (2015) Diaporthe rostrata, a novel ascomycete from Juglans mandshurica associated with walnut dieback. Mycol Prog 14:82
Fan X, Yang Q, Bezerra JD, Alvarez LV, Tian C (2018) Diaporthe from walnut tree (Juglans regia) in China, with insight of the Diaporthe eres complex. Mycol Prog 17:841–853
Feng XX, Chen JJ, Wang GR, Cao TT (2019) Diaporthe sinensis, a new fungus from Amaranthus sp. in China. Phytotaxa 425:259–268
Fuckel L (1867) Symbolae mycologicae. Beitrage zur Kenntniss der Rheinischen Pilze. Jahrbücher Des Nassauischen Vereins Für Naturkunde 23–24:1–459
Gao YH, Sun W, Su YY, Cai L (2014) Three new species of Phomopsis in Gutianshan nature reserve in China. Mycol Prog 13:111–121
Gao YH, Liu F, Cai L (2016) Unravelling Diaporthe species associated with Camellia. Syst Biodivers 14:102–117
Gao Y, Liu F, Duan W, Crous PW, Cai L (2017) Diaporthe is paraphyletic. IMA Fungus 8:153–187
Glass NL, Donaldson GC (1995) Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl Environ Microbiol 61:1323–1330
Gomes RR, Glienke C, Videira SI, Lombard L et al (2013) Diaporthe: a genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 31:1–41
Guarnaccia V, Crous PW (2017) Emerging citrus diseases in Europe caused by species of Diaporthe. IMA Fungus 8:317–334
Guarnaccia V, Crous PW (2018) Emerging citrus diseases in Europe, caused by species of Diaporthe. IMA Fungus 8:317–334
Guarnaccia V, Groenewald JZ, Woodhall J, Armengol J et al (2018) Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 40:135–153
Guerrero J, Perez S (2013) First report of Diaporthe australafricana caused stem canker and dieback in European hazelnut (Corylus avellana L.) in Chile. Plant Dis 97:1657–1657
Guo YS, Crous PW, Bai Q, Fu M et al (2020) High diversity of Diaporthe species associated with pear shoot canker in China. Persoonia 45:132–162
Hall T (2006) Bioedit 7.5.0.3. Department of Microbiology, North Carolina State University. http://www.mbio.ncsu.edu/BioEdit/Bioedit.html.
Harrington TC, Rizzo DM (1999) Defining species in the fungi. In: Worrall JJ (ed) Structure and dynamics of fungal populations. Springer, Dordrecht, pp 43–71
Hilário S, Gonçalves MFM (2023) Mechanisms underlying the pathogenic and endophytic lifestyles in Diaporthe: an omics-based approach. Horticulturae 9:423
Hilário S, Amaral IA, Gonçalves MFM, Lopes A, Santos L, Alves A (2020) Diaporthe species associated with twig blight and dieback of Vaccinium corymbosum in Portugal, with description of four new species. Mycologia 112:293–308
Hilário S, Santos L, Alves A (2021a) Diaporthe amygdali, a species complex or a complex species? Fungal Biol 125:505–518
Hilário S, Gonçalves MFM, Alves A (2021b) Using genealogical concordance and coalescent-based species delimitation to assess species boundaries in the Diaporthe eres complex. J Fungi 7:507
Hilário S, Santos L, Alves A (2021c) Diversity and pathogenicity of Diaporthe species revealed from a survey of blueberry orchards in Portugal. Agriculture 11:1271
Hongsanan S, Norphanphoun C, Senanayake IC, Jayawardena RS et al (2023) Annotated notes on Diaporthe species. Mycosphere 14:918–1189
Hu DM, Cai L, Hyde KD (2012) Three new ascomycetes from freshwater in China. Mycologia 104:1478–1489
Huang F, Udayanga D, Wang XH, Hou X et al (2015) Endophytic Diaporthe associated with Citrus: a phylogenetic reassessment with seven new species from China. Fungal Biol 119:331–347
Huang S, Xia J, Zhang X, Sun W (2021) Morphological and phylogenetic analyses reveal three new species of Diaporthe from Yunnan, China. MycoKeys 78:49–77
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Hyde KD, Nilsson RH, Alias SA, Ariyawansa HA et al (2014) One stop shop: backbones trees for important phytopathogenic genera: I. Fungal Divers 67:21–125
Hyde KD, Hongsanan S, Jeewon R, Bhat DJ et al (2016) Fungal diversity notes 367–492, taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 80:1–270
Hyde KD, Chaiwan N, Norphanphoun C, Boonmee S et al (2018) Mycosphere notes 169–224. Mycosphere 9:271–430
Hyde KD, Tennakoon DS, Jeewon R, Bhat DJ et al (2019) Fungal diversity notes 1036–1150: taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers 96:1–242
Hyde KD, Dong Y, Phookamsak R, Jeewon R et al (2020) Fungal diversity notes 1151–1276: Taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers 100:1–273
Hyde KD, Norphanphoun C, Ma J, Yang HD et al (2023) Mycosphere notes 387–412 – novel species of fungal taxa from around the world. Mycosphere 14:663–744
Jayawardena RS, Hyde KD, Wang S, Sun YR et al (2022) Fungal diversity notes 1512–1610: taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers 117:1–272
Jiang N, Voglmayr H, Tian CM (2018) New species and records of Coryneum from China. Mycologia 110:1172–1188
Kapli P, Lutteropp S, Zhang J, Kobert K et al (2017) Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33:1630–1638
Katoh K, Toh H (2010) Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26:1899–1900
Latorre BA, Elfar K, Espinoza JG, Torres R, Díaz GA (2012) First report of Diaporthe australafricana associated with stem canker on blueberry in Chile. Plant Dis 96:768
Lawrence DP, Travadon R, Baumgartner K (2015) Diversity of Diaporthe species associated with wood cankers of fruit and nut crops in northern California. Mycologia 107:926–940
Lesuthu P, Mostert L, Spies CFJ, Moyo P et al (2019) Diaporthe nebulae sp. nov. and first report of D. cynaroidis, D. novem, and D. serafiniae on grapevines in South Africa. Plant Dis 103:808–817
Liu JK, Chomnunti P, Cai L, Phookamsak R et al (2010) Phylogeny and morphology of Neodeightonia palmicola sp. nov. from palms. Sydowia 62:261–276
Liu JK, Hyde KD, Jones EBG, Ariyawansa HA et al (2015) Fungal diversity notes 1–110, taxonomic and phylogenetic contributions to fungal species. Fungal Divers 72:1–197
Liu F, Wang M, Damm U, Crous PW, Cai L (2016) Species boundaries in plant pathogenic fungi: a Colletotrichum case study. BMC Evol Biol 16:81
Lombard L, van Leeuwen GCM, Guarnaccia V, Polizii G et al (2014) Diaporthe species associated with Vaccinium, with specific reference to Europe. Phytopathol Mediterr 5:287–299
Luo M, Guo W, Zhao M, Manawasinghe IS et al (2022) Endophytic Diaporthe associated with Morinda officinalis in China. Journal of Fungi 8:806
Maharachchikumbura SSN, Hyde KD, Jones EBG, McKenzie EHC et al (2016) Families of Sordariomycetes. Fungal Divers 79:1–317
Maharachchikumbura SSN, Chen Y, Ariyawansa HA, Hyde KD et al (2021) Integrative approaches for species delimitation in Ascomycota. Fungal Divers 109:155–179
Manawasinghe IS, Dissanayake AJ, Li X, Liu M et al (2019) High genetic diversity and species complexity of Diaporthe associated with grapevine dieback in China. Front Microbiol 10:1936
Mapook A, Hyde KD, McKenzie EHC, Jones EBG et al (2020) Taxonomic and phylogenetic contributions to fungi associated with the invasive weed Chromolaena odorata (Siam weed). Fungal Divers 101:1–175
Marincowitz S, Crous PW, Groenewald JZ, Wingfield MJ (2008) Microfungi occurring on the Proteaceae in the fynbos. CBS Biodiversity Series 7:1–166. CBS Fungal Biodiversity Centre, Utrecht, Netherlands.
Minoshima A, Ono T, Truong HH, Sugawara Y, Hirooka Y (2020) First report of fruit rot on passion fruit caused by Diaporthe fructicola in Japan. Jpn J Phytopathol 86:97–101
Monkai J, Hongsanan S, Bhat DJ, Dawoud TM et al (2023) Integrative taxonomy of novel Diaporthe species associated with medicinal plants in Thailand. Journal of Fungi 9:603
Mostert L, Crous PW, Kang JC, Alan JL (2001) Species of Phomopsis and a Libertella sp. occurring on Grapevines with specific reference to South Africa: morphological, cultural, molecular and pathological characterization. Mycologia 93:146–167
Nitschke T (1870) Pyrenomycetes Germanici (2nd ed.). Eduard Trewendt, Germany, Breslau, pp 161–320.
Norphanphoun C, Gentekaki E, Hongsanan S, Jayawardena R et al (2022) Diaporthe: Formalizing species-group concepts. Mycosphere 13:752–819
Pereira DS, Hilário S, Gonçalves MEM, Phillips AJL (2023) Diaporthe species on palms: Molecular re-Assessment and species boundaries delimitation in the D. arecae species complex. Microorganisms 11: 2717.
Perera RH, Hyde KD, Dissanayake AJ, Jones EBG et al (2018) Diaporthe collariana sp. nov., with prominent collarettes associated with Magnolia champaca fruits in Thailand. Studies in Fungi 3:141–151
Petrović K, Riccioni L, Đorđević V, Balešević-Tubić S et al (2018) Diaporthe pseudolongicolla: The new pathogen on soybean seed in Serbia. Ratarstvo i Povrtarstvo 55:103–109
Phillips AJL (2003) Morphological characterization of Diaporthe foeniculacea and its Phomopsis anamorph on Foeniculum vulgare. Sydowia 55:274–285
Rayner RW (1970) A Mycological Colour Chart. Commonwealth Mycological Institute (Great Britain) & British Mycological Society.
Rehner SA, Uecker FA (1994) Nuclear ribosomal internal transcribed spacer phylogeny and host diversity in the coelomycetes Phomopsis. Can J Bot 72:1666–1674
Rosskopf EN, Charudattan R, DeValerio JT, Stall WM (2000) Field evaluation of Phomopsis amaranthicola, a biological control agent of Amaranthus spp. Plant Dis 84:1225–1230
Rossman AY, Adams GC, Cannon PF, Castlebury LA, Crous PW et al (2015) Recommendations of generic names in Diaporthales competing for protection or use. IMA Fungus 6:145–154
Rossman AY, Allen WC, Braun U, Castlebury LA et al (2016) Overlooked competing asexual and sexually typified generic names of Ascomycota with recommendations for their use or protection. IMA Fungus 7:289–308
Santos JM, Vrandecic K, Cosic J, Duvnjak T, Phillips AJ (2011) Resolving the Diaporthe species occurring on soybean in Croatia. Persoonia 27:9–19
Santos L, Alves A, Alves R (2017) Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe. PeerJ 5:e3120
Schubert K, Groenewald JZ, Braun U, Dijksterhuis J et al (2007) Biodiversity in the Cladosporium herbarum complex (Davidiellaceae, Capnodiales), with standardisation of methods for Cladosporium taxonomy and diagnostics. Stud Mycol 58:105–156
Senanayake IC, Jeewon R, Chomnunti P, Wanasinghe DN et al (2018) Taxonomic circumscription of Diaporthales based on multigene phylogeny and morphology. Fungal Divers 93:241–443
Sommer NF, Beraha L (1975) Diaporthe Actinidiae, A new species causing stem-end rot of Chinese Gooseberry Fruits. Mycologia 67:650–653
Stewart JE, Timmer LW, Lawrence CB, Pryor BM, Peever TL (2014) Discord between morphological and phylogenetic species boundaries: incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evol Biol 14:38
Summerell BA, Laurence MH, Liew EC, Leslie JF (2010) Biogeography and phylogeography of Fusarium: a review. Fungal Divers 44:3–13
Sun W, Huang S, Xia J, Zhang X, Li Z (2021) Morphological and molecular identification of Diaporthe species in south-western China, with description of eight new species. MycoKeys 77:65–95
Tan Y, Edwards J, Grice K, Shivas R (2013) Molecular phylogenetic analysis reveals six new species of Diaporthe from Australia. Fungal Divers 61:251–260
Taylor JW, Jacobson DJ, Kroken S, Ka T et al (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32
Tennakoon DS, Kuo CH, Maharachchikumbura SSN, Thambugala KM (2021) Taxonomic and phylogenetic contributions to Celtis formosana, Ficus ampelas, F. septica, Macaranga tanarius and Morus australis leaf litter inhabiting microfungi. Fungal Divers 108:1–215
Thambugala KM, Daranagama DA, Phillips AJL, Bulgakov TS et al (2016) Microfungi on Tamarix. Fungal Divers 82:239–306
Thompson SM, Tan YP, Young AJ, Neate SM et al (2011) Stem cankers on sunflower (Helianthus annuus) in Australia reveal a complex of pathogenic Diaporthe (Phomopsis) species. Persoonia 27:80–89
Thompson S, Tan Y, Shivas R, Neate S, Morin L et al (2015) Green and brown bridges between weeds and crops reveal novel Diaporthe species in Australia. Persoonia 35:39–49
Udayanga D, Liu XZ, Crous PW, McKenzie EHC, et al. (2012a) A multi-locus phylogenetic evaluation of Diaporthe (Phomopsis). Fungal Divers 56:157–171.
Udayanga D, Liu XZ, McKenzie EHC, Chukeatirote E, Hyde KD (2012b) Multi-locus phylogeny reveals three new species of Diaporthe from Thailand. Cryptog Mycol 33:295–309
Udayanga D, Castlebury LA, Rossman AY, Hyde KD (2014a) Species limits in Diaporthe: molecular re-assessment of D. citri, D. cytosporella, D. foeniculina and D. rudis. Persoonia 32:83–101
Udayanga D, Castlebury LA, Rossman AY, Chukeatirote E, Hyde KD (2014b) Insights into the genus Diaporthe: phylogenetic species delimitation in the D. eres species complex. Fungal Divers 67:203–229
Udayanga D, Castlebury LA, Rossman AY, Chukeatirote E, Hyde KD (2015) The Diaporthe sojae species complex: phylogenetic re-assessment of pathogens associated with soybean, cucurbits and other field crops. Fungal Biol 119:383–407
van Niekerk JM, Groenewald JZ, Farr DF, Fourie PH et al (2005) Reassessment of Phomopsis species on grapevines. Australas Plant Pathol 34:27–39
van Rensburg JCJ, Lamprecht SC, Groenewald JZ, Castlebury LA, Crous PW (2006) Characterisation of Phomopsis spp. associated with die-back of rooibos (Aspalathus linearis) in South Africa. Stud Mycol 55:65–74
Vrijmoed LLP, Hyde KD, Jones EBG (1994) Observations on mangrove fungi from Macau and Hong Kong, with the description of two new Ascomycetes: Diaporthe salsuginosa and Aniptodera haispora. Mycol Res 98:699–704
Wang X, Guo Y, Du Y, Yang Z et al (2021) Characterization of Diaporthe species associated with peach constriction canker, with two novel species from China. MycoKeys 80:77–90
Wang SY, McKenzie EHC, Phillips AJL, Li Y, Wang Y (2022) Taxonomy and multigene phylogeny of Diaporthales in Guizhou Province, China. J Fungi 8:1301
Wehmeyer LE (1933) The genus Diaporthe Nitschke and its segregates. Univ Mich Stud Sci Ser 9:1–349
White T, Bruns T, Lee S, Taylor FJRM et al (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: A guide to methods and applications. Academic Press 18:315–322
Williamson PM, Highet AS, Gams W, Sivasithamparam K, Cowling WA (1994) Diaporthe toxica sp. nov, the cause of lupinosis in sheep. Mycol Res 98:1364–1368
Wrona CJ, Mohankumar V, Schoeman MH, Tan YP et al (2020) Phomopsis husk rot of macadamia in Australia and South Africa caused by novel Diaporthe species. Plant Pathol (lond) 69:911–921
Wulandari NF, To-Anun C, Hyde KD, Duong LM et al (2009) Phyllosticta citriasiana sp. nov., the cause of Citrus tan spot of Citrus maxima in Asia. Fungal Divers 34:23–39
Xiao XE, Liu YD, Zheng F, Xiong T et al (2023) High species diversity in Diaporthe associated with citrus diseases in China. Persoonia 51:229–256
Yang Q, Fan XL, Du Z, Tian CM (2017) Diaporthe juglandicola sp. nov. (Diaporthales, As- comycetes), evidenced by morphological characters and phylogenetic analysis. Mycosphere 8:817–826
Yang Q, Fan XL, Guarnaccia V, Tian CM (2018) High diversity of Diaporthe species associated with dieback diseases in China, with twelve new species described. MycoKeys 39:97–149
Yang Q, Jiang N, Tian CM (2020) Three new Diaporthe species from Shaanxi Province, China. MycoKeys 67:1–18
Yang Q, Jiang N, Tian CM (2021) New species and records of Diaporthe from Jiangxi Province, China. MycoKeys 77:41–64
Zapata M, Palma MA, Aninat MJ, Piontelli E (2020) Polyphasic studies of new species of Diaporthe from native forest in Chile, with descriptions of Diaporthe araucanorum sp. nov., Diaporthe foikelawen sp. nov. and Diaporthe patagonica sp. nov. Int J Syst Evol Microbiol 70:3379–3390
Zhang K, Su YY, Cai L (2013) An optimized protocol of single spore isolation for fungi. Cryptogam, Mycol 34:349–356
Zhang JF, Liu JK, Hyde KD, Chen YY et al (2023) Ascomycetes from karst landscapes of Guizhou Province, China. Fungal Divers 122:1–160
Zhou YY, Zhang W, Wu LN, Zhang J et al (2023) Diversity of fungal communities associated with grapevine trunk diseases in China. Mycosphere 14:1340–1435
Acknowledgements
This study is supported by the National Natural Science Foundation of China (Grant No. 32350410401) and Joint Fund of the National Natural Science Foundation of China and the Karst Science Research Center of Guizhou Province (Grant No. U1812401). K.D. Hyde would thank the Chinese Research Fund (project no E1644111K1) entitled “Flexible introduction of high- level expert program, Kunming Institute of Botany, Chinese Academy of Sciences and Major science and technology projects and key R&D plans/programs, Yunnan Province (202202AE090001) and the High- level Talents in Zhongkai University of Agriculture and Engineering, grant no: J2201080102 and the Innovative team program of the Department of Education of Guangdong Province (2022KCXTD015 and 2022ZDJS020). K.D. Hyde also gratefully acknowledges the financial support of the Distinguished Scientist Fellowship Program of King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Handling Editor: Sinang Hongsanan.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Dissanayake, A.J., Zhu, JT., Chen, YY. et al. A re-evaluation of Diaporthe: refining the boundaries of species and species complexes. Fungal Diversity (2024). https://doi.org/10.1007/s13225-024-00538-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13225-024-00538-7