
Abstract
Trapelioid fungi constitute a widespread group of mostly crust-forming lichen mycobionts that are key to understanding the early evolutionary splits in the Ostropomycetidae, the second-most species-rich subclass of lichenized Ascomycota. The uncertain phylogenetic resolution of the approximately 170 species referred to this group contributes to a poorly resolved backbone for the entire subclass. Based on a data set including 657 newly generated sequences from four ribosomal and four protein-coding gene loci, we tested a series of a priori and new evolutionary hypotheses regarding the relationships of trapelioid clades within Ostropomycetidae. We found strong support for a monophyletic group of nine core trapelioid genera but no statistical support to reject the long-standing hypothesis that trapelioid genera are sister to Baeomycetaceae or Hymeneliaceae. However, we can reject a sister group relationship to Ostropales with high confidence. Our data also shed light on several long-standing questions, recovering Anamylopsoraceae nested within Baeomycetaceae, elucidating two major monophyletic groups within trapelioids (recognized here as Trapeliaceae and Xylographaceae), and rejecting the monophyly of the genus Rimularia. We transfer eleven species of the latter genus to Lambiella and describe the genus Parainoa to accommodate a previously misunderstood species of Trapeliopsis. Past phylogenetic studies in Ostropomycetidae have invoked “divergence order” for drawing taxonomic conclusions on higher level taxa. Our data show that if backbone support is lacking, contrasting solutions may be recovered with different or added data. We accordingly urge caution in concluding evolutionary relationships from unresolved phylogenies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Early concepts of the phylogenetic relationships of lichenized fungi drew heavily on the shape and gross attributes of ascomata, ascospores and thallus and the photobionts with which they associate (Watson 1929). Starting in the 1960s, detailed anatomical studies of the ascus (Letrouit-Galinou 1966; Hafellner 1984), ascomatal ontogeny (Letrouit-Galinou 1968) and secondary metabolite chemistry (Culberson 1969), as well as increased openness to the possibility of convergent evolution, led to a shake-up in the classification of lichenized fungi. One of the numerous enduring legacies of this era is the recognition that emerged in the 1970s and 1980s of the close relatedness of a group of genera with a characteristic non-amyloid, unitunicate ascus and well-defined apical cushion that came to be called the Agyrium- or Trapelia-type ascus (Hertel 1970). Using mainly ascus and ontogenetic characters, Lumbsch (1997) proposed uniting 16 of these genera into Lecanorales suborder Agyriinae, which was subsequently raised to the level of its own order, Agyriales (Lumbsch et al. 2001a). However, with the application of molecular phylogenetics to more members of this group it became apparent that ascus characters and ontogeny also exhibit convergent evolution, and that several of these genera are only distantly related, including Anzina and Elixia (Wedin et al. 2005), Miltidea (Widhelm and Lumbsch 2011) and not least the name-giving genus Agyrium (Lumbsch et al. 2007a). This latter finding resulted in the taxonomic orphaning of the genera remaining and led to several new taxonomic proposals, partly reflecting renewed attention to relationships with Baeomycetaceae (e.g., Lumbsch et al. 2007a; Lumbsch and Huhndorf 2010; Hodkinson and Lendemer 2011). The rump group can now be considered to consist of 11 genera (Trapeliaceae sensu Lumbsch and Huhndorf 2010): Amylora, Coppinsia, Lambiella (Spribille et al. 2014), Lithographa, Placopsis (encompassing Aspiciliopsis and Orceolina), Placynthiella, Ptychographa, Rimularia, Trapelia, Trapeliopsis and Xylographa (Sarea was recently excluded by Miadłikowska et al. 2014). Several of these genera were included in Lecanoromycetes subclass Ostropomycetidae at the time it was first recognized (Miadłikowska and Lutzoni 2004) and have since been routinely included in phylogenetic hypotheses of that subclass. For the purposes of the present discussion we will refer to this group as the trapelioid fungi (Fig. 1).

Diversity of trapelioid fungi in the broad sense, grouped by the major clades recovered here. a to f, Xylographaceae. a, Lithographa tesserata (Alaska, Spribille [=S] 38950, GZU; scale bar 0.5 mm); b, Ptychographa xylographoides (Scotland, Coppins 24229, GZU: 200 μm); c, Xylographa pallens (Austria, Resl 1143, GZU: 200 μm); d, Xylographa lagoi (Spain, S30267, GZU: 200 μm); e, Lambiella insularis (Montana, S/07.09.2012, GZU: 200 μm); f, Lambiella caeca (Alaska, S36295, GZU: 200 μm); g to k, Trapeliaceae. g, Rimularia limborina (Alaska, Fryday 10100, MSC: 200 μm); h, Trapeliopsis granulosa (Sweden, Nordin 7402, UPS: 500 μm); i, Placynthiella uliginosa (Montana, S/21.09.2013, GZU; 200 μm); j, Trapelia glebulosa (Montana, S/09.2013, photo courtesy of Tim Wheeler: ca. 1 mm); k, Placopsis cribellans (upper right) and P. lambii (bottom left; Alaska, S/09.2014, GZU: 1 mm); l, Baeomycetaceae. L, Parainoa subconcolor (Italy, Arnold Lich. Exs. 938, GZU: 500 μm), the only trapelioid species recovered outside of Xylographaceae and Trapeliaceae
Several characteristics suggest that trapelioid fungi are a promising study system for evolutionary biology of the lichen symbiosis, namely their role as pioneer colonizers (e.g., Jahns 1982; Ullmann et al. 2007; Raggio et al. 2012), their photobiont diversity (Voytsekhovich et al. 2011), and their substrate specificity (Spribille et al. 2008, 2014). Developing them as a model system however requires resolving evolutionary relationships that until now have been deeply entangled with other clades of Ostropomycetidae. Since its recognition as a subclass by Reeb et al. (2004), nearly all phylogenies of Ostropomycetidae have recovered a pattern of resolved terminal clades and an unresolved backbone (Lumbsch et al. 2005: mtSSU, nuLSU; Schmitt et al. 2005: mtSSU, nuLSU; Wedin et al. 2005: mtSSU, nuLSU; Lumbsch et al. 2007c: mtSSU, nuLSU; Schmitt et al. 2010: nuLSU, mtSSU, RPB1, MCM7; Lumbsch et al. 2012: mtSSU, nuLSU; Bendiksby and Timdal 2013: ITS, mtSSU, nuLSU; Otálora and Wedin 2013: mtSSU, RPB1, MCM7; Prieto and Wedin 2013: nuSSU, nuLSU, 5.8S, mtSSU, RPB1, MCM7 and Prieto et al. 2013, same loci). The pattern of persistent low backbone support is perhaps best visualized in the large-scale phylogeny of the group presented by Miadłikowska et al. (2014, Fig. 2). These results convinced us that any resolution of deep relationships of trapelioid fungi would require a taxon sampling that encompassed representatives of all key clades and more loci than anything sampled to date.

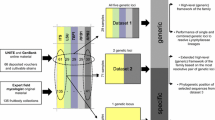
Phylogenetic hypotheses tested using the SOWH test. Locks represent constrained nodes. A: Hypothesis as in Lumbsch et al. (2007a) with Baeomycetaceae being sister to trapelioids, B: Hypothesis of a sister group relationship of trapelioids and Hymeneliaceae, C: Hypothesis of sister group relationship of trapelioids and Ostropales. D: Hypothesis obtained by Miadłikowska et al. (2014) with Ostropales and Arctomiaceae forming the crown group in Ostropomycetidae
Aside from trapelioid fungi, the Ostropomycetidae are dominated by two species-rich main groups, usually treated as orders, namely the Ostropales, which have almost always been recovered as monophyletic (Kauff and Lutzoni 2002; Miadłikowska et al. 2006, 2014; Lumbsch et al. 2007b; Prieto and Wedin 2013; Prieto et al. 2013), and the Pertusariales, which usually have not (e.g., Wedin et al. 2005; Lumbsch et al. 2007a, 2007b; Prieto et al. 2013; Prieto and Wedin 2013; but see Miadłikowska et al. 2014). Five smaller “floating clades” also feature in most studies: Arctomiaceae, Baeomycetaceae, Hymeneliaceae, Sarrameanaceae and Schaereriaceae. Phylogenetic hypotheses including trapelioid fungi repeatedly recover three recurring motifs that, though mostly lacking statistical support and in some cases forming polytomies, form the working basis for evolutionary hypotheses in this group:
-
1)
trapelioids are sister to the Baeomycetaceae, with or without the Hymeneliaceae (Wedin et al. 2005, as Agyriales; Lumbsch et al. 2007a, as Agyriaceae core group, and Lumbsch et al. 2007b, as Agyriales; Lumbsch et al. 2007c; Bendiksby and Timdal 2013);
-
2)
trapelioids are sister to the Ostropales or sandwiched between the Ostropales and Baeomyces + Arctomia (Miadłikowska et al. 2006; Prieto et al. 2013; Prieto and Wedin 2013);
-
3)
trapelioids are sister to the Ostropales + Arctomiaceae (Miadłikowska et al. 2014, as Trapeliales); this is the only study to present statistical support for multiple relationships.
The lack of support until now for sister group level relationships in the vicinity of trapelioid fungi makes it impossible to confidently infer order of divergence, and by extension character evolution, in this speciose group. A deeper locus sampling is in our view the only way a confident assessment of evolutionary relationships of trapelioid fungi can be advanced. We accordingly set out to assemble a large data set of trapelioid fungi as well as obtain an eight-locus sampling for each of the “floating clades” in Ostropomycetidae and selected outgroups. In so doing, we added hundreds of newly generated sequences and carefully curated published sequences to weed out mixed accessions (sequences of one species derived from different vouchers) that have weakened previous phylogenetic analyses. Our goals were: 1) to resolve as far as possible the backbone of the Ostropomycetidae; 2) test the support for rejecting alternative hypotheses about relationships that have been proposed to date for Trapeliaceae, Baeomycetaceae, Hymeneliaceae and Arctomiaceae; and 3) adjust the taxonomy to reflect some of the statistically significant evolutionary inferences derived from our analyses.
Material and methods
Assembly of taxon sample set
We designed our taxon sample to include representatives of all described orders of Ostropomycetidae and all available genera of trapelioid lichenized fungi. We drew upon two sources of DNA sequences. First we screened Genbank for specimens from which multiple loci had been sequenced; we did not permit mixed accessions (sequences attributed to one species but derived from different specimens) because the often dynamic understanding of species delimitations can lead to seemingly congruent sequences, if acquired from different isolates, actually deriving from different species. Second, because no Genbank samples had all eight loci targeted for this study, we ended up extracting DNA from fresh material from every taxonomic order of Ostropomycetidae. Consistent with our focus on trapelioid fungi we accorded most attention to the 11 genera of trapelioid lichenized fungi until now assigned to Trapeliaceae by Lumbsch and Huhndorf (2010; see Introduction). We also invested considerable sequencing effort in other groups within Ostropomycetidae. We did not undertake resampling of speciose families such as Graphidaceae, Megasporaceae and Pertusariaceae s.lat. that have been found to be monophyletic in the past (Mangold et al. 2008; Rivas Plata et al. 2013; Nordin et al. 2010; Schmitt and Lumbsch 2004; Schmitt et al. 2006, 2010). Instead, we tried to obtain as many loci as possible for several members of every major group, including where necessary from new isolates. Similarly, we generated multilocus data sets for single to multiple species in Lecanoromycetidae and Umbilicariomycetidae for use as outgroups. Newly generated sequences and used Genbank accessions are summarized in Table 1. Detailed information on isolated DNA vouchers, their obtained loci and NCBI accession numbers is provided in Online Resource 1.
DNA acquisition, polymerase chain reaction and Sanger sequencing
Samples from ascomata or thallus fragments were pulverized in a Retsch cell grinder in 1.5 ml Eppendorf tubes with a single 3 mm steel bead after freezing at −80 °C. Lysis buffer was applied directly to the sheared cells. Further extraction of genomic DNA was performed using the Qiagen DNeasy Plant Mini kit following the manufacturer’s instructions. For sparse specimen material the QIAmp DNA Investigator Kit was used. We eluted raw nucleic acids in 50 to 75 μL of elution buffer without RNAse treatment. Undiluted samples were used for downstream PCR reactions. For each sample, we sequenced as many as possible of eight commonly used gene fragments: the internal transcribed spacer regions 1 and 2 as well as the embedded 5.8S region of the ribosomal rDNA (hereafter ITS); the nuclear ribosomal large subunit (nuLSU); the nuclear ribosomal small subunit (nuSSU); the mitochondrial small ribosomal subunit DNA (mtSSU); parts of the largest and second largest subunit of the RNA polymerase II (RPB1 and RPB2, respectively); part of DNA replication licensing factor minichromosome maintenance complex 7 (MCM7); as well as a commonly used partial sequence of transcription elongation factor 1 alpha (EF1a). Primers and annealing temperatures used are listed in Table 2. PCR was performed using PuReTaq Ready-To-Go PCR beads. After checking the size of the obtained fragments on ethidium bromide-stained agarose gels, we purified PCR products using the AMPure XP bead clean-up protocol, or the Omega E.Z.N.A. Cycle Pure Kit according to manufacturer’s instructions. Purified PCR products were than sequenced by Microsynth (Switzerland).
Alignment
Sequence alignment was performed using MAFFT v7 (Katoh and Standley 2013). MAFFT allows the use of different alignment algorithms depending on the properties of input sequences (e.g., presence of unalignable introns). We used the –genafpair flag to align ribosomal ITS, nuSSU, nuLSU and mtSSU sequences and the –globalpair algorithm to align protein-coding MCM7, RPB1, RPB2 and EF1a sequences. We set MAFFT to run 10,000 iterations for each alignment. Alignments were manually checked for obvious errors and corrected when needed. Embedded sequence alignment and subsequent concatenation were performed in a single pipeline using custom Python scripts.
With a custom Python script we eliminated intron positions on the basis of the relative presence of nucleotides at each position in the alignment. We applied a cut-off value of 10 %, so that positions with more than 90 % missing data were excluded. This alignment was used for all subsequently performed phylogenetic analyses. To provide information on the completeness of our alignment we created a visualized alignment plot in which the percent completeness of each individual nucleotide position is graphically displayed relative to the number of isolates included in the alignment. The nucleotide completeness matrix was retrieved with a custom Python script and depicted using scripts written in the graphic programming language Processing 2. All used Python scripts have been released on the GitHub page of the first author under the repository phylo-scripts v0.1 (Resl 2015, https://github.com/reslp).
Phylogenetic analyses
We performed maximum likelihood (hereafter ML) as well as Bayesian inference (hereafter BI). The ML phylogenetic analysis was performed using RAxML v8.0.4 (Stamatakis 2014) and BI was carried out using MrBayes 3.2.2 (Ronquist and Huelsenbeck 2003). We created partitions for each gene fragment as well as for an intron present in RPB1 in the original alignment. Protein-coding genes were partitioned according to codon position. This a-priori selected scheme was used as input for PartitionFinder 1.1.1 (Lanfear et al. 2012) to optimize partitions and substitution models. As input parameters we selected linked branch lengths and the Bayesian Information Criterion (BIC) as optimality criterion in a greedy search. PartitionFinder retained ten partitions and chose GTRGAMMAI for each. To evaluate statistical node support we generated 1000 bootstrap replicates of the alignment using the fast bootstrap option of RAxML. We performed a maximum likelihood search to find the best scoring tree according to its log likelihood score (RAxML option –f a). To check for topological conflicts, we created single locus trees using RAxML. For each gene we used the GTRGAMMAI substitution model and generated 500 bootstrap replicates. The maximum likelihood search was performed in the same way as for the concatenated dataset. Topological conflict in single gene trees was assessed with the software compat.py (Kauff and Lutzoni 2003) for a cut-off bootstrap value of 70.
We performed BI to provide a second topological hypothesis of the concatenated dataset. As in the ML analysis we used the partitioning scheme selected by PartitionFinder. Parameters of the DNA substitution model for each partition were estimated by MrBayes using reversible jump MCMCMC as implemented in the command lset nst = 6. Bayesian phylogenetic analyses are known to have problems reaching stationarity when analyzing large datasets owing to the limitations of low run and chain numbers to adequately explore potential parameter space (Hackett et al. 2008). Preliminary Bayesian analyses of our dataset with two independent runs and four chains each failed to converge even after 100 million generations (mean deviation of split frequencies remained > 0.05) with the standard temperature factor of the heated chain set to 0.2. We substantially improved the diagnostic metrics (standard deviation of split frequencies, ESS values of parameter estimates of the model) of our analyses by performing four independent MCMCMC runs with eight chains each for 80 million generations. To provide better chain mixing we further set the temperature factor of the heated chain to 0.3 and increased the number of swaps to two. We used a 30 % relative burn-in (relburnin = yes burninfrac = 0.30) and the flag contype = halfcompat in sumt to create a majority rule consensus tree. We checked for convergence of the MCMCMC runs in terms of the obtained topology using the web version of AWTY (Wilgenbusch et al. 2004). We also investigated the convergence of the parameter estimates of the runs by Tracer 1.6 (Rambaut et al. 2014). In both cases we used a burn-in proportion of 30 %. The final tree was visualized in R using the ape package (R Development Core Team 2013).
Testing topological hypotheses
We performed topology tests on four alternative phylogenetic hypotheses specific to the immediate sister group relationships of trapelioid fungi (Fig. 2): A) the backbone topology of trapelioid fungi and Baeomycetaceae recovered by Lumbsch et al. (2007a), the 1:1 sister group relationship of trapelioid fungi to B) Hymeneliaceae and C) Ostropales, and finally D) the backbone obtained for trapelioid fungi and neighbouring groups by Miadłikowska et al. (2014). Two of these hypotheses (A, D) were formulated a priori but the others (B, C), as well as later specific alternative hypotheses constraining monophyly of two genera (see Discussion), were developed in part a posteriori after studying our own and past phylogenies. All tested scenarios focus on nodes that lack support in both phylogenetic reconstructions, BI and ML respectively. Bayesian phylogenetic methods are known to overestimate support from concatenated alignments relative to bootstrap methods applied in maximum likelihood analysis, which tend to be more conservative (e.g., Suzuki et al. 2002). Consequently, nodes that were supported in BI but not in ML were of particular interest to us. In keeping with a more conservative likelihood approach for hypothesis testing we applied the Swofford-Olsen-Waddell-Hillis (hereafter SOWH) test to each scenario. The SOWH test allows a direct comparison between an a posteriori obtained topology and a priori developed phylogenetic hypotheses (Swofford et al. 1996). It is thus more appropriate than the Approximately Unbiased (AU)- or Kishino-Hasegawa (KH)-tests, which assume strict a priori hypotheses (Goldman et al. 2000).
The SOWH tests were implemented in the SOWHAT pipeline (Church et al. 2014, https://github.com/josephryan/SOWHAT). The pipeline relies on RAxML for generating phylogenetic analyses and SeqGen (Rambaut and Grassly 1997) for creating simulated alignments. Statistical tests are performed using R. The test involves generating a null distribution of the differences in likelihood of the constrained and unconstrained topology by parametric sampling of simulated alignments that fit the original topology parameters (branch lengths, substitution model). The difference in log-likelihood of the constrained and unconstrained tree of the original alignment (test-statistic) is compared to the obtained null distribution of log-likelihood differences from simulated alignments with a one-sided t-test. The obtained p-value of the test indicates the probability that the observed difference in likelihood values would also be observed under H0 (no difference between both topologies). For each test we used RAxML in the PTHREADS version and employed a GTRGAMMA substitution model for all partitions as described above. Different numbers of trees were calculated depending on the minimum number needed for completing a null distribution relative to the data. We used the built-in convergence assessment algorithm (flag –stop) to halt the analysis when it reaches a point where subsequent sampling is unlikely to alter the likelihood distribution (Church et al. 2014). Statistical support to reject the alternative hypothesis was considered sufficient if p < 0.05. The specific Newick coding of topological constraints is provided in Online Resource 2.
Results
Acquired sequences
We obtained a total of 657 new sequences from 148 isolates including all nine trapelioid genera from which we had fresh material; only Amylora and Coppinsia could not be sampled. We acquired the most sequences for mtSSU and the fewest for RPB2 (Online Resource 1). We obtained five or more loci for 64 isolates. Together with 309 sequences from Genbank, we incorporated 966 sequences from 205 isolates into our alignment (Table 1; Online Resource 1). The raw alignment consisted of 20,999 positions. Following removal of sites with missing data exceeding threshold values, the final alignment used for phylogenetic analyses consisted of 8978 positions including introns in the ITS and nuLSU and nuSSU region as well as in the RPB1 gene (Fig. 3). The original alignment has been deposited at TreeBASE under study ID 16680.

Visualized alignment plot indicating percent completeness of nucleotides per alignment position (above) and the number of variable, parsimony-informative and conserved sites per gene fragment (below). x-axis: nucleotide position in alignment. y-axis: percent nucleotide completeness
Phylogenetic hypotheses
The best-scoring ML tree had a Ln of −184,362.3943. For BI we allowed the run to continue open-ended until the average deviation of split frequencies had stabilized under 0.016, which happened after 14 million generations. We then let the analysis run for another two million generations to ensure that the chains were not trapped in sub-optima of topology space. The final average standard deviation of split frequencies was 0.013827. The AWTY plots (Online Resource 3) show the posterior probabilities of splits over all pairs of independent MCMC runs indicating convergence of the topology. Tracer showed convergence of the LnL values of the tree (Online Resource 4) as well as for all parameters in the explored parameter space (effective sample size > 200; data not shown).
Tests for topological incongruence showed several disagreements between gene trees (Online Resource 5a–h). After manually investigating each conflict reported by compat.py most were found to be due to sequence gaps in either of the two trees or related to shallow relationships (e.g., affect species-level relationships). The remaining conflicts we found are summarized in Online Resource 6. Since removing those sequences did not affect nodal resolution, we retained them in the dataset.
ML and BI analyses of the concatenated dataset yielded similar topologies and we plot node support of both ML and BI analyses on the best-scoring ML topology (Fig. 4; all values in Online Resources 7–9). The Ostropomycetidae form a well-supported, reciprocally monophyletic clade with the outgroups (87%BS / 1.00PP support) if circumscribed to exclude Loxosporaceae and Schaereriaceae. All nine trapelioid genera from which we obtained sequences resolved as a monophyletic clade (100%BS / 1.00PP support; Fig. 4b), which in turn splits into two deeply divergent, reciprocally monophyletic clades. The only trapelioid found outside of this group was Trapeliopsis subconcolor, which was recovered within a strongly supported (100%BS / 1.00PP) aeomycetaceae. The latter forms a clade only supported in BI (60%BS / 1.00PP; together the “BAH clade”) including Arctomiaceae and Hymeneliaceae, each of which are independently strongly supported in both analyses (Fig. 4a). The BAH clade forms a supported sister group to trapelioids only in BI (45%BS / 0.95PP) Ostropales are resolved in a monophyletic clade (91%BS / 1.00PP support) that forms an unresolved sister group relationship with Protothelenellaceae. The split between the trapelioid/BAH clade and the Ostropales/Protothelenellaceae clade is supported in both analyses (73%BS / 0.98PP). The Pertusariales clade forms a monophyletic group (73%BS / 1.00PP) that is reciprocally monophyletic to the rest of Ostropomycetidae. The original Bayesian topology as well as all ML gene trees are provided in Online Resources 5a–h and 7–9.


Estimated tree of Ostropomycetidae obtained from concatenated maximum likelihood analysis of eight fungal gene fragments. Boostrap support values are plotted as boxes above, Bayesian posterior probabilities as boxes below nodes. Trapelioid groups are demarcated with colored boxes
Within trapelioids, the genus Rimularia s.lat was found to be polyphyletic, with most sampled species coming out in a monophyletic clade with Lambiella (97%BS / 1.00PP), while the type species, R. limborina, is recovered in a monophyletic clade sister to a well-supported clade including Placopsis, Trapelia, Trapeliopsis and Placynthiella with 100%BS / 1.00PP (Fig. 4b). Trapelia consists of four well-supported clades that form a paraphyletic assemblage with T. corticola as its most basal member (89%BS / 0.99PP). Placopsis is recovered as monophyletic but nested within Trapelia. The recently described Trapelia antarctica forms a well-supported sister group relationship with Placopsis. Trapeliopsis is monophyletic as currently circumscribed (100%BS / 1.00PP) with the exception of T. subconcolor, which comes out with high support in Baeomycetaceae and forms a well-supported clade with the Genbank-derived isolate X125 “Ainoa geochroa”, which is in fact also T. subconcolor (see Discussion). Within the Baeomycetaceae, Baeomyces is paraphyletic with Phyllobaeis.
Topology tests
Table 3 summarizes the results obtained by the individual SOWH test scenarios for four different topological scenarios that account for previously formulated phylogenetic hypotheses of sister group relationships of trapelioid clades (Fig. 2). Scenarios A and B tested for sister group relationships of trapelioid groups (trapelioids/Baeomycetaceae/Hymeneliaceae). Under the test for scenario A, a sister group relationship of trapelioids and Baeomycetaceae could not be rejected (p = 1). Under scenario B, a sister group relationship between trapelioids and Hymeneliaceae the SOWH test was not able to provide a significant solution given our data. Scenario C tested a sister group relationship of trapelioids and Ostropales, which is strongly rejected (p < 0.01). Scenario D, in which Ostropales/Arctomiaceae are sister to trapelioids, is rejected with high confidence given our dataset (p = 0).
Discussion
The sister group relationships of trapelioid fungi
Our phylogenetic hypothesis for Ostropomycetidae is the first to recover support for nearly the entire backbone of the subclass in both ML and BI. Amongst other things, it solidifies an emerging pattern, first observed by Miadłikowska et al. (2014) in a five-locus sample, in which a monophyletic Pertusariales is sister to the rest of the subclass. It confirms a monophyletic Ostropales as in previous studies, and is the first to recover a well-supported monophyletic group for a comprehensive sampling of trapelioid genera. In the process, it tightens a circle around three nodes that remain problematic inasmuch as they are lacking support in ML or BI and thus constitute the remaining destabilizing elements in the phylogeny: 1) the relationship between Baeomycetaceae and Arctomiaceae/Hymeneliaceae (the “BAH clade”); 2) the relationship between the latter two families (Arctomiaceae and Hymeneliaceae) individually; and 3) the immediate sister group relationship of trapelioids to the BAH clade. These three groups interact with each other and almost certainly account for most of the discrepancies in node support in our analyses.
Even without full resolution in our phylogenetic hypothesis, not all of the sister group relationships that have been hypothesized in previous studies are equally probable. The hypothesis of a sister group relationship to Ostropales can be rejected with p < 0.01 given the taxon and locus sampling used here. Similarly, a topology consistent with the hypothesis in which trapelioids are sister to a combined Ostropales + Arctomiaceae and these in turn sister to Hymeneliaceae and Baeomycetaceae, is impossible to obtain with our data set and can be ruled out (p = 0). This leaves the first and original set of hypotheses based on molecular data, namely a sister group relationship to Baeomycetaceae and/or Hymeneliaceae, with or without Arctomiaceae. Constraining trapelioids to form a single monophyletic sister group relationship with either Baeomycetaceae or Hymeneliaceae yields a likelihood distribution not significantly different from the unconstrained topology, meaning that the null hypothesis cannot be rejected (Table 3; we did not test the 1:1 sister group relationship to Arctomiaceae because of the small sample).
Major groups of trapelioid fungi
The nine genera of trapelioids resolve into two reciprocally monophyletic clades representing Lithographa, Ptychographa, Xylographa and Lambiella, on the one hand, and Rimularia, Placynthiella, Trapeliopsis, Trapelia, and Placopsis, on the other. The first grouping includes mostly species with linearized, hysteriothecial ascomata (though round ascomata also occur, e.g., in Lambiella caeca: Fig. 1f) and we recognize this group as the family Xylographaceae (see below). The other represents mainly species with rounded ascomata, the Trapeliaceae in the original sense of Hertel (1970). The split between Xylographaceae and Trapeliaceae runs through the middle of the genus Rimularia as used by current authors, as well as through the family Rimulariaceae (Hafellner 1984; Hertel and Rambold 1990). The split echoes earlier suspicions by Hertel (1984) that Rimularia consists of disparate elements. Rimularia s.str. (around the type species R. limborina) forms a basal group with Trapeliaceae, though supported only in the Bayesian analysis. The majority of species sampled thus far go to Xylographaceae, where the name Lambiella was established for L. psephota by Hertel (1984). The polyphyly of Rimularia as defined to date is well supported and statistically beyond doubt (result of parametric topology test: p = 0). Notably, Lambiella in its expanded definition adopted here itself splits into deeply diverging lineages, with a clade for the impavida group, a clade for the insularis group and two isolated branches supporting the type species of Lambiella, L. psephota, as well as L. sphacelata; overall, however, it is monophyletic.
Within the Trapeliaceae, the genus Trapelia is strongly paraphyletic with Placopsis nested within Trapelia as currently understood. The paraphyly also extends to the species level, with taxa such as T. coarctata and T. glebulosa recovered in disparate clades. The genus is the subject on ongoing character evolution studies (K. Schneider, in prep.).
Novelties in non-trapelioid Ostropomycetidae
A collateral consequence of expanding taxon and locus sampling is the resolution of several relationships that have long been unstable in Ostropomycetidae, beyond the sister group relationships of the trapelioid genera. The position of Schaereriaceae, represented by S. corticola and/or S. fuscocinerea, has typically been plotted as the first divergence in Ostropomycetidae, though always without support (Wedin et al. 2005; Miadłikowska et al. 2006; Lumbsch et al. 2007a, b). Ours is the first phylogeny to include the type species, Schaereria cinereorufa (Hafellner 1984). Miadłikowska et al. (2014) included Schaereria together with Loxospora in Sarrameanaceae, though they conceded that the anatomical evidence made this seem unlikely. We also provide sequences of the type species of Loxospora, L. elatina, for the first time, confirming its close relationship to other species placed in that genus, and retain it in Loxosporaceae in absence of evidence supporting its relationship to Sarrameana. Though statistically testing this was outside the scope of this study, both of our phylogenetic hypotheses suggest a close relationship of Schaereria to Loxospora could be rejected, as well as their inclusion in an otherwise monophyletic Ostropomycetidae. The Pezizalean-like ascus of Schaereria (Hafellner 1984; Lumbsch 1997) has few if any parallels in this subclass. Similarly, the unstable position of Loxospora even in a six-locus sample suggests an isolated position.
The recovery of Anzina as sister to Protothelenella at the base of Ostropales sheds light on another heretofore unstable element in the phylogeny of Ostropomycetidae. Anzina was repeatedly postulated to belong to Trapeliaceae based on similarities in ascus structure, conidiogenesis and secondary metabolites (Scheidegger 1985; Lumbsch 1997). Both Anzina and Protothelenella were sequenced and recovered as sister to Ostropales in separate studies in 2005 (Anzina: Wedin et al. 2005; Protothelenella: Schmitt et al. 2005). Protothelenella was subsequently recovered on a polytomy together with Arctomiaceae, Schaereriaceae and Ostropales by Lumbsch et al. (2007a) and even on a polytomy with what are here called trapelioid fungi by Lumbsch et al. (2007b, 2007c). Curiously neither genus was included in a later overview of secondary delichenization in Ostropales (Baloch et al. 2010). Lumbsch et al. (2012) recovered both on a supported branch in a two locus (mtSSU, nuLSU) phylogeny of Ostropomycetidae. Our results appear to lend strong support to the monophyly of Anzina and Protothelenella and further suggest that the synonymization of Thrombiaceae and Protothelenellaceae by Schmitt et al. (2005) should be revisited in future sampling at the base of the Ostropales.
Insights into lower level relationships in trapelioid fungi
-
1)
We found no support for rejecting the placement of Anamylopsora in Baeomycetaceae. Anamylopsora was originally assigned to Baeomycetales on the basis of morphological characters, but in its own family, Anamylopsoraceae. In describing the family, Lumbsch et al. (1995) pointed out similarities in ascomatal ontogeny, excipular structures and the form of conidia of A. pulcherrima with Baeomycetaceae. However, differences in the ascus apical apparatus, the structure of the cortex and the production of benzyl esters led Lumbsch et al. (1995) to the conclusion that similarities between Anamylopsora and Baeomycetaceae must be due to convergent evolution. Because all heretofore published sequences of A. pulcherrima were derived from a single individual (Lumbsch et al. 2001b, 2005), we generated sequences from four loci from a second specimen. Orthologous DNA sequences closely match those obtained by Lumbsch et al. (2001b) and group in a monophyletic Baeomycetaceae.
-
2)
We recover Ainoa in Baeomycetaceae and found one set of previous Genbank accessions to contain errors. The genus Ainoa was erected by Lumbsch et al. (2001b) with the type species Ainoa geochroa and an ITS sequence of that species from Slovakia, deposited as Trapelia geochroa (AF274078). The next set of sequences deposited in Genbank came from a specimen of A. mooreana from the Czech Republic (AY212828 and others; Schmitt et al. 2003). We recover these sequences in Baeomycetaceae with strong support. The classification of the genus has however become confused by the introduction of DNA sequences under the name Ainoa geochroa that are recovered in disparate parts of Ostropomycetidae. These were uploaded to Genbank by Lumbsch et al. (2007a, 2007b; number DQ871006 and other loci) based on a specimen collected in Ecuador. We have since studied the specimen (Z. Palice 8600, F!) and determined that it is not Ainoa geochroa, but rather Trapeliopsis subconcolor, previously known from South America from Colombia and Venezuela (Hertel 1977, 1981).
We sequenced eight loci from another set of Ainoa mooreana from Japan and three from Trapeliopsis subconcolor from China, and are now able to triangulate. The Japanese A. mooreana fully matches the Czech specimen and its ITS is 100 % identical to the original, Slovakian ITS sequence of A. geochroa published by Lumbsch et al. (2001b). The sequences from the Ecuadorian specimen are however heterogeneous and doubtfully originate from the same fungus. The nuLSU sequence (DQ871006) differs from Dibaeis baeomyces (AF279385) by only 15 nucleotide positions and most probably belongs to an unknown Icmadophilaceae. Excluding nuLSU, the sequences of mtSSU and RPB1 from the Ecuadorian specimen place it in Baeomycetaceae in a monophyletic group with Trapeliopsis subconcolor from China, in agreement with its morphology and chemistry. Furthermore, the Ecuadorian nuLSU sequence does not match Chinese T. subconcolor nuLSU; we included both sequences in the nuLSU gene tree (Online Resource 5c) to demonstrate this point. We suspect that a mix-up occurred with the nuLSU sequence of the Ecuadorian specimen and recommend that this sequence (DQ871006) be flagged as a likely error in Genbank. As for Trapeliopsis subconcolor, we propose that this species, which is not closely related to Trapeliopsis, be treated in a new genus (see Taxonomic changes).
-
3)
Trapeliopsis aeneofusca and T. gelatinosa are mutually paraphyletic and can be considered synonyms (the older name is T. gelatinosa Flörke 1809). Purvis and Smith (2009) already suspected that T. aeneofusca is only a pigment-deficient morph of T. gelatinosa.
-
4)
Our phylogeny confirms a close relationship between Trapeliopsis glaucopholis, T. steppica and T. wallrothii as suspected by McCune et al. (2002; see also Printzen and McCune 2004). We refrain from recommending synonymization of these taxa as they may represent incipient speciation, but our results clearly highlight the need for work on species delimitation in this group. In this context, the recently described T. gymnidiata from the Canary Islands (Aptroot and Schumm 2012) and Madeira (C. Printzen, pers. obs.) also merits attention. T. wallrothii is the oldest name in the complex and also the type of the genus (Hafellner 1984).
-
5)
We recover Trapeliopsis colensoi, T. haumanii, T. glaucolepidea and T. precrenata as part of a single highly supported species complex, a close relationship noted already by Galloway (2007). The separation of the mainly Southern Hemisphere T. colensoi from the widespread T. glaucolepidea is weakly supported by our phylogeny but our study was not designed to test whether they should be maintained as distinct. The similarity of T. glaucolepidea and T. percrenata has been noted in the past (e.g., Coppins and James 1984) and their distinctness questioned (Purvis and Smith 2009). We found them mutually paraphyletic and consider them synonyms, echoing the results of Palice and Printzen (2004).
What we can and cannot say with our data
Our study includes more loci and specifically more protein-coding sequences than any previous study of the group (protein-coding loci constitute 38 % of our sequence data as opposed to 22 % for Ostropomycetidae in e.g., Miadłikowska et al. 2014). A reality of Sanger sequencing in non-model organisms is that it is impossible to obtain a full sequence sample for every isolate extracted, especially in poorly known taxonomic groups with trace amounts of DNA. This inevitably leads to a “long tail” of isolates for which partial sequence data are available. Where these isolates are from species for which other, complete sequence sets are available, we have excluded them. The practice of including taxa with large amounts of missing data and their effect on the accuracy of phylogenetic reconstructions is still under debate (for a summary see Wiens and Morrill 2011). Evidence from simulation and empirical studies shows that the impact of missing data is dependent on the phylogenetic method used and correlates with the number of characters (Wiens 2003; Dunn et al. 2003; Wiens and Morrill 2011). A consensus has emerged that distance-based phylogenetic methods (e.g., neighbour joining) and small character numbers (under 500 nucleotide positions; see Wiens and Morrill 2011) may contribute to inaccuracies in phylogenetic estimations. Even so, maximum likelihood and Bayesian phylogenetic reconstructions have been shown to be robust even in the presence of large amounts of missing data (up to 90 %; Wiens 2003) when the overall number of characters is large enough (e.g., 2000; Wiens and Moen 2008). Since our own dataset (total length of used alignment: 8978) substantially exceeds the total number of nucleotides proposed by Wiens and Moen (2008) and our data inclusion threshold for taxa with missing loci leads to concatenated sequences well above 500 nucleotides we included them in our dataset. This is consistent with evidence showing that such sequences can not only be placed accurately but also make a net positive contribution to phylogenetic analyses (Wiens and Tiu 2012; Jiang et al. 2014).
The practical interpretation of our results extends to two areas. The first of these concerns evolutionary relationships, regardless of how they are named. A legitimate question is that if our study explicitly rules out topologies acquired in past studies, how can we be sure that our own topology is not overtaken by more sampling? Our approach to this is to restrict our interpretation to relationships that are supported and/or for which alternative hypotheses can be rejected. The limitations of this approach are that it is easier to identify and reject unsupported hypotheses than propose practical resolutions for relationships that continue to be unresolved. This is exemplified by the way our data can be mined to reject or not reject sister group relationships of the trapelioids. Miadłikowska et al. (2014) argued that groups such as Arctomiales and Hymeneliales, though themselves poorly supported in their analyses, deserve recognition because they are “flanked” by well supported monophyletic groups. This explicitly assumes that the “order of divergence” of clades (“following the evolutionary split of Baeomycetales and preceding the split of Trapeliales”) is significant, despite lack of statistical support at the corresponding nodes. Our testing of the Miadłikowska et al. (2014) “order of divergence” hypothesis shows it is not only unsupported but in fact impossible to obtain given our data set (p = 0). Similarly we could reject other “sister group, but unsupported” relationships, such as that of trapelioids to Ostropales, which have been obtained several times in the literature. What we cannot do given our current data, however, is establish the sister group relationship of trapelioids with certainty. Several lines of evidence suggest molecular data will ultimately establish a statistically sound link between trapelioids and one or more of the three families in the BAH clade (see Results).
The second area affected by our results is how to name the orders of Ostropomycetidae. The “order of divergence” mentioned above was also inferred to be taxonomically consequential and led to erection of two orders, which under recent classification schemes makes for no fewer than five orders in Ostropomycetidae that consist of only a single family each (Arctomiales, Arctomiaceae; Baeomycetales, Baeomycetaceae; Hymeneliales, Hymeneliaceae; Sarrameanales, Sarrameanaceae; and Trapeliales, Trapeliaceae). No universal rules stipulate what constitutes an order, and there is no single correct solution. Nor is a solution necessary; the Code of Nomenclature explicitly provides for taxa of uncertain position (Art. 3.1, note 1; McNeill et al. 2012). However, we note that we recover a topological hypothesis in which Arctomiaceae, Baeomycetaceae and Hymeneliaceae form a single clade, an hypothesis that at the same time is unsupported and cannot be rejected with our data. All of these have been recently recognized as orders in their own right. We could, theoretically, adopt the name Trapeliales for the trapelioids (Trapeliaceae/Xylographaceae) as proposed by Hodkinson and Lendemer (2011), as it constitutes a third major monophyletic group in Ostropomycetidae following Ostropales and Pertusariales. The alternative hypothesis, that trapelioids from a natural phylogenetic group with the BAH clade, cannot be rejected using our data. Because no other hypothesis receives greater support, we adopt a broad view of the unresolved relationships in this sector of Ostropomycetidae and for practical reasons will treat all five families as Baeomycetales s.lat.. We note that this solution is similar to those of two earlier phylogenies (Wedin et al. 2005; Lumbsch et al. 2007a, b), and broadly consistent with long-running anatomy-based hypotheses (e.g., Hertel 1970; Lumbsch et al. 1995; Lumbsch 1997). A similar problem, though with fewer possible solutions, is whether to subsume the family Rimulariaceae (Hertel and Rambold 1990), typified through Rimularia s.str., into Trapeliaceae. We know little about what additional diversity may be uncovered in the clade we here call Rimularia s.str., and currently the genus is reciprocally monophyletic to the rest of Trapeliaceae. A family Rimulariaceae would have no apomorphies following the removal of Lambiella and Lithographa, which were originally included in it (Hertel and Rambold 1990), and we see little reason to maintain it as distinct from Trapeliaceae.
Another area that could affect future phylogenies is taxon sampling. Changes in taxon sampling can mediate large changes in inference, especially when the added taxa represent evolutionary “missing links” (Wiens and Tiu 2012). Specific to resolving the evolutionary relationships of trapelioid fungi, we were unable to sample several genera and species groups that may contribute to resolving future trees. Both Amylora (Rambold 1994) and Coppinsia (Lumbsch and Heibel 1998) are currently placed in Trapeliaceae (Lumbsch and Huhndorf 2010) but we could not obtain fresh material for sequencing. Ascus anatomy and thalline amyloidy suggest that Amylora in particular may represent a link between Hymeneliaceae and the Trapeliaceae/Xylographaceae clade; Rambold (1994) noted similarities to Rimularia in the broad sense. Another group of interest is the recently described genus Cameronia, the two species of which have muriform ascospores (Kantvilas 2012). We examined Cameronia ITS and a short fragment of mtSSU from a previous study (Lumbsch et al. 2012) but ultimately excluded it from our final analysis because it was below our data inclusion threshold. However, our initial results suggest Cameronia is related to Hymeneliaceae (data not shown). Another group that needs better sampling is Lithographa. This genus is heterogeneous as currently circumscribed and also includes two species with (sub)muriform ascospores (Fryday and Coppins 2007). However, many of the unsampled species of Lithographa occur in remote regions of the southern hemisphere. Finally, the origins of the cyanobacterium-associated Arctomiaceae will be especially interesting to clarify given the rare nature of this symbiosis in Ostropomycetidae, and the discovery of disparate lineages found to belong there (Otálora and Wedin 2013; Spribille and Muggia 2013).
Taxonomic changes: new synonyms and combinations
Baeomycetaceae Dumort., Anal. fam. pl. (Tournay): 71 (1829) (MB80510)
= Anamylopsoraceae Lumbsch & Lunke in Lumbsch et al., Pl. Syst. Evol. 198: 285 (1995), syn. nov. (MB81979)
Trapeliaceae M. Choisy ex Hertel, Deutsche Bot. Ges., N.F. 4: 181 (1970) (MB81480)
= Rimulariaceae Hafellner, Nova Hedw. Beih. 79: 331 (1984), syn. nov. (MB81354)
Xylographaceae Tuck., Synopsis N. Am. Lich. Part II: 110 (1888) (MB81529)
= Lithographaceae Poelt, Ahmadjian & Hale, The Lichens: 626 (1974), nom. inval. Articles 36.1, 39.1 (MB81651)
The relationship between Lambiella, Lithographa, Ptychographa and Xylographa repeats the pattern recovered by Spribille et al. (2014); our present sampling further shows a highly supported sister group relationship of core genera of Trapeliaceae. In his original circumscription of Xylographaceae, Tuckerman (1888, as the family “Xylographei”) included the genera Agyrium and Xylographa as united by an “innate” (immersed) thallus and rounded to lirellate fruiting bodies that are pale to blackening (Tuckerman 1888). Watson (1929) interpreted the family to include Lithographa, Ptychographa and Encephalographa (now recognized as an Arthoniomycete) and dropped the inclusion of Agyrium. The family has otherwise seldom been used. We propose resurrecting the family Xylographaceae as distinct from Trapeliaceae to accommodate the genera Lambiella, Lithographa and Ptychographa and Xylographa (Agyrium has been established to be not closely related, Lumbsch et al. 2007a). Hertel (1970) already recognized the differences of the thick walled ascus with a well developed tholus of Xylographa and that of Trapeliaceae concluding that no close relationship exists between these groups. Here we show that Xylographaceae also includes round to broadly angular fruiting bodies (Lambiella). Our phylogenetic results further allow us to reject the proposal by Poelt (1974) that lirellate species with carbonized excipula, which he called Lithographaceae, are isolated from the lirellate genus Xylographa (which he placed in Agyriaceae).
The new genus Parainoa
As discussed above, Trapeliopsis subconcolor is more closely related to Ainoa than to Trapeliopsis, but it also does not cluster with Ainoa and differs from the latter in the production of depsidones, rather than tridepsides, as secondary metabolites. This species has been classified in both Trapelia (Hertel 1973) and Trapeliopsis (Hertel 1981) and the latter classification, and its relatedness to the east Asian T. hainanensis, was last considered to be beyond doubt (Hertel 1981). Though described from northern Italy (Anzi 1862), recent material of T. subconcolor has not been reported to our knowledge from Europe and the species appears to have two centres of distribution in south and east Asia and the Neotropics (Hertel 1977). T. subconcolor was compared to Ainoa (as Trapelia mooreana) by Hertel (1977) and differs in its creamish white papillate thallus (orangish and smooth in Ainoa), the more yellowish hypothecium, the conglutinated paraphyses and presence of stictic acid in the thallus, as opposed to gyrophoric acid and associated substances in Ainoa species (Hertel and Leuckert 1969, as Trapelia torellii). In T. subconcolor, gyrophoric acid may be present or absent in the ascomata (Hertel 1977). An SOWH test rejects the hypothesis of monophyly of Ainoa and T. subconcolor with p = 0 (we did not test for monophyly of Trapeliopsis subconcolor with Trapeliopsis because our topology leaves little room for that hypothesis). We refrain from combining the above-mentioned T. hainanensis into Parainoa at this time as study of two isotypes (Hertel, Lecideaceae Exs. 59, GZU!, PRA!) shows an exciple structure of tightly interwoven hyphae reaching almost to the surface of the exciple, and the presence of an incipient “stalk” in the hypothecium, recalling Baeomyces. The species needs further study and preferably also DNA work to compare it to Baeomyces s.lat. but we doubt it is congeneric with Parainoa subconcolor.
Parainoa Resl & T. Sprib., gen. nov. (MB810870)
Similar to Ainoa but differing in containing depsidones, similar to Baeomyces but differing in the complete lack of a differentiated, extended hypothecial stalk for the ascoma.
Typus generis: Parainoa subconcolor (Anzi) Resl & T. Sprib., comb. nov. (MB810871). Basionym: Biatora subconcolor Anzi, Comm. Soc. Crittogam. Ital. 1(3): 151 (1862). Type: ITALY. Prov. Sondrio, in castanetis inter pagum Rodolo et prata della Corna, Anzi, Lich. Langob. 163 (M, lectotype, FH, isolectotype, studied by Hertel 1977). Thin layer chromatography revealed stictic acid in four specimens (Arnold Lich. Exs. 938, GZU; Palice 8354, F; Palice 8600, F; and Aptroot 55,969, PRA).
Etymology: a nod to its past confusion with and occurrence near Ainoa in our phylogenetic hypothesis; also with reference to the problematic specimen of P. subconcolor from the Andean Páramo that was long confused with Ainoa (see Discussion).
New combinations in Lambiella
With the following new combinations Lambiella contains 10 species compared to four confirmed for Rimularia using molecular methods. A posteriori analysis confirms some differences between Rimularia s.str. and Lambiella, notably the development of depsidones in Lambiella. Ascus apical apparatus may also differ between the two groups of species, with all species of Rimularia s.str. developing a thin, vertical, non-amyloid tube that is absent in Lambiella species (Hertel and Rambold 1990). Although both genera occur on bare acidic rock, Lambiella so far accounts for all cases of occurrence on other substrates. We acknowledge our present analysis leaves 18 mostly southern hemisphere species of Rimularia s.lat. in limbo, but the status of most of these species cannot be easily resolved without a detailed taxonomic study of the entire group and acquisition of fresh material, often of very rare species. The task is complicated by the possibility that several species, especially R. subconcava from Central Asia and R. michoacanensis from Mexico (Timdal 2002), may not belong to either clade in the strict sense.
Lambiella caeca (J. Lowe) Resl & T. Sprib., comb. nov. (MB810862) Basionym: Lecidea caeca J. Lowe, Lloydia 2: 245 (1939). ≡ Rimularia caeca (J. Lowe) Rambold & Printzen, Mycotaxon 44: 454 (1992).
Lambiella furvella (Nyl. ex Mudd) M. Westb. & Resl, comb. nov. (MB810863) Basionym: Lecidea furvella Nyl. ex Mudd, Brit. Lich.: 207 (1861). ≡ Rimularia furvella (Nyl. ex Mudd) Hertel & Rambold, Mitt. Bot. Staatssamml. München 23: 391 (1987).
Lambiella fuscosora (Muhr & Tønsberg) M. Westb. & Resl, comb. nov. (MB810864) Basionym: Rimularia fuscosora Muhr & Tønsberg, Nordic J. Bot. 8: 649 (1989).
Lambiella globulosa (Coppins) M. Westb. & Resl, comb. nov. (MB810865) Basionym: Rimularia globulosa Coppins, Bibl. Lich. 78: 45 (2001).
Lambiella gyrizans (Nyl.) M. Westb. & Resl, comb. nov. (MB810866) Basionym: Lecidea gyrizans Nyl., Not. Sällsk. Fauna Fl. Fenn. Förh., n.s., 2: 231 (1861). ≡ Rimularia gyrizans (Nyl.) Hertel & Rambold, Bibl. Lich. 38: 173 (1990).
Lambiella hepaticola (Kantvilas & Coppins) Resl & T. Sprib., comb. nov. (MB810867) Basionym: Rimularia hepaticola Kantvilas & Coppins, Bibl. Lich. 78: 41 (2001). Recently acquired DNA sequence data (not shown) confirm that this species belongs in Lambiella.
Lambiella impavida (Th.Fr.) M. Westb. & Resl, comb. nov. (MB810868) Basionym: Lecidea impavida Th.Fr., Kongl. Svenska Vetensk. Acad. Handl. ser. 2, 7(2): 42 (1867). ≡ Rimularia impavida (Th.Fr.) Hertel & Rambold, Mitt. Bot. Staatssamml. München 23: 391 (1987).
Lambiella sphacelata (Th.Fr.) M. Westb. & Resl, comb. nov. (MB810869) Basionym: Lecidea sphacelata Th.Fr., Lichenogr. Scand. 2: 445 (1874). ≡ Rimularia sphacelata (Th.Fr.) Hertel & Rambold, Bibl. Lich. 38: 185 (1990).
References
Anzi M (1862) Manipulus lichenum variorum vel novorum, quos in Langobardia et Etruria collegit et enumeravit. Commentario della Societa Crittogamologica Italiana 1:130–166
Aptroot A, Schumm F (2012) A new terricolous Trapelia and a new Trapeliopsis (Trapeliaceae, Baeomycetales) from Macaronesia. Lichenologist 44:449–456
Baloch E, Lücking R, Lumbsch HT, Wedin M (2010) Major clades and phylogenetic relationships between lichenized and non-lichenized lineages in Ostropales (Ascomycota: Lecanoromycetes). Taxon 59:1483–1494
Bendiksby M, Timdal E (2013) Molecular phylogenetics and taxonomy of Hypocenomyce sensu lato (Ascomycota, Lecanoromycetes) - extreme polyphyly and morphological convergence. Taxon 62:940–956
Church SH, Ryan JF, Dunn CW (2014) Automation and evaluation of the SOWH test of phylogenetic topologies with SOWHAT. BioRxiv. doi:10.1101/005264
Coppins BJ, James PW (1984) New or interesting British lichens V. Lichenologist 16:241–264
Culberson CF (1969) Chemical and botanical guide to lichen products. University of North Carolina Press, Chapel Hill
R Development Core Team (2013) R: a language and environment for statistical computing, reference index version 3.0.2. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org
Dunn KA, McEachran JD, Honeycutt RL (2003) Molecular phylogenetics of myliobatiform fishes (Chondrichthyes: Myliobatiformes), with comments on the effects of missing data on parsimony and likelihood. Mol Phylogenet Evol 27:259–270
Fryday AM, Coppins BJ (2007) A second species of Lithographa with submuriform ascospores. Lichenologist 39:245–250
Galloway DJ (2007) Flora of New Zealand lichens, Revised second edition. Manaaki Whenua Press, Lincoln, New Zealand
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes –application to the identification of mycorrhizae and rusts. Mol Ecol 2(2):113–118
Gargas A, Taylor JW (1992) Polymerase chain reaction (PCR) primers for amplifying and sequencing 18S rDNA from lichenized fungi. Mycologia 84:589–592
Goldman N, Anderson JP, Rodrigo AG (2000) Likelihood-based tests of topologies in phylogenetics. Syst Biol 49:652–670
Hackett SJ et al (2008) A phylogenomic study of birds reveals their evolutionary history. Science 320:1763–1768
Hafellner J (1984) Studien in Richtung einer natürlicheren Gliederung der Sammelfamilien Lecanoraceae und Lecideaceae. In: Hertel H, Oberwinkler F (eds) Beiträge zur Lichenologie. Festschrift J. Poelt, vol 79, Beiheft zur Nova Hedwigia. J. Cramer, Vaduz, pp 241–371
Hertel H (1970) Trapeliaceae - eine neue Flechtenfamilie. Vorträge aus dem Gesamtgebiet der Botanik, N.F. Deut Bot Ges 4:171–185
Hertel H (1973) Beiträge zur Kenntnis der Flechtenfamilie Lecideaceae V. Herzogia 2:479–515
Hertel H (1977) Gesteinsbewohnende Arten der Sammelgattung Lecidea (Lichenes) aus Zentral-, Ost- und Südasien. Khumbu Himal 6(3):145–378
Hertel H (1981) Beiträge zur Kenntnis der Flechtenfamilie Lecideaceae VIII. Herzogia 5:449–463
Hertel H (1984) Über saxicole, lecideoide Flechten der Subantarktis. In: Hertel H, Oberwinkler F (eds) Beiträge zur Lichenologie. Festschrift J. Poelt, vol 79, Beiheft zur Nova Hedwigia. J. Cramer, Vaduz, pp 399–499
Hertel H, Leuckert C (1969) Über Flechtenstoffe und Systematik einiger Arten der Gattungen Lecidea, Placopsis und Trapelia mit C+ rot reagierendem Thallus. Willdenowia 5:369–383
Hertel H, Rambold G (1990) Zur Kenntnis der Familie Rimulariaceae (Lecanorales). Bibl Lichenologica 38:145–189
Hodkinson BP, Lendemer JC (2011) The orders of Ostropomycetidae (Lecanoromycetes, Ascomycota): recognition of Sarrameanales and Trapeliales with a request to retain Pertusariales over Agyriales. Phytologia 93:407–412
Hofstetter V, Miadlikowska J, Kauff F, Lutzoni F (2007) Phylogenetic comparison of protein-coding versus ribosomal RNA-coding sequence data: A case study of the Lecanoromycetes (Ascomycota). Mol Phylogenet Evol 44:412–426
Jahns HM (1982) The cyclic development of mosses and the lichen Baeomyces rufus in an ecosystem. Lichenologist 14:261–265
Jiang W, Chen SY, Wang H, Wiens JJ (2014) Should genes with missing data be excluded from phylogenetic analyses? Mol Phylogenet Evol 80:308–318
Kantvilas G (2012) Cameronia (lichenized Ascomycetes), a remarkable new alpine genus from Tasmania. Lichenologist 44:91–100
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kauff F, Lutzoni F (2002) Phylogeny of the Gyalectales and Ostropales (Ascomycota, Fungi): among and within order relationships based on nuclear ribosomal RNA small and large subunits. Mol Phylogenet Evol 25:138–156
Kauff F, Lutzoni M (2003) Compat.py - a program to detect topological conflict between supported clades in phylogenetic trees, available online at http://www.lutzonilab.net/pages/download.shtml. Accessed 10 October 2014
Lanfear R, Calcott B, Ho SY, Guindon S (2012) Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Letrouit-Galinou MA (1966) Recherches sur l’ontogenie et l’anatomie comparées des apothécies de quelques Discolichens. Revue Bryologique et Lichenologique 34:413–588
Letrouit-Galinou MA (1968) The apothecia of the discolichens. Bryologist 71:297–327
Liu YJ, Whelen S, Hall BD (1999) Phylogenetic relationships among ascomycetes: evidence from an RNA polymerase II subunit. Mol Biol Evol 16:1799–1808
Lumbsch HT (1997) Systematic studies in the suborder Agyriineae (Lecanorales). J Hattori Bot Lab 83:1–73
Lumbsch HT, Heibel E (1998) Coppinsia minutissima, a new genus and species in the Agyriaceae from the British Isles. Lichenologist 30:95–101
Lumbsch HT, Huhndorf SM (2010) Myconet volume 14. Part One. Outline of Ascomycota—2009. Part Two. Notes on ascomycete systematics. Nos. 4751—5113. Fieldiana Life Earth Sci 1:1–64
Lumbsch HT, Lunke T, Feige GB, Huneck S (1995) Anamylopsoraceae - a new family of lichenized ascomycetes with stipitate apothecia (Lecanorales: Agyriineae). Plant Syst Evol 198:275–286
Lumbsch HT, Schmitt I, Döring H, Wedin M (2001a) Molecular systematics supports the recognition of an additional order of Ascomycota: the Agyriales. Mycol Res 105:16–23
Lumbsch HT, Schmitt I, Döring H, Wedin M (2001b) ITS sequence data suggest variability of ascus types and support ontogenetic characters as phylogenetic discriminators in the Agyriales (Ascomycota). Mycol Res 105:265–274
Lumbsch HT, del Prado R, Kantvilas G (2005) Gregorella, a new genus to accommodate Moelleropsis humida and a molecular phylogeny of Arctomiaceae. Lichenologist 37:291–302
Lumbsch HT, Schmitt I, Mangold A, Wedin M (2007a) Ascus types are phylogenetically misleading in Trapeliaceae and Agyriaceae (Ostropomycetidae, Ascomycota). Mycol Res 111:1133–1141. doi:10.1016/j.mycres.2007.06.016
Lumbsch HT, Schmitt I, Lücking R, Wiklund E, Wedin M (2007b) The phylogenetic placement of Ostropales within Lecanoromycetes (Ascomycota) revisited. Mycol Res 111:257–267
Lumbsch HT, Archer AW, Elix JA (2007c) A new species of Loxospora (lichenized Ascomycota: Sarrameanaceae) from Australia. Lichenologist 39:509–517
Lumbsch HT, Kantvilas G, Parnmen S (2012) Molecular data support placement of Cameronia in Ostropomycetidae (Lecanoromycetes, Ascomycota). MycoKeys 5:31–44
Mangold A, Martin MP, Lücking R, Lumbsch HT (2008) Molecular phylogeny suggests synonymy of Thelotremataceae within Graphidaceae (Ascomycota: Ostropales). Taxon 57:476–486
Matheny PB, Liu YJ, Ammirati JF, Hall BD (2002) Using RPB1 sequences to improve phylogenetic inference among mushrooms (Inocybe, Agaricales). Amer J Bot 89:688–698
McCune B, Camacho F, Ponzetti J (2002) Three new species of Trapeliopsis on soil in western North America. Bryologist 105:78–85
McNeill J (2012) International code of nomenclature for algae, fungi, and plants (Melbourne code). Regnum Vegetabile 154. Koeltz Scientific Books, Koenigstein
Miadłikowska J, Lutzoni F (2004) Phylogenetic classification of peltigeralean fungi (Peltigerales, Ascomycota) based on ribosomal RNA small and large subunits. Am J Bot 91:449–464
Miadłikowska J et al (2006) New insights into classification and evolution of the Lecanoromycetes (Pezizomycotina, Ascomycota) from phylogenetic analyses of three ribosomal RNA- and two protein-coding genes. Mycologia 98:1088–1103
Miadłikowska J et al (2014) A multigene phylogenetic synthesis for the class Lecanoromycetes (Ascomycota): 1307 fungi representing 1139 infrageneric taxa, 317 genera and 66 families. Mol Phylogenet Evol 79:132–168
Nordin A, Savić S, Tibell L (2010) Phylogeny and taxonomy of Aspicilia and Megasporaceae. Mycologia 102:1339–1349
Otálora M, Wedin M (2013) Collema fasciculare belongs in Arctomiaceae. Lichenologist 45:295–304
Palice Z, Printzen C (2004) Genetic variability in tropical and temperate populations of Trapeliopsis glaucolepidea: evidence against long-range dispersal in a lichen with disjunct distribution. Mycotaxon 90:43–54
Poelt J (1974) Classification. In: Ahmadjian V, Hale ME (eds) The lichens. Academic, New York and London, pp 599–632
Prieto M, Wedin M (2013) Dating the diversification of the major lineages of Ascomycota (Fungi). PLoS One 8(6), e65576
Prieto M, Baloch E, Tehler A, Wedin M (2013) Mazaedium evolution in the Ascomycota (Fungi) and the classification of mazaediate groups of formerly unclear relationship. Cladistics 29:296–308
Printzen C, McCune B (2004) Trapeliopsis. In: Nash TH III, Ryan BD, Diederich P, Gries C, Bungartz F (eds) Lichen Flora of the Greater Sonoran Desert Region, vol 2, Lichens Unlimited. Arizona State University, Tempe, Arizona, pp 538–541
Purvis OW, Smith CW (2009) Trapeliopsis. In: Smith CW, Aptroot A, Coppins BJ, Fletcher A, Gilbert OL, James PW, Wolseley PA (eds) The lichens of Great Britain and Ireland. The British Lichen Society, London, pp 908–910
Raggio J, Green TGA, Crittenden PD, Pintado A, Vivas M, Pérez-Ortega S, de los Ríos A, Sancho LG (2012) Comparative ecophysiology of three Placopsis species, pioneer lichens in recently exposed Chilean glacial forelands. Symbiosis 56:55–66
Rambaut A, Grassly NC (1997) Seq-Gen: an application for the monte carlo simulation of DNA sequence evolution along phylogenetic trees. Comput Appl Biosci 13:235–238
Rambaut A, Suchard MA, Xie D, Drummond AJ (2014) Tracer v1.6, Available from http://beast.bio.ed.ac.uk/Tracer. Accessed 07 November 2014
Rambold G (1994) Amylora, a new genus in the Rimulariaceae. Bull Soc Linnéenne Provence 45:343–348
Reeb V, Lutzoni F, Roux C (2004) Contribution of RPB2 to multilocus phylogenetic studies of the euascomycetes (Pezizomycotina, Fungi) with special emphasis on the lichen-forming Acarosporaceae and evolution of polyspory. Mol Phylogenet Evol 32:1036–1060
Rehner SA, Buckley E (2005) A Beauveria phylogeny inferred from nuclear ITS and EF1-α sequences: evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 97:84–98. doi:10.3852/mycologia.97.1.84
Resl P (2015) phylo-scripts: python scripts for phylogenetics. release v0.1. available at: http://github.com/reslp. doi: 10.5281/zenodo.15983
Rivas Plata E, Parnmen S, Staiger B, Mangold A, Frisch A, Weerakoon G, Hernández MJE, Cáceres MES, Kalb K, Sipman HJM, Common RS, Nelsen MP, Lücking R, Lumbsch HT (2013) A molecular phylogeny of Graphidaceae (Ascomycota, Lecanoromycetes, Ostropales) including 428 species. MycoKeys 6:55–94
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Scheidegger C (1985) Systematische Studien zur Krustenflechte Anzina carneonivea (Trapeliaceae, Lecanorales). Nova Hedwigia 41:191–218
Schmitt I, Lumbsch HT (2004) Molecular phylogeny of the Pertusariaceae supports secondary chemistry as an important systematic character set in lichen-forming ascomycetes. Mol Phylogenet Evol 33:43–55
Schmitt I, Lumbsch HT, Søchting U (2003) Phylogeny of the lichen genus Placopsis and its allies based on Bayesian analyses of nuclear and mitochondrial sequences. Mycologia 95:827–835
Schmitt I, Mueller G, Lumbsch HT (2005) Ascoma morphology is homoplaseous and phylogenetically misleading in some pyrenocarpous lichens. Mycologia 97:362–374
Schmitt I, Yamamoto Y, Lumbsch HT (2006) Phylogeny of Pertusariales (Ascomycotina): resurrection of Ochrolechiaceae and new circumscription of Megasporaceae. J Hattori Bot Lab 100:753–764
Schmitt I, Crespo A, Divakar PK, Fankhauser JD, Herman-Sackett E, Kalb K, Nelsen MP, Nelson NA, Rivas-Plata E, Shimp AD, Widhelm T, Lumbsch HT (2009) New primers for single-copy genes useful in fungal phylogenetics and systematics. Persoonia 23:35–40
Schmitt I, Fankhauser JD, Sweeney K, Spribille T, Kalb K, Lumbsch HT (2010) Gyalectoid Pertusaria species form a sister-clade to Coccotrema (Ostropomycetidae, Ascomycota) and comprise the new lichen genus Gyalectaria. Mycology 1:75–83
Schneider K, Resl P, Westberg M, Spribille T (2015) A new, highly effective primer pair to exclude algae when amplifying nuclear large ribosomal subunit (LSU) DNA from lichens. Lichenologist (in press)
Spribille T, Muggia L (2013) Expanded taxon sampling disentangles evolutionary relationships and reveals a new family in Peltigerales (Lecanoromycetidae, Ascomycota). Fungal Divers 58:171–184
Spribille T, Thor G, Bunnell FL, Goward T, Björk CR (2008) Lichens on dead wood: species-substrate relationships in the epiphytic lichen floras of the Pacific Northwest and Fennoscandia. Ecography 31:741–750
Spribille T, Resl P, Ahti T, Pérez-Ortega S, Tønsberg T, Mayrhofer H, Lumbsch HT (2014) Molecular systematics of the wood-inhabiting, lichen-forming genus Xylographa (Baeomycetales, Ostropomycetidae) with eight new species. Symbolae Botanicae Upsalienses 37(1):1–87
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. doi:10.1093/bioinformatics/btu033
Suzuki Y, Glazko GV, Nei M (2002) Overcredibility of molecular phylogenies obtained by Bayesian phylogenetics. Proc Natl Acad Sci U S A 99:16138–16143
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996) Phylogenetic inference. In: Hillis DM, Motitz C, Mable BK (eds) Molecular systematics, 2nd edn. Sinauer Associates, Sunderland, MA, pp 407–514
Timdal E (2002) Three squamulose species of Rimularia (Lecanorales). Bryologist 105:219–224
Tuckerman E (1888) A synopsis of the North American lichens. Part. II. Comprising the Lecideacei, and (in part) the Graphidacei. E. Anthony & Sons, New Bedford, MA. 176 pp
Ullmann I, Bannister JM, Bannister P (2007) The Placopsis trachyderma-Raoulia-community, a special type of biological soil crusts in the braided rivers of southern New Zealand. Flora 202:687–694
Vilgalys R, Hester M (1990) Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J Bacteriol 172:4238–4246
Voytsekhovich A, Dymytrova L, Nadyeina O (2011) Photobiont composition of some taxa of the genera Micarea and Placynthiella (Lecanoromycetes, lichenized Ascomycota) from Ukraine. Folia Cryptogamica Estonica 48:135–148
Watson W (1929) The classification of lichens. Part II. New Phytol 28:85–116
Wedin M, Wiklund E, Crewe A, Döring H, Ekman S, Nyberg Å, Schmitt I, Lumbsch HT (2005) Phylogenetic relationships of Lecanoromycetes (Ascomycota) as revealed by analyses of mtSSU and nLSU rDNA sequence data. Mycol Res 109:159–172
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sinsky JJ, White TJ (eds) PCR Protocols: a guide to Methods and Applications. Academic Press New York, pp 315–322
Widhelm T, Lumbsch HT (2011) The phylogenetic placement of Miltideaceae inferred from ribosomal DNA sequence data. Bibl Lichenologica 106:365–373
Wiens JJ (2003) Missing data, incomplete taxa, and phylogenetic accuracy. Syst Biol 52:528–538
Wiens JJ, Moen DS (2008) Missing data and the accuracy of Bayesian phylogenetics. J Syst Evol 46:307–314
Wiens JJ, Morrill MC (2011) Missing data in phylogenetic analysis: reconciling results from simulations and empirical data. Syst Biol 60:719–731
Wiens JJ, Tiu J (2012) Highly incomplete taxa can rescue phylogenetic analyses from the negative impacts of limited taxon sampling. PLoS One 7(8), e42925. doi:10.1371/journal.pone.0042925
Wilgenbusch JC, Warren DL, Swofford DL (2004) AWTY: a system for graphical exploration of MCMC convergence in Bayesian phylogenetic inference. http://ceb.csit.fsu.edu/awty. Accessed 06 November 2014
Zoller S, Scheidegger C, Sperisen C (1999) PCR primers for the amplification of mitochondrial small subunit ribosomal DNA of lichen-forming ascomycetes. Lichenologist 31:511–516
Acknowledgments
We would like to thank the numerous individuals who provided specimens for DNA sequencing for this study, including A. Acton, A. Aptroot, C. Björk, B. Coppins, G. Kantvilas, J. McCarthy, B. McCune, L. Muggia, O. Peksa, S. Pérez-Ortega, T. Tønsberg, P. van den Boom and the curators of F, GZU, O and UPS. Thanks go to Walter Obermayer, Graz, for performing thin layer chromatography on several specimens. Fernando Fernández-Mendoza and Martin Grube provided helpful comments at earlier stages of this project. We also thank Joseph Ryan for help with troubleshooting the SOWHAT analyses. DNA sequencing of Alaskan specimens was funded in part by a materials contribution from the Tongass National Forest, U.S. Department of Agriculture, courtesy of K. Dillman, for which we are grateful. The project was funded by the Austrian Science Foundation (FWF grant P25237, “Evolution of Substrate Specificity in Lichens”). The work by MW was financed by the Swedish Taxonomy Initiative (Svenska Artprojektet, administered by the Swedish Species Information Centre/ArtDatabanken). CP gratefully acknowledges financial support through the program “LOEWE- Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz” of the Hessen Ministry of Higher Education, Research, and the Arts. ZP thanks for the support by the Czech Academy of Science (AV0Z60050516, RVO 67985939) and the Minsitry of Education, Youth and Sports of the Czech Republic.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 5590 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Resl, P., Schneider, K., Westberg, M. et al. Diagnostics for a troubled backbone: testing topological hypotheses of trapelioid lichenized fungi in a large-scale phylogeny of Ostropomycetidae (Lecanoromycetes). Fungal Diversity 73, 239–258 (2015). https://doi.org/10.1007/s13225-015-0332-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-015-0332-y