
Abstract
In this study, three possibly smuggled marble statues of an unknown origin, two human torsi (a female and a male) and a small head, were subjected to molecular analyses. The aim was to reconstruct the history of the storage of each single statue, to infer the possible relationship among them, and to elucidate their geographical shift. A genetic strategy, comprising metagenomic analyses of the 16S ribosomal DNA (rDNA) of prokaryotes, 18S rDNA of eukaryotes, as well as internal transcribed spacer regions of fungi, was performed by using the Ion Torrent sequencing platform. Results suggest a possible common history of storage of the two human torsi; their eukaryotic microbiomes showed similarities comprising many soil-inhabiting organisms, which may indicate storage or burial in land of agricultural soil. For the male torso, it was possible to infer the geographical origin, due to the presence of DNA traces of Taiwania, a tree found only in Asia. The small head displayed differences concerning the eukaryotic community, compared with the other two samples, but showed intriguing similarities with the female torso concerning the bacterial community. Both displayed many halotolerant and halophilic bacteria, which may indicate a longer stay in arid and semi-arid surroundings as well as marine environments. The microbiomes retrieved from each statue showed to be very specific, but some individual members showed to be biological markers for the geographical regions through which the pieces traveled and for the conditions under which they were stored.
Similar content being viewed by others


Introduction
Each piece of art harbors a specific and absolute unique pile of information about its own history. Every step in the history of an object leaves its own genetic information: the plant from which a canvas or paper was made, the organic binder used to prepare a paint, the animal from which a parchment was made, and the hands by which an object was manufactured and through which it was passed during its history (Bower et al. 2010; O’Sullivan et al. 2016). In addition to the information given by the material, each object carries a specific microbiome (Teasdale et al. 2017), mainly consisting of bacteria and fungi. The microflora again can be used as an indicator for the geographical and climatic regions through which a piece of art traveled, the conditions under which it might have been stored, or the soil in which it might have been buried. Microbes—as humans—leave a specific mark on and in an object: their unique fingerprint represented by DNA (Burger et al. 2000).
Nowadays, using high-throughput molecular methods, it is possible to extract and to analyze even smallest traces of DNA from an object, to amplify these fragments, to visualize, and by the help of bioinformatics and international databases, to assign single molecules of DNA to a certain plant, a bacterium, and a fungus or even to humans (Fu et al. 2013). The compilation of these data results in an individual “biological pedigree.” The biochemical and molecular methods applied are the same that are in use in forensics—for example to identify an offender—or in medicine, where the microbiome of the human skin is currently in the focus of research (Schommer and Gallo 2013).
To date, molecular analyses applied on cultural heritage studies have mainly focused on the identification of microbial communities associated with specific biodeterioration phenomena displayed by different materials. Such studies have enabled the implementation of conservative and disinfecting treatments as well as the monitoring during and after the conservation treatments of valuable objects (Sterflinger and Piñar 2013). Recently, the ongoing scientific and technological progress has led to metagenomics, metatranscriptomics, metaproteomics, and metabolomics, which are giving a more complete overview of the present microorganisms, their activity, and the expressed proteins and produced metabolites in a specific environment. These state-of-the-art methodologies are now developing at a very fast pace, and next-generation sequencing (NGS) methods are becoming applicable in the field of cultural heritage (Adamiak et al. 2017, 2018), allowing to answer the two most basic questions in microbial ecology: “who is out there?” and “what are they doing?”
Nevertheless, the biological information contained in pieces of art has the potential to answer many other interesting questions that may arise when dealing with cultural heritage objects. The state-of-the-art genetic analyses can, for instance, provide amazing insights into the geographical origins of artifacts (geolocation), the selection of materials at the time of manufacturing, the storage conditions, the species composition of materials, and the use history of an object (Bower et al. 2010; Teasdale et al. 2017). These answers may help to understand many open questions in a variety of fields, such as archeology, history, restoration, philology, and last but not least, criminology. In some cases, curators and custom officers are confronted with valuable smuggled objects of an unknown origin. In these cases, analyses that could enable to make a grouping of some of the objects, as well as to infer the conditions of their storage and their geographical shift, could help to reconstruct the history of these objects, a kind of time travel.
In this study, three possibly smuggled marble statues of an unknown origin were transitorily deposited in the Museum of Art History (KHM) in Vienna, Austria. Curators and conservators of the museum put many efforts to infer the origin and the possible relationship among the three statues through petrological and morphological analyses, without success. Therefore, they considered the possibility of constituting an interdisciplinary team, recruiting microbiologists and bioinformaticians, in order to include a genetic strategy, which could provide novel insights into these questions. Metagenomic analyses of the 16S ribosomal DNA (rDNA) of prokaryotes, 18S rDNA of eukaryotes, as well as internal transcribed spacer (ITS) regions of fungi were performed by using the Ion Torrent sequencing platform, and the generated data were used to infer the storage history and the geographical shift of the samples, as well as to consider the possible relationship among them.
Material and methods
Sampling
Three statues of unknown origin, two of them representing human torsi, one female (S1) and one male (S2), and the third one representing a small young girl head (S3), were deposited in the KHM, Vienna, Austria (Fig. 1). Samples were taken from the surface of the three statues with the help of sterile scalpels and needles by gently scratching. Approximately 40–60 mg of material, mainly powdered material, dust, dirt, and even some textile fibers, was collected independently from four different points of each statue without damaging the marble stone. The material collected from each point was immediately placed into sterile Eppendorf tubes. Later, in the laboratory, the material collected from each point of the same statue was pooled in order to obtain about 160–250 mg of material for DNA extraction analyses per statue. DNA extraction was performed at the next day of sampling.

The three antique marble statues. a S1, female torso. b S2, male torso. c S3, small head
Quality assurance (QA), in order to avoid any kind of contamination or bias, was performed by using only high-quality reagents for DNA extraction, including single-use manufactured vials to avoid cross-contamination as well as high-quality reagents for down steps, as PCR amplification and sequencing. All reagents used for library preparation and sequencing are tested and single packet/reaction, to avoid contaminations. In addition, the steps of (1) DNA extraction and amplification, (2) the DNA library preparation, and (3) the sequencing were performed in three separated laboratories dedicated only and exclusively to one of the mentioned steps. DNA extraction was performed in a laminar flow bank only used for ancient DNA (aDNA) preparations, and PCR reactions were performed in a PCR workstation equipped with UV lamps.
DNA extraction
Total DNA was isolated directly from the powdered material collected and pooled from each statue (160–250 mg/statue, as mentioned above) using the FastDNA SPIN Kit for soil (MP Biomedicals, Illkirch, France) as recommended by the manufacturers. The DNA concentrations were assessed by using the Qubit 2.0 fluorometer (Thermo Fisher Scientific, MA, USA), which uses fluorescent dyes to determine the concentration of nucleic acids, with the Qubit dsDNA HS Assay Kit.
PCR amplification
All PCR reactions were executed in a Bio-Rad C1000 Thermal Cycler. PCR reactions performed to amplified eukaryotes and fungi were carried out with the 2× PCR Master Mix from Promega (Vienna, Austria) (50 units/ml of Taq DNA Polymerase supplied in an appropriate reaction buffer (pH 8.5), 400 μM dNTPs, 3 mM MgCl2) diluted to one time, to which 12.5 pmol/μl of each primer (stock 50 pmol/μl; VBC-Biotech, Vienna, Austria) and 400 μg/ml BSA (stock 20 mg/ml; Roche Diagnostics GmbH, Basel, Switzerland) were added.
Quality control (QC) was performed in order to exclude the possibility of cross-contamination, and in each PCR reaction, a negative control was included (no DNA template).
For the amplification of the eukaryotic 18S rDNA, fragments targeting the V4 region were amplified with the primers 528F (GCGGTAATTCCAGCTCCAA) and 706R (AATCCRAGAATTTCACCTCT). The thermocycling program was as follows: 5-min denaturation at 95 °C, followed by 35 cycles of 1-min denaturation at 95 °C, 1-min annealing at 53 °C, and 1-min extension at 72 °C. Five-minute denaturation at 72 °C was performed as a final extension step. Reactions were performed in 2× 50 μl, and a 3.5-μl DNA template was added to each reaction tube. The duplicate amplicons of each sample (2× 50 μl) were pooled, and 7 ml was visualized on 2% (w/v) agarose gels, stained in an ethidium bromide solution (10 mg/ml), and documented using a UVP documentation system (Bio-Rad Transilluminator; Universal Hood, CA, USA). The remaining PCR reaction was purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany).
For the amplification of fungal ITS regions, fragments of 450–600 bp, corresponding to the ITS1 and the ITS2 regions, and the 5.8S ribosomal RNA (rRNA) gene situated between them were amplified with the primer pairs ITS1 forward and ITS4 reverse (White et al. 1990). The thermocycling program was as follows: 5-min denaturation at 95 °C, followed by 35 cycles of 1-min denaturation at 95 °C, 1-min annealing at 55 °C, and 1-min extension at 72 °C. Five-minute denaturation at 72 °C was performed as a final extension step. Thereafter, the obtained PCR products were further amplified in a nested PCR using the forward primer ITS1 and the reverse primer ITS2. The cycling scheme was the same as before, but the annealing T was 58 °C, and the reactions were performed in 2× 50 μl volume containing each 3.5 μl of template DNA. The duplicate amplicons of each sample (2× 50 μl) were pooled and visualized as mentioned above. The remaining PCR reaction was purified using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany).
For the amplification of the prokaryotic 16S rDNA, the DNA was amplified using the Ion 16S Metagenomics Kit (Thermo Fisher Scientific, MA, USA). The kit includes two primer sets that selectively amplify the corresponding hypervariable regions of the 16S region in bacteria (primer set V2-4-8 and primer set V3-6,7-9). After amplification, the reactions obtained from each sample with the two primer sets were pooled and mixed with Agencourt AMPure XP reagent (Beckman Coulter, CA, USA) for purification, using a magnetic rack as described by the manufacturers.
The concentration of all the purified amplicons (prokaryotes, fungi, and eukaryotes) was assessed with the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, MA, USA).
Library construction and quantification
DNA libraries were constructed using the Ion Plus Library Kit for the AB Library Builder System (Thermo Fisher Scientific, MA, USA) following the library preparation protocol for short amplicons, provided by the manufacturers. The amplicons were processed as barcoded libraries, by using the P1 adapter and the barcoded A adapters provided in the Ion Xpress Barcode Adapter Kit. The adapters were diluted according to the amount of DNA input, as recommended by the manufacturers.
The resulting fungal and eukaryotic DNA libraries were subsequently quantified by qPCR using the Ion Library Quantitation Kit (Thermo Fisher Scientific, MA, USA) and the prokaryotic DNA libraries using the Ion Universal Library Quantitation Kit (Thermo Fisher Scientific, MA, USA). Reactions were performed as described by the manufacturers, to calculate the dilution factor of each library for a final concentration of 50 pmol. After individual quantitation, the barcoded libraries of each sample (amplified with prokaryote-, eukaryote-, and fungus-specific primers) were pooled in equimolar amounts to ensure equal representation of each barcoded library in the sequencing run.
Template preparation and sequencing
Template preparation was performed by the Ion Chef System (Thermo Fisher Scientific, MA, USA) using the Ion PGM Hi-Q Chef Kit and the sequencing by using the Ion Personal Genome Machine (PGM) using the Ion PGM Hi-Q Sequencing Kit with the Ion 318 Chip v2 following the instructions of the manufacturers (Thermo Fisher Scientific, MA, USA). All barcoded libraries amplified with the prokaryote primer sets were pooled in one chip, and all barcoded libraries amplified with eukaryote- and fungus-specific primers were pooled in a second chip.
Data analyses
Raw reads were trimmed and filtered with cutadapt (Martin 2011). After trimming, only reads longer than 100 nt and containing at least one primer were kept. The prokaryotic reads were first grouped based on their hypervariable region (HVR) of origin. The HVR was inferred by running cmscan (Nawrocki and Eddy 2013) with the prokaryotic 16S rRNA model from Rfam (Kalvari et al. 2018) on each read and comparing the start and end position of the match with the corresponding HVR position in the Rfam model. Reads mapping outside of the HVR were discarded while the other reads were trimmed and filtered with cutadapt similar to what was done for the eukaryotic and fungal reads.
Taxonomy assignment was made with DADA based on the protocol published in Callahan et al. (2016). For the prokaryotic reads, the protocol was applied on each group of HVR separately as well as on all HVR at the same time.
Geographical assignment
For each genus found in the samples, the geographic spread was plotted by fetching information from the Global Biodiversity Information Facility. Geographical assignment of the species was done with the help of the R rgbif package (Chamberlain and Boettiger 2017).
Nucleotide sequence accession number
The nucleotide sequences were deposited in the GenBank database under the accession number PRJNA506995:
-
Statue 1 (SAMN10477674): fungi1 (SRX5063102), RUN: FungiTO1.fastq (SRR8245025); bacteria1 (SRX5063103) RUN: bac1RfamAllClean.fastq (SRR8245024); eukaryote1 (SRX5063105) RUN: Euk4TrimmedOnly.fastq (SRR8245022)
-
Statue 2 (SAMN10477675): fungi2 (SRX5063107), RUN: FungiTO2.fastq (SRR8245020); bacteria2 (SRX5063104) RUN: bac2RfamAllClean.fastq (SRR8245023); eukaryote2 (SRX5063106) RUN: Euk5TrimmedOnly.fastq (SRR8245021)
-
Statue 3 (SAMN10477695): fungi3 (SRX5063108) RUN: FungiTO3.fastq (SRR8245019); bacteria3 (SRX5063109) RUN: bac3RfamAllClean.fastq (SRR8245018); eukaryote3 (SRX5063110) RUN: Euk6TrimmedOnly.fastq (SRR8245017)
Data availability
All data generated or analyzed during this study are included in this published article.
Results and discussion
In this study, we investigated the microbiomes of three antique marble statues of unknown origin. Two different theories arose, the first arguing that the statues might have been found under the rubble of an old building and the second arguing that they could have a criminal background of being smuggled. In order to provide novel insights into the origin of the three statues, we applied a genetic strategy including metagenomic analyses, by using the Ion Torrent sequencing platform, to test the suitability and the potential of the strategy to retrieve the maximal genetic information contained on the surface of the statues. The resulting information was used to reconstruct the history of the storage of each single object, to infer the possible relationship among the three statues, and to elucidate their geographical shift. To this end, DNA libraries, targeting seven variable regions of the 16S rDNA fragments of prokaryotes, as well as a variable region of the 18S rDNA of eukaryotes and the ITS regions of fungi, were performed from each statue and further analyzed. To our knowledge, this is the first metagenomic approach in cultural heritage studies targeting so many regions in one single sample. The results showed to be very complex, and the challenge in this study, as in other “omics” studies, was the interpretation of the results. Bioinformatic pipelines were optimized in order to get results from lower taxonomic levels, as genus and species, what is not always possible when using universal primers.
DNA extraction, yield, and amplification procedure
The DNAs were directly extracted from the pooled powdered material collected from each statue and yielded a concentration of 11.7 ng/μl, 0.054 ng/μl, and 0.5 ng/μl for S1–S3, respectively. DNAs were subjected to PCR analyses using all the three primer sets (prokaryotes, eukaryotes, and fungi) described in the section “Material and methods.” All PCR reactions showed positive results when using the eukaryote-specific primer pair, the fungal ITS-specific primer pair, as well as the Ion 16S Metagenomics Kit.
Sequencing analyses
Two sequencing runs were carried out; the first run was performed with the three pooled barcoded libraries derived from the amplicons obtained with the Ion 16S Metagenomics Kit of all three samples, generating 4,307,464 total reads, with an average of 1,305,936 reads per sample (supplementary Table 1). The second run was performed with the six pooled barcoded libraries derived from the amplicons obtained with eukaryote- and fungus-specific primers of all three samples, generating 4,195,209 total reads, with an average of 585,818 reads per sample (supplementary Table 2).
Eukaryotic community
The DNAs obtained from the three statues were processed for eukaryotic and fungal metagenomic analyses, respectively. The obtained DNA libraries were barcoded and pooled as mentioned in the section “Material and methods” and were sequenced in a single run, generating a total of 830 Mb.
Results derived from the eukaryotic DNA libraries revealed similarities between the two torsi (S1 and S2), both displaying a low biodiversity concerning the amount of detected phyla, namely Ascomycota, Basidiomycota, and Embryophyta (or land plants) (Fig. 2). In contrast, the small head (S3) showed a much higher diversity and some other additional phyla were detected in lower proportions (Figs. 2 and 3a). Even if the main targeted organisms in all three samples were fungi, it is worth noting that those phyla present in lower relative proportions showed to be important biological markers for specific environments.

Relative abundance of the eukaryotic community at the phylum level. S1, female torso; S2, male torso; S3, small head

Venn diagrams showing the OTUS shared by all three samples. S1, female torso; S2, male torso; S3, small head. a OTUs derived from eukaryotic DNA libraries. b OTUs derived from prokaryotic DNA libraries
The phylum Chlorophyta (green algae) accounted for 8% of the eukaryotic sequences retrieved from S3, and this phylum was negligible in the other samples (Fig. 4). Members belonging to three of the four recognized classes within the Chlorophyta were identified in S3: the freshwater or terrestrial Chlorophyceae (97%) and Trebouxiophyceae (3%), as well as the Ulvophyceae (0.5%). The latter group is best known as marine macroalgae (green seaweeds) in coastal ecosystems (Cocquyt et al. 2010). Within the Chlorophyceae class, the genera Heterochlamydomonas (82%), Fasciculochloris (3%), Chlorosarcinopsis (3%), Chlorosarcina (4%), and Chlorococcum (5%) were identified. All of them have been described as terrestrial species (McLean and Trainor 1965), but some of them are also found in freshwater and marine environments and as aerial isolates. Chlorococcum sp. has been previously described as one of the dominant organisms contributing to biodeterioration of stone monuments in Korea (Klochkova et al. 2006). Within the Trebouxiophyceae class, Trebouxia was the dominant genus. Members of this genus are the most widespread photobionts in lichens but are only sporadically found in soil or bark outside of a lichen. They all appear to be desiccation tolerant; i.e., they can survive drying to water contents of below 10% (Carniel et al. 2015) and, thus, are a good indicator for an osmophilic environment.

Krona charts displaying the relative abundance of Embryophyta (land plants) at the genus level. a S1, female torso. b S2, male torso. c S3, small head
The phylum Cercozoa showed to be present only in S3, with a relative abundance (4% of eukaryotes). This phylum embraces numerous ancestrally biciliate zooflagellates, euglyphid, and other amoebae (Cavalier-Smith and Chao 2003). The Cercozoa contribute to a great biomass in soil (Urich et al. 2008) but are also abundant in every marine habitat. A high percentage of these sequences could not be identified at lower taxonomic levels (data not shown). However, 29% of the sequences affiliated with members of the family Cercomonadidae, which are found in soil, salt, brackish, and fresh water (Mylnikov and Karpov 2004); 3% with members of the order Glissomonadida; and 1% with the Marimonadida, namely with the genus Pseudopirsonia, which are marine, naked, ancestrally gliding amoeboflagellates and flagellates (Howe et al. 2011).
Some additional eukaryotic phyla were detected only in S3, as the phyla Ochrophyta and Chytridiomycota (0.3% and 0.8% of total eukaryotic reads, respectively). Even though they were detected in very low proportions, they represented important ecological markers in this sample. The phylum Ochrophyta is a major line of eukaryotes, most of them algae, which are a primary component of plankton (Patterson 1989). Several genera were identified, such as Xanthonema, Spumella, Eustigmatos, and Chlamydomyxa, all reported to be common in fresh waters. The phylum Chytridiomycota, often called chytrids, are mainly aquatic organisms (Barr 1990; Zhang et al. 2016), found in peats, bogs, rivers, ponds, springs, and ditches. The presence of these two phyla in S3 supports the suspect that this sample was in contact with water or even a marine environment.
Other sequences retrieved in very low proportion, but reinforcing the hypothesis that S3 was in contact with water, are sequences related with the Dinophyceae class of the dinoflagellates (0.06% of eukaryotic reads). Dinoflagellates constitute one of the main groups of marine and freshwater protists (Gómez 2012). In terms of a number of species, dinoflagellates are one of the largest groups of marine eukaryotes, and some species are endosymbionts of marine animals. In addition, we detected some parasites, as members of the Ichthyosporea class (0.04% of eukaryotes), namely the genus Ichthyophonus, which species are known to be unicellular eukaryotic parasites of marine and freshwater fishes (Ragan et al. 1996), and members of the Tubulinea of the Amoebozoa (0.03% of eukaryotes), which are free-living amoebae (FLA). Within this group, we identified the genus Vermamoeba, which infects various organisms, as freshwater fishes (Milanez et al. 2017).
The Embryophyta (or land plants) showed to be present in all three samples in relatively low proportions (2%, 1%, and 0.3% of eukaryotes for S1–S3, respectively), but they represented important biological markers. In fact, all samples showed signs of storage in contact with agricultural soils, where the most detected land plants were crop plants.
The order Poales was dominant in all three samples (77%, 65%, and 52% of Embryophyta in S1–S3, respectively) (Fig. 4), and it was possible to identify plants of the genus Aegilops in all samples (48%, 61%, and 32% of Embryophyta in S1–S3, respectively), which are commonly known as goat-grasses. The statue S1 was also in contact with other crop plants, as members of the Zea genus (29%). This genus includes large grasses, such as Zea mays, commonly known as maize or corn, which is one of the most important crops for human societies all over the world. In addition, we could detect in this statue DNA of plants of the Glycine genus (order Fabales, 23%), of which the best-known species is the soybean. The majority of the Glycine species are found only in Australia, but the soybean’s native range is in East Asia.
In S2, we could detect DNA from some other crop plants, such as the genus Triticum, or common wheat (4%), which are widely cultivated for their cereal grains and are a worldwide staple food. In addition, this sample revealed traces of DNA from plants of the genus Capsicum (or peppers) of the family Solanaceae (4%), which are cultivated worldwide, but with the highest production in East Asia. Also DNA of plants of the genus Cucumis was found in this sample (4%), which is the major group of angiosperms and includes gherkins, melons, and cucumbers, as examples.
S3 differed from the other two samples and showed the presence of one of the largest subtribes of temperate grasses, namely plants of the genus Loliinae (20%). In addition, plants of the genus Brassica (order Brassicales, 31%), which are informally known as cabbages, or mustard plants, were detected in this sample. The genus Brassica is known for its important agricultural and horticultural crops and includes species and varieties commonly used for food, such as broccoli, cauliflower, cabbage, choy sum, rutabaga, and turnip, and some seeds used in the production of canola oil and the condiment mustard. The genus is native to Western Europe, the Mediterranean, and temperate regions of Asia. Finally, 17% of sequences of Embryophyta were related to the order Malvales, namely to the genus Malva. This genus consists of about 25–30 species of herbaceous annual, biennial, and perennial plants and is widespread throughout the temperate, subtropical, and tropical regions of Africa, Asia, and Europe.
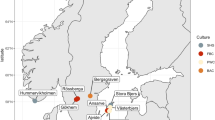
Nevertheless, the most relevant finding when analyzing the DNA of land plants was the relative abundance of DNA (24% of total reads of land plants) of the genus Taiwania in S2. Taiwania, with the single living species Taiwania cryptomerioides, is a large coniferous tree in the cypress family Cupressaceae. It is native and endemic to Eastern Asia, growing in the mountains of central Taiwan and locally in Southwest China, adjoining Myanmar, and northern Vietnam (according to the Global Biodiversity Information Facility). The outliers outside Asia are located in botanical gardens (supplementary Fig. 1). This tree was in very high demand in the past for timber, particularly for temple building and making furniture and coffins (“Chinese coffin tree”). Therefore, the species was decreasing and, nowadays, has legal protection in China and Taiwan (Hayata 1906). The detection of DNA of Taiwania tree on the surface of S2 enabled the geolocation of this statue, as staying or being transported through Asia.
Fungal community
As mentioned in the section “Eukaryotic community,” the main targeted organisms in the eukaryotic 18S rDNA libraries were fungi, accounting for 98% and 99% of the total reads for S1 and S2, respectively, and a lower proportion (86.6%) for S3. The phylum Ascomycota accounted for 92%, 82%, and 86%, while the phylum Basidiomycota represented 6%, 17%, and 0.6% of all reads in the eukaryotic DNA libraries for S1–S3, respectively. It is worth mentioning that data derived from18S rRNA sequence analyses, even if showing a higher diversity of fungi than ITS analyses, are not variable enough to infer identification at taxonomic levels lower than genus. Therefore, analyses of fungal ITS regions were implemented to identify fungal species.
Ascomycota
Within the Ascomycota, it was possible to infer similarities, but also differences at lower taxonomic levels among the three samples (Fig. 5). The order Eurotiales (97%, 93%, and 90% of Ascomycota reads for S1–S3, respectively), with the genus Aspergillus, showed to be dominant in all three samples. ITS analyses enabled to identify fungi at the species level. The species Aspergillus sydowii was present in all three samples. This species is a saprophytic fungus found mainly in soil, but it has been recently proven to be present in dust and indoor environments (Visagie et al. 2014). Aspergillus proliferans was present in S1 and S2; this species has been widely found in indoor environments and food (Chen et al. 2017). Aspergillus penicillioides was present only in S1. This fungus is typically found on substrates with low water activity, such as dried food. It is also known as a causal agent of foxing on paper and books and of brown spots on ancient Egyptian paintings (Koestler 2003). Aspergillus ruber was present only in S2. This fungus has been described as plant pathogen, i.e., in red peppers (Ham et al. 2016), and their presence in S2 may correlate with the presence of plants of the genus Capsicum (or peppers) also detected in this sample. S3 differed from the other two samples, and the dominant species was Aspergillus microcysticus, which was isolated from a Savannah soil in Somalia (Samson et al. 2011). Members of the genus Penicillium were detected surprisingly only in S1, with the species Penicillium glandicola and Penicillium citrinum. These species have been detected growing in association with plants, such as cereals (Vinokurova et al. 2003), which were also identified as dominant land plants in this sample. Nevertheless, both species have been recently identified as causative agents of the biodeterioration on rock surfaces (Ogórek et al. 2016; Boniek et al. 2017).

Relative abundance of the Ascomycota community at the order level. S1, female torso; S2, male torso; S3, small head
The order Onygenales was negligible in S1 and S2 but accounted for 6% of the Ascomycota in S3 (Fig. 5), in which fungal members of the family Gymnoascaceae were all identified. This family includes species of some genera (as Gymnoascus) that have been found in sediments of salt lakes, compost, and cornfield soils and described as halophilic fungi (Zhou et al. 2016). Interestingly, only S3 showed the presence of members of the order Chaetothyriales, even though in a very low proportion (0.03% of Eurotiomycetes class).
Members of the order Sordariales showed to be represented in all three samples (Fig. 5) but dominated in S2 (0.3%, 6%, and 2% of Ascomycota reads for S1–S3, respectively). ITS analyses revealed that all members of this order belonged to the family Chaetomiaceae, with Chaetomium as the dominant genus in S1 and S3. Chaetomium species are capable of colonizing various substrates and are well known for their ability to degrade cellulose. The species Chaetomium murorum was identified in S1; this species has been identified in geographically widely distributed indoor environments (Wang et al. 2016). In contrast, the genus Thielavia with the species Thielavia basicola dominated in S2. This species is a phytopathogenic fungus, known to attack the roots of at least 100 angiosperm plants (Alam et al. 2012), as members of the family Cucurbitaceae, which correlates with some of the land plant sequences retrieved from this sample.
Members of the order Microascales could be identified in S1 and S3 (0.4% and 2% of Ascomycota) but were absent in S2 (Fig. 5). ITS analyses allowed identifying the species Microascus verrucosus in S1. The genus Microascus comprises species commonly isolated from soil, decaying plant material, and indoor environments, but few species are also recognized as opportunistic pathogens.
The order Hypocreales was detected in all three samples, accounting for 0.1%, 0.5%, and 0.3% of Ascomycota (Fig. 5), with Gibberella and Fusarium as the dominant genera. Both genera comprise a large variety of plant pathogens (rice and grain) with worldwide distribution. Gibberella intricans was identified only in S1, which is an opportunistic pathogen of durians (Sivapalan et al. 1998). The durian is the fruit of several tree species belonging to the genus Durio, abundant in Southeast Asia. Fusarium sp. and, in less proportion, Acremonium sp. were identified only in S3. Acremonium contains many species that are saprophytic, which was isolated from dead plant material and soil. The presence of members of the order Hypocreales in all three samples seems to correlate with the presence of plants and as normal inhabitants of soils.
The order Capnodiales was present in S1 and S2 and made up 1% and 0.1% of Ascomycota, respectively (Fig. 5). ITS sequencing allowed to infer lower taxonomic levels and showed differences between the samples. The genus Cladosporium, with species of Cladosporium halotolerans and Cladosporium sphaerospermum, was detected only in S1. Both species have been found in hypersaline aqueous habitats worldwide and are described as halo- or osmotolerant as a recurrent feature. Both species have been isolated from hypersaline water (Zalar et al. 2007). This finding is in line with the many halotolerant-halophilic bacteria detected in this sample. In contrast, the genus Vermiconia of the Extremaceae family, with the species Vermiconia calcicola, was identified in S2. This species is an extremotolerant rock-inhabiting black fungus, which was isolated and described for the first time in marble monuments in Italy (Isola et al. 2016). Surprisingly, in S3, members of the order Capnodiales were detected only when analyzing ITS regions, and two species, namely V. calcicola and Extremus antarcticus, were identified. The latter species was originally isolated from rock samples in the Antarctica (Quaedvlieg et al. 2014) but also isolated and identified as a rock-inhabiting black fungus in Italian monumental sites (Isola et al. 2016).
The order Pleosporales accounted for 0.5%, 0.01%, and 0.3% of Ascomycota in S1–S3, respectively (Fig. 5). Further analyses of ITS regions allowed identifying Alternaria alternata as the dominant species in S1, in addition to Alternaria chlamydospora. Alternaria alternata causes leaf spot and other diseases on over 380 host species of plant. Interestingly, A. chlamydospora is primarily found in salty soils (Mouchacca 1973). Some additional species were identified in S1 in lower proportions, such as Didymella urticicola, Phoma saxea, Stagonosporopsis sp., and Preussia sp. All of them are plant pathogenic, saprobic, and endophytic species (Chen et al. 2015). S2 and S3 showed much less diversity inside this order, being Phoma saxea the only species identified in S2 and as the dominant species in S3.
Deeper analyses of 18S rDNA sequences enabled to detect the presence of other fungal taxa, even if in very low proportions. In this way, members of the order Dothideales represented 0.01% of the Ascomycota in S2; members of the Pezizomycetes class were identified in S1 and S3 (0.3% and 0.07% of Ascomycota, respectively). The genus Pseudombrophila was detected in S1, which is found on dead wood, soil, or rotten plant remains and on substrates often contaminated with urine by animals. In contrast, in S3, all sequences affiliated with the genus Tuber. The true truffles belong to this genus, one of the few ectomycorrhizal Ascomycetes, which are highly valued as delicacies (Mello et al. 2006). In addition, members of the Saccharomycetales were detected in S2 and S3 (0.2% and 0.03% of Ascomycota, respectively). The genus Cyberlindnera with the species Cyberlindnera jadinii was detected only in S2. This fungus has been used since the early 1900s as fodder yeast for livestock and as a dietary supplement for humans.
Basidiomycota
Within the Basidiomycota, the majority of the sequences affiliated with members of the Agaricomycetes class (99% of Basidiomycota for S1 and S2 and 78% for S3, data not shown), in which it was not possible to infer lower taxonomic levels in S1 and S2, but in sample S3. The latter sample showed the presence of relatives of the genera Pleurotus and Pluteus (order Agaricales) commonly called oyster, abalone, or tree mushrooms. In addition, S3 showed that 22% of the Basidiomycota sequences were affiliated with the order Malasseziales (Exobasidiomycetes class), namely with species of the genus Malassezia. This genus comprises yeast species that are part of the normal skin microbiota, but some species can also be involved in skin disorders (Velegraki et al. 2015). Malassezia spp. have not yet been cultured from the environment; nevertheless, metagenomic studies have identified Malassezia phylotypes from marine habitats (Amend 2014).
In addition, other basidiomycetes detected in very low proportion allowed inferring additional differences among the three samples, as members of the Microbotryomycetes class, only detected in S1 (0.6% of Basidiomycota). ITS analyses further enabled to assign these sequences to the genus Rhodotorula, namely to Rhodotorula kratochvilovae. Rhodotorula is common environmental yeast isolated from different ecosystems and environments, including sites with extreme conditions. In contrast, members of the order Ustilaginales (0.5% of Basidiomycota) were detected only in S2. Members of this order are plant pathogens and are known as the smut fungi, which are the causal agent of corn smut disease (Stirnberg and Djamei 2016). Their presence in this sample may be associated with the detected corn plants (Zea).
Prokaryotic community
First, it is worth mentioning that in this study, we applied for the first time in cultural heritage studies a metagenomic strategy implying the amplification of seven of the nine variable regions of the 16S rDNA of prokaryotes, simultaneously. The Ion 16S Metagenomics Kit (Thermo Fisher Scientific) was used. This kit takes advantage of using two primer pools to amplify seven of the nine variable regions of the prokaryotic 16S rDNA (V2, V3, V4, V6, V7, V8, and V9). The results (supplementary Fig. 2) clearly highlighted that some variable regions overestimate specific phyla, as it was very evident for the V9 region, which almost uniquely amplified the phylum Proteobacteria. In contrast, other variable regions underestimated or even completely missed specific phyla, as the V4, which was not targeting the phylum Planctomycetes in S3 (supplementary Fig. 2c). This fact is of great relevance, because the V4 region is the one that is standardly used in most of the metagenomic approaches performed with the Illumina sequencing platform. In summary, these results show that the targeting of specific variable regions of the 16S rDNA gene can influence the results and remark the necessity to target more than one variable region simultaneously, in order to diminish the bias caused when analyzing single hypervariable regions of the 16S rDNA (Fouhy et al. 2016).
All three samples were processed for 16S rDNA metagenomic sequencing, generating a total of 774 Mb. Computational analyses were performed, merging the results obtained from all targeted variable regions, and revealed 22 bacterial phyla in all three marble samples. Nevertheless, only eight phyla showed to count for at least 0.5% of the total reads in at least one sample. At the level of phyla, differences could be observed among the three samples (Figs. 3b and 6); members of the Actinobacteria phylum showed to be dominant in S1, representing 43% of the total sequences and 32% and 29% of total sequences for S2 and S3, respectively. In contrary, the Firmicutes phylum dominated in S2 and S3, with 62% and 55% of the total sequences, respectively, representing 35% of sequences for S1. The Proteobacteria phylum showed to be also representative in all three samples (15%, 6%, and 12% for S1–S3, respectively). Interestingly, the phyla Bacteroidetes and Acidobacteria accounted for 2% and 1% of the total reads only in S1, and the phylum Tectomicrobia accounted for 1% of total reads only in S3. The phylum Chloroflexi accounted for 1% and 0.7% of total reads for S1 and S3, respectively, and the phylum Planctomycetes for 0.7% of total reads in S1 and S3, and the latter phyla were negligible in S2.

Relative abundance of the bacterial community at the phylum level. S1, female torso; S2, male torso; S3, small head
Results derived from prokaryotes supported those obtained from eukaryotes, and sequences derived from all samples showed strong evidence of contact with agricultural soils. All samples showed high abundance of Actinobacteria (Fig. 6). Members of this phylum are widespread in soils throughout the world and are successful colonizers of different extreme environments. However, important differences were observed among the three samples concerning the biodiversity of this phylum (supplementary Fig. 3). S2 showed the lowest biodiversity within the Actinobacteria, and only members of the order Micromonosporales (76% of Actinobacteria), Frankiales (17% of Actinobacteria), and Micrococcales (2% of Actinobacteria) were represented with relatively high abundance in this sample. Within the order Micromonosporales, the genus Micromonospora dominated, accounting for 49% of all Actinobacteria, followed by the genera Plantactinospora (10%) and Verrucosispora (3%). Bacteria belonging to the Micromonospora genus have been widely isolated from mangrove soils, where they represent high populations (Hong et al. 2009). The genus Plantactinospora represents mainly bacteria associated with plant tissues, such as endophytic Actinomycetes which was isolated from Camptotheca acuminata Decne, a Chinese traditional medicine plant endemic to Southwest China (Zhu et al. 2012). Species of the genus Verrucosispora have been isolated from peat bog and deep-sea sediments but also from mangrove sediments in China (Xi et al. 2012). Within the Frankiales, the only detected genus was Blastococcus (16% of Actinobacteria). Bacteria of this genus are commonly isolated from the surface of marble and calcareous stones (Urzì et al. 2004).
In contrast, S1 and S3 showed a much higher biodiversity concerning the phylum Actinobacteria. At least seven orders could be detected within the Actinobacteria class in all two samples with relatively high abundance, at least in one of them. Some orders were present in only one sample, such as the order Corynebacteriales, which made up 23% of the phylum Actinobacteria in S1, which were almost negligible in the other two samples (supplementary Fig. 3). The Corynebacterium_1 group showed to be dominant (17% of Actinobacteria). Members of this group have been described mainly in swine manure (Song et al. 2017). At the species level, it was possible to identify the species Corynebacterium maris (5% of Actinobacteria), which are halotolerant bacteria isolated for the first time from the coral Fungia granulosa in the Gulf of Eilat, Israel (Ben-Dov et al. 2009), and in lower proportion in Corynebacterium efficiens, which have been isolated from soil and vegetables (Fudou et al. 2002). Two additional genera were detected within this order in S1, namely Mycobacterium and Dietzia (each representing 2% of Actinobacteria), and both were isolated from aquatic and terrestrial habitats, as well as from clinical material (Koerner et al. 2009).
Bacteria of the order Micrococcales dominated in S1 (16% of the phylum Actinobacteria) but were also present in S2 and S3 (2% and 5% of Actinobacteria, respectively). The genera Brachybacterium and Kocuria were the most dominant in S1 (each 3% of Actinobacteria), followed by the genera Citricoccus and Curtobacterium (each 1%), in addition to other genera with relatively lower proportions as Arthrobacter and Pseudarthrobacter (both 0.7% of Actinobacteria) as well as Agromyces (0.5%). Species of the genus Brachybacterium have been isolated from poultry deep litter, stool samples, soils, sediments, and seawater (Collins et al. 1988). Bacteria belonging to all abovementioned genera are typically species found in soils. Kocuria and Citricoccus have been widely isolated from soils, and some species are halotolerant and alkalitolerant (Li et al. 2005; Wang et al. 2015). The Curtobacterium genus has a global distribution with a predominant presence in soil ecosystems and is described mainly in association with plants, especially in the phyllosphere (Chase et al. 2016). Agromyces are soil actinomycetes with formation of true mycelia (Zgurskaya et al. 1992). In contrast, the most dominant genus in S3 was Arthrobacter, with the species Arthrobacter crystallopoietes. This species has been described as desiccation-resistant bacteria, and this capability may explain their numerical dominance in soil (Boylen 1973).
Bacteria of the order Streptomycetales were present in relatively high abundance in S1 (14% of the phylum Actinobacteria) and S3 (9%). The genus Streptomyces dominated in both samples (13% and 8%, respectively). Bacteria belonging to this genus are ubiquitous in soils and marine sediments and are commonly found in the rhizosphere or inside plant roots (Rey and Dumas 2017). The species Streptomyces albiaxialis was detected only in S1, which is a thermotolerant and halotolerant Streptomyces (Kuznetsov et al. 1992). In addition, it was also possible to detect in a very low proportion members of the genus Streptacidiphilus. The genus Streptacidiphilus is composed of acidophilic actinomycetes and has a major role in the turnover of organic matter in acidic habitats (Kim et al. 2003).
The order Streptosporongiales was better represented in S1 and S3 (6% and 7% of the phylum Actinobacteria, respectively), with the genus Nocardiopsis as dominant in S1 and Actinomadura in S3. Nocardiopsis is a bacterial genus that produces some antimicrobial compounds and occurs mostly in saline and alkaline soils (Bennur et al. 2014). Bacteria of the genus Actinomadura are commonly isolated from soils (Zhao et al. 2015).
The order Propionibacteriales accounted for 3% and 7% of the phylum Actinobacteria in S1 and S3, respectively. The most represented genera in both samples were Nocardioides and Microlunatus, and both genera are common bacteria in soils (Kämpfer et al. 2010; Wang et al. 2017). Finally, the order Pseudonocardiales accounted for 4% and 6% of the phylum Actinobacteria in S1 and S3, respectively, in which Pseudonocardia is the dominant genus. In both samples, it was possible to detect the species Pseudonocardia zijingensis, which was isolated from a soil sample collected from Zijing Mountain, Yunnan Province, China (Huang et al. 2002).
Members of the Thermoleophilia class were detected in S1 and S3 (24% and 23% of the phylum Actinobacteria, respectively). Uncultivated bacteria of the Solirubrobacterales Elev-16S-1332 group as well as cultivated bacteria of the genera Solirubrobacter and Gaiella showed to be dominant in both samples, which are bacteria commonly isolated from soil (Singleton et al. 2003) and water. In addition, members of the class Acidimicrobiia were detected in both samples (3% of the phylum Actinobacteria in each sample) where it was possible to infer only the genus Lamia, which members have been isolated from marine environments (Kurahashi et al. 2009). Curiously, members of the Rubrobacteria class were only detected in a relatively high proportion (23% of the phylum Actinobacteria) in S3, where it was possible to identify only the genus Rubrobacter. Unfortunately, it was not possible to infer at the species level, but species of this genus have been widely detected in terrestrial habitats, as stone monuments usually subjected to salt weathering (Otlewska et al. 2017) as well as in marine environments, and some of them are known to be halotolerant, thermotolerant, and radiotolerant (Egas et al. 2014).
Interestingly, members of other phyla found in significant proportions only in S1 strongly indicated that the soil being in contact with this statue must have been from animal farm. For example, members of the phylum Bacteroidetes were present only in this sample, accounting for 2% of bacteria (Fig. 6), with mainly species of Bacteroides. Members of Bacteroides make up the most substantial portion of the mammalian gastrointestinal flora and play a fundamental role in the processing of complex molecules to simpler ones in the host intestine (Wexler 2007); the species Bacteroides barnesiae was identified, which has been isolated from the cecum of a healthy chicken (Lan et al. 2006). In addition, members of the family Rikenellaceae, with the Rikenellaceae_RC9_gut group and the genus Alistipes, as well as members of the family Prevotellaceae were identified. Members of these two families are often found in the gastrointestinal tract of different animals and humans (Rosenberg 2014). Other bacteria that could be indicators of a contact with soils of animal farms belonged to the phylum Acidobacteria, accounting for 1% of bacteria in S1 (Fig. 6), which are among the most common bacteria in soils worldwide. Nevertheless, the main Acidobacteria group detected in S1 was subgroup 6, which members are related to those detected in pasture soils (Navarrete et al. 2015).
In addition, sequences related to the phylum Firmicutes, retrieved in significant proportions only from S1, confirmed the suspicion that this statue must have been in contact with animal farm soil (supplementary Fig. 3). Sequences affiliated to the order Clostridiales were found in a relatively high proportion (8% of Firmicutes). Within this order, the most dominant members comprised the Christensenellaceae_R-7 group, which includes uncultured bacteria mainly found in rumen, as well as members of the genera Romboutsia, Intestinibacter, and Clostridium and of the families Ruminococcaceae and Lachnospiraceae, all of them were identified in the gut microbiota of human and other animals (Gerritsen et al. 2014; Meehan and Beiko 2014). Sequences related to the order Lactobacillales were also detected in high proportion in this sample (12% of Firmicutes). The most detected genus was Lactobacillus (3% of Firmicutes), which are a major part of the group of lactic acid bacteria (LAB) and in humans and constitute a significant component of the microbiota at a number of body sites, such as the digestive, urinary, and genital systems. At lower taxonomic levels, it was possible to identify the species Lactobacillus kitasatonis, Lactobacillus reuteri, Lactobacillus pontis, and Lactobacillus salivarius, among the other unidentified species. Several genera within the family Carnobacteriaceae were also detected, such as Atopostipes and Carnobacterium (each 3% of Firmicutes). At the level of species, the only one known species (Atopostipes suicloacale) was identified in this study. This species has been previously isolated from an underground swine manure storage pit (Cotta et al. 2004). Members of the genus Carnobacterium are ubiquitous LAB, which are isolated from cold and temperate environments, and predominate in a range of foods, including fish, meat, and some dairy products (Leisner et al. 2007). Other members of the family Enterococcaceae could be detected even in low proportions (0.2% of Firmicutes), with the species Enterococcus cecorum, which is known as a commensal in the intestines of mammals and birds, but it has been described as an emerging pathogen in poultry industry worldwide (Dolka et al. 2017). Sequences related to the order Erysipelotrichales (Firmicutes), which species are known to be common in the gut microbiome of animals, were also detected in this sample (4% of Firmicutes), with the genus Turicibacter as the sole representative (Auchtung et al. 2016).
In contrast, S2 and S3 showed to have a lower biodiversity of Firmicutes and were dominated by members of the order Bacillales (supplementary Fig. 3), which represent 97% and 82% of Firmicutes, while this order accounted for 76% of Firmicutes in S1. Nevertheless, the detected genera showed to be different among the samples. S2 and S3 showed a strong dominance of Bacillus (39% and 23% of Firmicutes, respectively). At the level of species, Bacillus timonensis and Bacillus subtilis were detected in S2. Bacillus timonensis has been detected in human fecal flora (Kokcha et al. 2012), and B. subtilis is commonly found in the gastrointestinal tract of ruminants and humans and is widely used as a probiotic feed supplement for farm animals (Wang et al. 2018). In addition, this species collectively inhabits the rhizosphere, where it contributes to the promotion of plant growth (Hirooka 2014).
In S3, it was possible to detect a wider range of species of Bacillus. Some of them accounted for relatively high proportions, such as Bacillus niacini and Bacillus longiquaesitum, both isolated from soils (Nagel and Andreesen 1991), but the latter one was also commonly isolated from microbial communities associated with truffles (Mello et al. 2013). It is worth noting that traces of DNA of truffles were identified in this sample (genus Tuber). Other species were detected in this sample, such as Bacillus nealsonii, which produces endospores that are resistant to UV, gamma-radiation, salinity, and desiccation (Venkateswaran et al. 2003). In lower proportions, Bacillus humi, Bacillus benzoevorans, and Bacillus pumilus were detected as well, all of them most commonly isolated from soils (Heyrman et al. 2005) and known to be plant growth-promoting bacilli (Yadav et al. 2011).
The genus Anoxybacillus was additionally detected in S2 (2% of Firmicutes). Many species of this genus are known to be alkaliphilic and thermophilic and commonly occur in geothermal springs, manure, and milk processing plants (Yamamoto et al. 2006). Other detected genera in S3 were Fictibacillus (2% of Firmicutes) and Falsibacillus (1%), both commonly isolated from marine environments (Dastager et al. 2014).
In contrast, S1 showed a much lower proportion of Bacillus (7% of Firmicutes) and the only identified species was Bacillus alkalitelluris, an alkaliphilic bacterium isolated from soil in Korea (Lee et al. 2008). The genus Oceanobacillus was also detected in this sample in a relatively high proportion (4% of Firmicutes). Oceanobacillus species have been isolated from diverse environmental samples, including deep-sea sediment cores and salt fields; fermented shrimp paste samples, soy sauce production equipment; and traditional Korean fermented food. We could identify some species, such as Oceanobacillus iheyensis, an extremely halotolerant species isolated from deep-sea sediment (Lu et al. 2001); Oceanobacillus profundus, a halotolerant bacterium, isolated from the deep surface of a sediment core in Korea (Kim et al. 2007a); and Oceanobacillus picturae, isolated for the first time from a mural painting (Heyrman et al. 2003). Other genera identified in lower proportions (0.9–0.2% of Firmicutes), such as Virgibacillus, Falsibacillus, Fictibacillus, Pontibacillus, Terribacillus, and Marinococcus, further supported the hypothesis that statue 1 was in contact with a saline environment, in which many of the species of all detected genera were able to grow in moderate concentrations of NaCl and were isolated from saline environments.
S1 showed actually a higher proportion of members of the Planococcaceae family (39% of Firmicutes), which belong to the genus Planomicrobium (32% of Firmicutes). At the level of species, Planomicrobium okeanokoites showed to be dominant. This species has been isolated from the marine muds and from soils in China (Nakagawa et al. 1996). Other species detected in lower proportions were Planomicrobium glaciei (Zhang et al. 2009), which was isolated from a glacier in Northwest China, and Planomicrobium chinense (Dai et al. 2005), which was isolated from a coastal sediment of the Eastern China Sea, China. Both species are able to growth at moderate salinities (0–10% NaCl). The genera Planococcus and Sporosarcina were also detected, but in lower proportions (1% of Firmicutes). Members of the Planococcaceae were less dominant in S2 and S3 (3% and 10% of Firmicutes, respectively), and the main genera detected in these samples differed from those in S1. Bacteria of the genus Domibacillus were dominant in both samples (1% and 5% of Firmicutes in S2 and S3, respectively), in addition to Sporosarcina (0.2% and 2% of Firmicutes, respectively). At the level of species, Sporosarcina soli was detected in S2 and Sporosarcina koreensis in S3. Both species were isolated for the first time from soils in Korea, and both tolerate moderate (5–7%) NaCl concentrations (Kwon et al. 2007). The genus Chungangia (1% of Firmicutes) was only present in S2. Other genera detected in lower proportions in S3 were Paenisporosarcina, Lysinibacillus, and Bhargavaea.
Members of the Staphylococcaceae were present in relatively high abundance only in S1 (14% of Firmicutes) which were negligible in the other two samples. The genus Salinicoccus showed to be dominant (10% of Firmicutes) in this sample. It was possible to identify the species Salinicoccus kekensis, a moderate halophilic bacterium isolated from a salt lake in China (Gao et al. 2010), as well as other species, such as Salinicoccus halodurans, Salinicoccus kunmingensis, and Salinicoccus roseus, which were isolated from saline soils and a salt mine in China (Chen et al. 2007; Wang et al. 2008). The genus Jeotgalicoccus accounted, in addition, for 3% of Firmicutes in S1. Bacteria of this genus are halotolerant to halophilic bacteria. The species Jeotgalicoccus psychrophilus was identified, which was isolated from the Korean fish sauce jeotgal (Yoon et al. 2003). The genus Staphylococcus was also found in this sample, even if in less proportion, with the species Staphylococcus saprophyticus. This species is part of the normal human flora that colonizes the perineum, rectum, urethra, cervix, and gastrointestinal tract but has also been found as a common gastrointestinal flora in pigs and cows (Ehlers and Merrill 2018).
Members of the Paenibacillaceae were present in relatively high proportions in all three samples, with the dominance of the genus Paenibacillus (2%, 12%, and 5% of Firmicutes, respectively). Paenibacillus spp. are found in soil, often associated with plant roots. These rhizobacteria promote plant growth and can be exploited for use in agriculture. Nevertheless, some differences were observed among the three samples concerning the detected species. Paenibacillus alginolyticus and Paenibacillus harenae were detected in S1 and S3. The first one was isolated from soils and the second one from desert sand in China (Jeon et al. 2009). In S2, the detected species were Paenibacillus lautus, which has been isolated from soil and human intestinal tract, and Paenibacillus glucanolyticus, which is known to degrade cellulose, hemicellulose, and lignin (Mathews et al. 2014). The genus Cohnella was also detected in all three samples (0.5%, 0.2%, and 5% of Firmicutes, respectively). Members of this genus are highly cellulolytic bacteria, which can be found in a variety of habitats. At the level of species, it was possible to detect Cohnella saccharovorans only in S3. This species was isolated from ginseng soils (Choi et al. 2016). Additional genera were detected in S1, such as Saccharibacillus, with the species Saccharibacillus deserti, which was isolated from desert soil in China (Sun et al. 2016).
Members of the Alicyclobacillaceae, with the genus Effusibacillus, were detected in relatively high abundance only in S2 (6% of Firmicutes). Unfortunately, it was not possible to infer at the level of species. The genus Exiguobacterium was detected only in S1 (2% of Firmicutes), in which it was possible to identify the species Exiguobacterium acetylicum. This species has been isolated from soils and expressed multiple plant growth promotion attributes such as phosphate solubilization (Selvakumar et al. 2010). Finally, concerning the phylum Firmicutes, some sequences affiliated with uncultivated bacilli in S2 and S3, as the uncultivated clone TSCOR001-H18 (2% of Firmicutes sequences in each sample), which has been detected on rice paddy soils in Japan (Ishii et al. 2009).
Proteobacteria was the third phylum most represented in all three samples (15%, 6%, and 12% of total reads for S1–S3, respectively; see Fig. 6). Nevertheless, some differences were observed among the three samples (supplementary Fig. 3). The Alphaproteobacteria class was dominant on S1 and S3, accounting for 40% and 76% of Proteobacteria, respectively, whereas it accounted for only 18% of Proteobacteria reads of S2. Some additional differences were noticed at lower phylogenetic levels. The order Rhizobiales dominated on S1 (13% of Proteobacteria) and accounted for 4% and 8% of the Proteobacteria in S2 and S3, respectively. Within this order, the genera Mesorhizobium (3% of Proteobacteria), Bradyrhizobium, and Neorhizobium (both 0.6% of Proteobacteria) were present in S1, all of them were nitrogen-fixing endosymbionts associated with roots of legumes. It is worth mentioning that members of the Fabales (leguminous plants) were detected in this sample. Another dominant genus in S1 was Devosia (1% of Proteobacteria). Species of Devosia are commonly isolated from rhizospheric soils (Mohd Nor et al. 2017). In contrast, the dominant genus in S2 and S3 was Microvirga. This genus comprises mainly soil bacteria, and some species are root nodule symbiotic bacteria that fix nitrogen in different plant hosts (Ardley et al. 2012).
The Sphingomonadales were present in S1 and S2 (9% and 2% of Proteobacteria, respectively) and almost negligible in S3. The most dominant genus in both samples was Sphingomonas. Members of this genus are pigmented bacteria found in various environments, including both aqueous (freshwater and seawater) and terrestrial habitats, plant root systems, clinical specimens, and others. Nevertheless, it was possible to infer differences at the level of species. Sphingomonas mucosissima and Sphingomonas jaspsi were identified in S1, being the first species isolated from biological soil crust collected from sandy arid soils (Reddy and Garcia-Pichel 2007), and S. jaspsi was isolated from freshwater samples collected in Japan (Asker et al. 2007). In contrast, Sphingomonas kaistensis and Sphingomonas humi were detected in S2, both previously isolated from soils in South Korea (Kim et al. 2007b; Yi et al. 2010).
The Rhodobacterales accounted for 6%, 7%, and 3% of Proteobacteria in S1–S3, respectively. Samples S1 and S2 showed certain similarities, as the most dominant genus in both samples was Paracoccus. Paracoccus siganidrum was identified in both samples, and this species was isolated from the fish gastrointestinal tract (Liu et al. 2013). In addition, Paracoccus sanguinis was identified only in S1 and Paracoccus yeei only in S2. Both species have been identified as clinical specimens (McGinnis et al. 2015). The genus Rubellimicrobium was also identified in both samples. Species of this genus are reddish bacteria commonly isolated from soils (Riedel et al. 2014). In contrast, in S3, the genus Amaricoccus showed to be dominant. Species of this genus have been isolated from activated sludge (Maszenan et al. 1997).
Members of the Caulobacterales were detected in S1 and S2, but absent in S3. The most dominant genus in both samples was Brevundimonas, and it was possible to identify Brevundimonas vesicularis in S1. Brevundimonas spp., and in particular B. vesicularis, are emerging global opportunistic pathogens (Ryan and Pembroke 2018).
Sample S3 differed significantly from the other two samples, concerning the Alphaproteobacteria group, and the dominant order was the Rhodospirillales (65% of Proteobacteria), of which most reads match the Rhodospirillales MSB-1E8 group (53% of Proteobacteria). Members of this group are all uncultivated bacteria, which have been found on the microbiome of seabirds and corals (Grottoli et al. 2018). The genus Skermanella was also dominant in this sample (5% of Proteobacteria). It was not possible to infer at the level of species, but species of this genus have been identified as airborne bacteria in Asian dust, as well as isolated from freshwater and soils in extreme environments, as deserts in China (Zhang et al. 2015). In addition, the Candidatus_Alysiosphaera group and the genus Craurococcus accounted each for 3% of Proteobacteria in this sample, and the latter one was isolated from soils (Saitoh et al. 1998). This order accounted for 9% and 3% of Proteobacteria in S1 and S2, respectively. The group Rhodospirillales MSB-1E8 was also identified in S1, but in lower proportion.
The Betaproteobacteria class showed to be dominant in S2 (51% of Proteobacteria) and accounted for 21% and 6% of Proteobacteria in S1 and S3, respectively (supplementary Fig. 3). However, some differences were observed at the family and genus levels, being the Oxalobacteraceae, with the genus Massilia dominant on S1. Species of Massilia, even if known as pathogens, have been also isolated from soils in China (Zhang et al. 2006). The family Burkholderiaceae, with the genus Ralstonia, dominated on S2. Species of this genus are well-known plant pathogens, and their significant dominance in S2 may correlate with the presence of plants detected in this sample. Sample S2 showed the highest biodiversity of Betaproteobacteria, and additional genera, such as Aquabacterium, Pelomonas, and Achromobacter, were identified. Species of all these genera have been isolated from water (fresh and marine) and soils. Sample S3 comprises also species of Ralstonia, in addition to members of the family Nitrosomonadaceae. All cultivated representatives of the last family are lithoautotrophic ammonia oxidizers that exert control over nitrification by oxidizing ammonia to nitrate. They therefore play major roles in the control of the nitrogen cycle in terrestrial, freshwater, and marine environments.
The Gammaproteobacteria class accounted for 33% and 27% of Proteobacteria in S1 and S2, respectively, and for significantly less proportion in S3 (3%). Members of the Pseudomonadales were present in S1 and S2, with the genus Pseudomonas being dominant in S1 and the genus Acinetobacter in S2. Pseudomonas are commonly isolated from soils. The species Pseudomonas pseudoalcaligenes, detected in S1, has been isolated from plant rhizospheres and is an alkaliphilic cyanide-degrading bacterium (Luque-Almagro et al. 2011). In addition, the genus Alkanindiges, with the species Alkanindiges illinoisensis, an alkane-degrading bacterium isolated from oilfield soils (Bogan et al. 2003), was detected only S1.
The order Enterobacterales showed to be relevant only in S1 (12% of Proteobacteria), less represented in S2 (3% of Proteobacteria), and absent in S3. Many members of this order are a normal part of the gut flora found in the intestines of humans and other animals. The significant presence of these bacteria in S1 supports, once more, the hypotheses that this statue was in contact with farm soil. The genus Pantoea was dominant in S1 and S2. The species Pantoea agglomerans and Pantoea ananatis were identified in S1. P. agglomerans is a ubiquitous bacterium commonly isolated from plant surfaces, seeds, fruit, and animal or human feces. P. ananatis is a bacterium with the ability to colonize and interact with hosts in both the plant and animal kingdoms and was found in a wide range of natural environments, as part of the epiphytic and endophytic flora of various plant hosts, and in the insect gut (De Maayer et al. 2014). In addition, members of Enterobacter, Escherichia/Shigella, Cronobacter, and Salmonella were detected only in S1 and Buttiauxella only in S2.
The order Xanthomonadales accounted for 5% of Proteobacteria in S1 and was very low in S2 and S3 (0.5% and 0.8% of Proteobacteria, respectively). The genera Steroidobacter, with the species Steroidobacter flavus, and Acidibacter were identified in S1 and S3. The detected S. flavus is a microcystin-degrading strain, isolated from forest soil in China (Gong et al. 2016). The genus Acidibacter was recently described as acidophilic, mesophilic, and capable of reducing ferric iron (Falagán and Johnson 2014). In addition, the genera Stenotrophomonas and Lysobacter were identified in S1 and S2. The genus Lysobacter accommodates non-fruiting, gliding bacteria commonly isolated from soils. At lower taxonomic levels, the species Lysobacter niastensis was identified. This species was originally isolated from greenhouse soil cultivated with cucumber in Korea (Weon et al. 2007). It is worth noting that plants of the genus Cucumis were detected in S2. Within the genus Stenotrophomonas, Stenotrophomonas maltophilia was detected. This species is ubiquitous in aqueous environments, soil, and plants, but it is also an emerging multidrug-resistant global opportunistic pathogen (Brooke 2012).
The Salinisphaerales showed to be present only in S3, with members of the Salinisphaeraceae family. Unfortunately, it was not possible to infer lower taxonomic levels. This family comprises two genera: Salinisphaera and Halophibacterium. All members of the family are halophilic or halotolerant and have been isolated from marine/oceanic and high-salinity environments (Vetriani et al. 2014).
The Deltaproteobacteria accounted for 14% of Proteobacteria in S3, in contrast to the lower proportions detected in S1 and S2 (6% and 4%, respectively). Members of the orders Desulfurellales and Myxococcales accounted each for 6% of Proteobacteria in S3. It is worth mentioning that members of Desulfurellales were negligible in S1 and S2. This order comprises exclusively the family Desulfurellaceae. Members of this family are all strictly anaerobic and moderately thermophilic bacteria, found mostly in warm-hot sulfur-containing anoxic environments, such as hydrothermal springs and sediments, and are often in association with microbial mats (Greene 2014). All the reads matched with the uncultured genus Desulfurellaceae H16, previously detected in sediments of lakes (Ruuskanen et al. 2018). Members of the order Myxococcales were also represented in S1 and S2 (5% and 3% of Proteobacteria). The genera Haliangium and Sorangium were present in all three samples, with the first one being dominant in S1 and the second in S2 and S3. Species of Haliangium are known as marine myxobacteria and are halotolerant (Albataineh and Stevens 2018). The genus Sorangium, with the detected species Sorangium cellulosum, plays an important role in soil ecology by its ability to degrade cellulosic materials. In addition, S1 showed the presence of the Myxococcales Blrii 41 group, which comprises uncultivated bacteria that have been identified in rice field soils (Hori et al. 2007) as well as the genera Vulgatibacter, Anaeromyxobacter, and Polyangium, all of them being commonly isolated from soils and rhizosphere, and specifically from saline-alkaline soils (Zhang et al. 2013b). These genera were also present in S3, but absent in S2. The order Bdellovibrionales accounted for 1% of Proteobacteria in S3 and was present at lower proportions in S1 and S2. In S1 and S3, this order was composed exclusively of members of the OM27_clade, which comprises uncultivated bacteria involved in nutrient cycles in the ocean (Orsi et al. 2016).
Some additional phyla were detected in S1 and S3 but were absent in S2 (Fig. 6). Even though they were detected in low proportions, they represented important ecological markers in these two samples. The phylum Chloroflexi (1% and 0.8% of total reads in S1 and S3, respectively) contains bacteria with various metabolic features and is one of the most common and diverse bacterial phyla in sponges with many sponge-specific lineages (Schmitt et al. 2011). Nevertheless, it is worth noting that all detected sequences belonging to this phylum affiliated with uncultivable microorganisms of the no-rank groups Gitt-GS-136, TK10, S085, JG30-KF-CM45, and KD4-96. These groups have been detected in bat guano (De Mandal et al. 2015) and in manure- and fertilizer-treated soils (Zhang et al. 2013a); thus, they seem to be biological markers indicating a contact with agricultural land.
The phylum Planctomycetes (0.7% of total reads in both S1 and S3) comprises mainly aquatic bacteria found in samples of brackish, marine, and fresh waters. The family Planctomycetaceae accounted for 83% of Planctomycetes in both samples; within this family, it was possible to identify some genera in both samples, such as Gemmata, Zavarzinella, and Singulisphaera, all of them being isolated from acid bogs, swamps, and wetlands, and the genus Planctomyces was detected only in S3. The family Tepidisphaeraceae was present in both samples. Members of this family have been isolated from microbial mats (Kovaleva et al. 2015).
Finally, the phylum Tectomicrobia was detected only in S3 (1% of bacteria; see Fig. 6). This phylum consists of uncultured members comprising at least three discrete phylogenetic clades. The largest one encompasses all Entotheonella sequences sensu stricto, and in this study, half of the reads matching this phylum belonged to the candidate genus Entotheonella, which are widely distributed in marine sponges and seawater (Wilson et al. 2014).
Giving answers to the questions posed
The complex data obtained in this metagenomic study show that all three statues own a very specific microbiome, with a biodiversity that differs with respect to all groups studied (bacteria, fungi, plants, protozoa, etc.). Nevertheless, their microbiomes enable to answer some of the posed questions:
Were the statues stored individually or together during their recent history? Moreover, could all three objects have been stored under building dust and rubble?
A period of common storage in the recent past can be accepted cautiously only for the two torsi (S1 [female] and S2 [male]), but not for the girl’s head (S3), which differs considerably from the other two objects mainly in terms of eukaryotes. Nevertheless, in terms of bacteria, S1 and S3 harbor intriguing similarities, such as the presence of many halotolerant and halophilic bacteria, which could indicate a common origin in the past.
There is no definitive indication that all three pieces would have been stored together for a longer period of time under house dust or building rubble, as previously supposed by custom officers. Typical indicators for this environment would be species of Aspergillus and Penicillium (Pitt and Hocking 2009). Very common Aspergillus species described in indoor include Aspergillus calidoustus, Aspergillus flavus, Aspergillus fumigatus, Aspergillus penicillioides, Aspergillus restrictus, Aspergillus versicolor, Aspergillus westerdijkiae, and more recently, A. sydowii (Visagie et al. 2014). Among the Penicillium species, commonly reported species from indoor surveys include Penicillium expansum, Penicillium digitatum, Penicillium italicum, Penicillium brevicompactum, Penicillium chrysogenum, Penicillium commune, Penicillium glabrum, Penicillium olsonii, Penicillium oxalicum, and Penicillium rubens.
In our study, Penicillium spp. were only detected in S1, but completely absent in S2 and S3. Furthermore, the identified species P. glandicola and P. citrinum are commonly growing in association with plants, such as cereals. Thus, their presence is more in line with the hypothesis that statue S1 was stored in an agricultural soil. In addition, some species of Aspergillus could be markers of house dust, such as the identified A. proliferans in S1 and S2, which has been widely found in indoor environments, and A. penicillioides only in S1, which is typically found in indoor air and house dust and on substrates with low water activity. Nevertheless, these species were not the dominant ones among the identified Aspergilli. The most dominant species in samples S1 and S2 was A. sydowii, which has been identified in indoor environments and dust in countries of Asia and South America, but not in European countries (Visagie et al. 2014), where it is mainly found as a saprophytic fungus in soils. In S2, A. ruber, which is a plant pathogen, was relevant. Therefore, a common storage in a household or in building rubble is not considered the most plausible hypothesis for the statues in the recent past. The data strongly pointed out that most probably both torsi were stored or even buried temporarily together in agricultural soils or even animal farm soils.
In contrast, the girl’s head differed considerably from the other two samples and clearly showed a primarily longer stay in arid and semi-arid surroundings, but in any case, a contact or even its submersion in seawater. The fungal and eukaryotic flora differs considerably from the other two objects.
What is the long-term storage history of the three statues?
To answer this question, we have focused on the following biological markers that strongly point out some specific environments where the statues could have been stored:
-
i.
Biological markers indicating a prolonged contact with agricultural soils, such as land plants, plant-associated and phytopathogenic fungi, as well as typical soil and plant-associated bacteria (found in all three samples; see supplementary Table 3).
-
ii.
Biological markers indicating a storage in courtyard, stables, or animal farm, such as those bacteria which are typical inhabitants of the gastrointestinal tract of animals, rumen, or manure (mainly in S1, but also in low proportion in S2; see supplementary Table 4).
-
iii.
Biological markers indicating a contact with saline or arid environments, such as halotolerant and halophilic fungi and bacteria isolated from desert soils, sandy arid soils, saline soils, or even salt mines (in S1 and S3; see supplementary Table 5)
-
iv.
Biological markers indicating a contact with aquatic and/or marine environments, including members of the Chlorophyta (green algae), Ochrophyta (brown seaweeds), amoeboflagellates, and flagellates of the phylum Cercozoa, Dinophyceae (Dinoflagellates), and some fungi of the phylum Chytridiomycota, indicating an aquatic environment in S3. In addition, some bacteria indicated contact with aquatic environments for S1 and S3 (see supplementary Table 6).
Is it possible to infer a geographical origin or a geographical shift?
The eukaryotic community of objects S1 and S2 is similar and can allow the conclusion that both objects have been stored and/or transported together in the recent past. Both objects have references to a stay in China or contact with materials from China (e.g., wood of packaging/containers/packaging crates). In S2, the detection of Taiwania sequences allowed a real geolocation of this sample, because this tree is native and endemic to Eastern Asia. The additional geolocation of an object due to the detection of bacteria uniquely or frequently isolated from one country may be used carefully, due to the ubiquity of bacteria in different geographical locations with similar environmental conditions. Nevertheless, it is important to point out that many of the identified bacteria in this study, mainly in S1, were described in environmental samples in Asia and mainly in China.
Conclusion
Our data showed the potential of the applied genetic strategy to answer interesting questions that may arise when dealing with cultural heritage objects and that may not be related with the state of conservation, but more with the history of the object. It is worth mentioning that the challenge was to retrieve, among the immense amounts of data, those referring to unique groups of organisms that could be considered as specific of one sample or as biological markers of a specific environment.
All three statues showed each a very specific microbiome. Nevertheless, each of the kingdoms analyzed gave hints for the following environments, however with some differences between the three statues: (a) a contact with arid or saline environment, (b) a period spent in agricultural soil and/or animal farm, and (c) a period spent in fresh, brackish, or sea water. A specialty was the clear affiliation of the male torso with a tree endemic to Taiwan.
Summarizing and based on the genetic fingerprints of the three objects, conclusions can be drawn about the storage of the objects: The female and male torsi both indicate storage in agricultural soils, with the female torso being the most probably stored even in an animal farm. The eukaryotic communities of both objects are similar and can allow the conclusion that both objects have been stored or transported together in the recent past. Both objects have references to a stay in China or contact with materials from China (e.g., packaging/containers/packaging crates). The girl’s head differs considerably from the two other samples concerning the eukaryotic community but shows intriguing similarities with the female torso concerning the bacterial community, displaying many halotolerant and halophilic bacteria. This fact may indicate a longer stay in arid and semi-arid surroundings as well as in marine environments in the past.
In summary, these strategies offer an immense potential and reveal more information than we could have imagined before. Nevertheless, it is important to note that these techniques pose a challenge concerning the interpretation of the immense amount of data generated.
References
Adamiak J, Bonifay V, Otlewska A, Sunner JA, Beech IB, Stryszewska T, Kańka S, Oracz J, Zyźelewicz D, Gutarowska B (2017) Untargeted metabolomics approach in halophiles: understanding the biodeterioration process of building materials. Front Microbiol 8:2448
Adamiak J, Otlewska A, Tafer H, Lopandic K, Gutarowska B, Sterflinger K, Piñar G (2018) First evaluation of the microbiome of built cultural heritage by using the Ion Torrent next generation sequencing platform. Int Biodeterior Biodegrad 131:11–18
Alam M, Dharni S, Abdul-Khaliq, Srivastava SK, Samad A, Gupta MK (2012) A promising strain of Streptomyces sp. with agricultural traits for growth promotion and disease management. Indian J Exp Biol 50:559–568
Albataineh H, Stevens DC (2018) Marine myxobacteria: a few good halophiles. Mar Drugs 16:209
Amend A (2014) From dandruff to deep-sea vents: Malassezia-like fungi areecologically hyper-diverse. PLoS Pathog 10:e1004277
Ardley JK, Parker MA, De Meyer SE, Trengove RD, O’Hara GW, Reeve WG, Yates RJ, Dilworth MJ, Willems A, Howieson JG (2012) Microvirga lupini sp. nov., Microvirga lotononidis sp. nov. and Microvirga zambiensis sp. nov. are alphaproteobacterial root-nodule bacteria that specifically nodulate and fix nitrogen with geographically and taxonomically separate legume hosts. Int J Syst Evol Microbiol 62:2579–2588
Asker D, Beppu T, Ueda K (2007) Sphingomonas jaspsi sp. nov., a novel carotenoid-producing bacterium isolated from Misasa, Tottori, Japan. Int J Syst Evol Microbiol 57:1435–1441
Auchtung TA, Holder ME, Gesell JR, Ajami NJ, Duarte RT, Itoh K, Caspi RR, Petrosino JF, Horai R, Zárate-Bladés CR (2016) Complete genome sequence of Turicibacter sp. strain H121, isolated from the feces of a contaminated germ-free mouse. Genome Announc 4:e00114–e00116
Barr DJS (1990) Phylum Chytridiomycota. In: Handbook of Protoctista. Margulis, Corliss, Melkonian, & Chapman (eds). Jones & Barlett, Boston, pp 454–466
Ben-Dov E, Ben Yosef DZ, Pavlov V, Kushmaro A (2009) Corynebacterium maris sp. nov., a marine bacterium isolated from the mucus of the coral Fungia granulosa. Int J Syst Evol Microbiol 59:2458–2463
Bennur T, Kumar AR, Zinjarde S, Javdekar V (2014) Nocardiopsis species as potential sources of diverse and novel extracellular enzymes. Appl Microbiol Biotechnol 98:9173–9185
Bogan BW, Sullivan WR, Kayser KJ, Derr KD, Aldrich HC, Paterek JR (2003) Alkanindiges illinoisensis gen. nov., sp. nov., an obligately hydrocarbonoclastic, aerobic squalane-degrading bacterium isolated from oilfield soils. Int J Syst Evol Microbiol 53:1389–1395
Boniek D, de Castro Mendes I, Paiva CAO, de Paula Lana UG, Dos Santos AFB, de Resende Stoianoff MA (2017) Ecology and identification of environmental fungi and metabolic processes involved in the biodeterioration of Brazilian soapstone historical monuments. Lett Appl Microbiol 65:431–438
Bower MA, Campana MG, Checkley-Scott C, Knight B, Howe CJ (2010) The potential for extraction and exploitation of DNA from parchment: a review of the opportunities and hurdles. J Inst Conserv 33:1–11
Boylen CW (1973) Survival of Arthrobacter crystallopoietes during prolonged periods of extreme desiccation. J Bacteriol 113:33–37
Brooke JS (2012) Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev 25:2–41
Burger J, Hummel S, Herrmann B (2000) Palaeogenetics and cultural heritage. Species determination and STR-genotyping from ancient DNA in art and artefacts. Thermochim Acta 365:141–146
Callahan BJ, Sankaran K, Fukuyama JA, McMurdie P, Holems SP (2016) Bioconductor workflow for microbiome data analysis: from raw reads to community analyses [version 2; referees: 3 approved]. F1000Research 5:1492
Carniel FC, Zanelli D, Bertuzzi S, Tretiach M (2015) Desiccation tolerance and lichenization: a case study with the aeroterrestrial microalga Trebouxia sp. (Chlorophyta). Planta 242:493–505
Cavalier-Smith T, Chao EY (2003) Phylogeny and classification of phylum Cercozoa (Protozoa). Protist 154:341–358
Chamberlain SA, Boettiger C (2017) R Python, and Ruby clients for GBIF species occurrence data. PeerJ Preprints 5:e3304v1. https://doi.org/10.7287/peerj.preprints.3304v1
Chase AB, Arevalo P, Polz MF, Berlemont R, Martiny JBH (2016) Evidence for ecological flexibility in the cosmopolitan genus Curtobacterium. Front Microbiol 7:1874
Chen YG, Cui XL, Pukall R, Li HM, Yang YL, Xu LH, Wen ML, Peng Q, Jiang CL (2007) Salinicoccus kunmingensis sp. nov., a moderately halophilic bacterium isolated from a salt mine in Yunnan, South-West China. Int J Syst Evol Microbiol 57:2327–2332
Chen Q, Jiang JR, Zhang GZ, Cai L, Crous PW (2015) Resolving the Phoma enigma. Stud Mycol 82:137–217
Chen AJ, Hubka V, Frisvad JC, Visagie CM, Houbraken J, Meijer M, Varga J, Demirel R, Jurjević Ž, Kubátová A, Sklenář F, Zhou YG, Samson RA (2017) Polyphasic taxonomy of Aspergillus section Aspergillus (formerly Eurotium), and its occurrence in indoor environments and food. Stud Mycol 88:37–135
Choi JH, Seok JH, Jang HJ, Cha JH, Cha CJ (2016) Cohnella saccharovorans sp. nov., isolated from ginseng soil. Int J Syst Evol Microbiol 66:1713–1717
Cocquyt E, Verbruggen H, Leliaert F, De Clerck O (2010) Evolution and cytological diversification of the green seaweeds (Ulvophyceae). Mol Biol Evol 27:2052–2061
Collins MD, Brown J, Jones D (1988) Brachybacterium faecium gen. nov. sp. nov., a coryneform bacterium from poultry deep litter. Int J Syst Evol Microbiol 38:45–48
Cotta MA, Whitehead TR, Collins MD, Lawson PA (2004) Atopostipes suicloacale gen. nov., sp. nov., isolated from an underground swine manure storage pit. Anaerobe 10:191–195
Dai X, Wang YN, Wang BJ, Liu SJ, Zhou YG (2005) Planomicrobium chinense sp. nov., isolated from coastal sediment, and transfer of Planococcus psychrophilus and Planococcus alkanoclasticus to Planomicrobium as Planomicrobium psychrophilum comb. nov. and Planomicrobium alkanoclasticum comb. nov. Int J Syst Evol Microbiol 55:699–702
Dastager SG, Mawlankar R, Srinivasan K, Tang SK, Lee JC, Ramana VV, Shouche YS (2014) Fictibacillus enclensis sp. nov., isolated from marine sediment. Antonie Van Leeuwenhoek 105:461–469
De Maayer P, Chan WY, Rubagotti E, Venter SN, Toth IK, Birch PR, Coutinho TA (2014) Analysis of the Pantoea ananatis pan-genome reveals factors underlying its ability to colonize and interact with plant, insect and vertebrate hosts. BMC Genomics 15:404. https://doi.org/10.1186/1471-2164-15-404
De Mandal S, Zothansanga, Panda AK, Bisht SS, Kumar NS (2015) First report of bacterial community from a bat guano using Illumina next-generation sequencing. Genom Data 4:99–101
Dolka B, Chrobak-Chmiel D, Czopowicz M, Szeleszczuk P (2017) Characterization of pathogenic Enterococcus cecorum from different poultry groups: broiler chickens, layers, turkeys, and waterfowl. PLoS One 12:e0185199
Egas C, Barroso C, Froufe HJ, Pacheco J, Albuquerque L, da Costa MS (2014) Complete genome sequence of the radiation-resistant bacterium Rubrobacter radiotolerans RSPS-4. Stand Genomic Sci 9:1062–1075
Ehlers S, Merrill SA (2018) Staphylococcus saprophyticus. In: StatPearls [Internet]. Treasure Island: StatPearls; https://www.ncbi.nlm.nih.gov/books/NBK482367/
Falagán C, Johnson DB (2014) Acidibacter ferrireducens gen. nov., sp. nov.: an acidophilic ferric iron-reducing gammaproteobacterium. Extremophiles 18:1067–1073
Fouhy F, Clooney AG, Stanton C, Claesson MJ, Cotter PD (2016) 16S rRNA gene sequencing of mock microbial populations—impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol 16:123 http://www.biomedcentral.com/1471-2164/15/404
Fu Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, Pääbo S (2013) DNA analysis of an early modern human fromTianyuan Cave, China. Proc Natl Acad Sci U S A 110:2223–2227
Fudou R, Jojima Y, Seto A, Yamada K, Kimura E, Nakamatsu T, Hiraishi A, Yamanaka S (2002) Corynebacterium efficiens sp. nov., a glutamic-acid-producing species from soil and vegetables. Int J Syst Evol Microbiol 52:1127–1131
Gao M, Wang L, Chen SF, Zhou YG, Liu HC (2010) Salinicoccus kekensis sp. nov., a novel alkaliphile and moderate halophile isolated from Keke Salt Lake in Qinghai, China. Antonie Van Leeuwenhoek 98:351–357
Gerritsen J, Fuentes S, Grievink W, van Niftrik L, Tindall BJ, Timmerman HM, Rijkers GT, Smidt H (2014) Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int J Syst Evol Microbiol 64:1600–1616
Gómez F (2012) A checklist and classification of living dinoflagellates (dinoflagellata, alveolata). Oceánides 27:65–140
Gong ZL, Zhang CF, Jin R, Zhang YQ (2016) Steroidobacter flavus sp. nov., a microcystin-degrading Gammaproteobacterium isolated from soil. Antonie Van Leeuwenhoek 109:1073–1079
Greene AC (2014) The family Desulfurellaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes. Springer, Berlin ISBN 978-3-642-39043-2
Grottoli AG, Dalcin Martins P, Wilkins MJ, Johnston MD, Warner ME, Cai W-J et al (2018) Coral physiology and microbiome dynamics under combined warming and ocean acidification. PLoS One 13:e0191156
Ham H, Kim S, Kim MH, Lee S, Hong SK, Ryu JG, Lee T (2016) Mycobiota of ground red pepper and their aflatoxigenic potential. J Microbiol 54:832–837
Hayata B (1906) On Taiwania, a new genus of Coniferae from the island of Formosa. J Linn Soc Bot 37:330–331
Heyrman J, Logan NA, Busse HJ, Balcaen A, Lebbe L, Rodriguez-Diaz M et al (2003) Virgibacillus carmonensis sp. nov., Virgibacillus necropolis sp. nov. and Virgibacillus picturae sp. nov., three novel species isolated from deteriorated mural paintings, transfer of the species of the genus Salibacillus to Virgibacillus, as Virgibacillus marismortui comb. nov. and Virgibacillus salexigens comb. nov., and emended description of the genus Virgibacillus. Int J Syst Evol Microbiol 53:501–511
Heyrman J, Rodríguez-Díaz M, Devos J, Felske A, Logan NA, De Vos P (2005) Bacillus arenosi sp. nov., Bacillus arvi sp. nov. and Bacillus humi sp. nov., isolated from soil. Int J Syst Evol Microbiol 55:111–117
Hirooka K (2014) Transcriptional response machineries of Bacillus subtilis conducive to plant growth promotion. Biosci Biotechnol Biochem 78:1471–1484
Hong K, Gao AH, Xie QY, Gao H, Zhuang L, Lin HP, Yu HP, Li J, Yao XS, Goodfellow M, Ruan JS (2009) Actinomycetes for marine drug discovery isolated from mangrove soils and plants in China. Mar Drugs 7:24–44
Hori T, Noll M, Igarashi Y, Friedrich MW, Conrad R (2007) Identification of acetate-assimilating microorganisms under methanogenic conditions in anoxic rice field soil by comparative stable isotope probing of RNA. Appl Environ Microbiol 73:101–109
Howe AT, Bass D, Scoble JM, Lewis R, Vickerman K, Arndt H, Cavalier-Smith T (2011) Novel cultured protists identify deep-branching environmental DNA clades of Cercozoa: new genera Tremula, Micrometopion, Minimassisteria, Nudifila, Peregrinia. Protist 162:332–372
Huang Y, Wang L, Lu Z, Hong L, Liu Z, Tan GYA, Goodfellow M (2002) Proposal to combine the genera Actinobispora and Pseudonocardia in an emended genus Pseudonocardia, and description of Pseudonocardia zijingensis sp. nov. Int J Syst Evol Microbiol 52:977–982
Ishii S, Yamamoto M, Kikuchi M, Oshima K, Hattori M, Otsuka S, Senoo K (2009) Microbial populations responsive to denitrification-inducing conditions in rice paddy soil, as revealed by comparative 16S rRNA gene analysis. Appl Environ Microbiol 75:7070–7078
Isola D, Zucconi L, Onofri S, Caneva G, de Hoog GS, Selbmann L (2016) Extremotolerant rock inhabiting black fungi from Italian monumental sites. Fungal Divers 76:75–96
Jeon CO, Lim JM, Lee SS, Chung BS, Park DJ, Xu LH, Jiang CL, Kim CJ (2009) Paenibacillus harenae sp. nov., isolated from desert sand in China. Int J Syst Evol Microbiol 59:13–17
Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, Bateman A, Finn RD, Petrov AI (2018) Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res 46(D1):D335–D342. https://doi.org/10.1093/nar/gkx1038
Kämpfer P, Young CC, Busse HJ, Chu JN, Schumann P, Arun AB, Shen FT, Rekha PD (2010) Microlunatus soli sp. nov., isolated from soil. Int J Syst Evol Microbiol 60:824–827
Kim SB, Lonsdale J, Seong CN, Goodfellow M (2003) Streptacidiphilus gen. nov., acidophilic actinomycetes with wall chemotype I and emendation of the family Streptomycetaceae (Waksman and Henrici (1943) AL) emend. Rainey et al. 1997. Antonie Van Leeuwenhoek 83:107–116
Kim YG, Choi DH, Hyun S, Cho BC (2007a) Oceanobacillus profundus sp. nov., isolated from a deep-sea sediment core. Int J Syst Evol Microbiol 57:409–413
Kim MK, Schubert K, Im WT, Kim KH, Lee ST, Overmann J (2007b) Sphingomonas kaistensis sp. nov., a novel alphaproteobacterium containing pufLM genes. Int J Syst Evol Microbiol 57:1527–1534
Klochkova TA, Kang SH, Cho GY, Pueschel CM, West JA, Kim GH (2006) Biology of a terrestrial green alga, Chlorococcum sp. (Chlorococcales, Chlorophyta), collected from the Miruksazi stupa in Korea. Phycologia 45:349–358
Koerner RJ, Goodfellow M, Jones AL (2009) The genus Dietzia: a new home for some known and emerging opportunist pathogens. FEMS Immunol Med Microbiol 55:296–305
Koestler RJ (2003). Art, biology, and conservation: biodeterioration of works of art. New York: Metropolitan Museum of Art. p. 285. ISBN 978-1-58839-107-0
Kokcha S, Mishra AK, Lagier JC, Million M, Leroy Q, Raoult D, Fournier PE (2012) Non contiguous-finished genome sequence and description of Bacillus timonensis sp. nov. Stand Genomic Sci 6:346–355
Kovaleva OL, Merkel AY, Novikov AA, Baslerov RV, Toshchakov SV, Bonch-Osmolovskaya EA (2015) Tepidisphaera mucosa gen. nov., sp. nov., a moderately thermophilic member of the class Phycisphaerae in the phylum Planctomycetes, and proposal of a new family, Tepidisphaeraceae fam. nov., and a new order, Tepidisphaerales ord. nov. Int J Syst Evol Microbiol 65:549–555
Kurahashi M, Fukunaga Y, Sakiyama Y, Harayama S, Yokota A (2009) Iamia majanohamensis gen. nov., sp. nov., an actinobacterium isolated from sea cucumber Holothuria edulis, and proposal of Iamiaceae fam. nov. Int J Syst Evol Microbiol 59:869–873
Kuznetsov VD, Zaitseva TA, Vakulenko LV, Filippova SN (1992) Streptomyces albiaxialis sp. nov.—a new petroleum hydrocarbon-degrading species of thermo- and halotolerant Streptomyces. Microbiology 61:62–67
Kwon SW, Kim BY, Song J, Weon HY, Schumann P, Tindall BJ, Stackebrandt E, Fritze D (2007) Sporosarcina koreensis sp. nov. and Sporosarcina soli sp. nov., isolated from soil in Korea. Int J Syst Evol Microbiol 57:1694–1698
Lan PT, Sakamoto M, Sakata S, Benno Y (2006) Bacteroides barnesiae sp. nov., Bacteroides salanitronis sp. nov. and Bacteroides gallinarum sp. nov., isolated from chicken caecum. Int J Syst Evol Microbiol 56:2853–2859
Lee JC, Lee GS, Park DJ, Kim CJ (2008) Bacillus alkalitelluris sp. nov., an alkaliphilic bacterium isolated from sandy soil. Int J Syst Evol Microbiol 58:2629–2634
Leisner JJ, Laursen BG, Prévost H, Drider D, Dalgaard P (2007) Carnobacterium: positive and negative effects in the environment and in foods. FEMS Microbiol Rev 31:592–613
Li WJ, Chen HH, Zhang YQ, Kim CJ, Park DJ, Lee JC, Xu LH, Jiang CL (2005) Citricoccus alkalitolerans sp. nov., a novel actinobacterium isolated from a desert soil in Egypt. Int J Syst Evol Microbiol 55:87–90
Liu Y, Xie QY, Hong K, Li L, Zhao YM, Tang YL, An JY, Zhu PP, Xu CH (2013) Paracoccus siganidrum sp. nov., isolated from fish gastrointestinal tract. Antonie Van Leeuwenhoek 103:1133–1139
Lu J, Nogi Y, Takami H (2001) Oceanobacillus iheyensis gen. nov., sp. nov., a deep-sea extremely halotolerant and alkaliphilic species isolated from a depth of 1050 m on the Iheya Ridge. FEMS Microbiol Lett 205:291–297
Luque-Almagro VM, Blasco R, Martínez-Luque M, Moreno-Vivián C, Castillo F, Roldán MD (2011) Bacterial cyanide degradation is under review: Pseudomonas pseudoalcaligenes CECT5344, a case of an alkaliphilic cyanotroph. Biochem Soc Trans 39:269–274
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12
Maszenan AM, Seviour RJ, Patel BK, Rees GN, McDougall BM (1997) Amaricoccus gen. nov., a gram-negative coccus occurring in regular packages or tetrads, isolated from activated sludge biomass, and descriptions of Amaricoccus veronensis sp. nov., Amaricoccus tamworthensis sp. nov., Amaricoccus macauensis sp. nov., and Amaricoccus kaplicensis sp. nov. Int J Syst Bacteriol 47:727–734
Mathews SL, Pawlak JJ, Grunden AM (2014) Isolation of Paenibacillus glucanolyticus from pulp mill sources with potential to deconstruct pulping waste. Bioresour Technol 164:100–105
McGinnis JM, Cole JA, Dickinson MC, Mingle LA, Lapierre P, Musser KA, Wolfgang WJ (2015) Paracoccus sanguinis sp. nov., isolated from clinical specimens of New York State patients. Int J Syst Evol Microbiol 65:1877–1882
McLean RJ, Trainor FR (1965) Fasciculochloris, a new chlorosphaeraceaen alga from a Connecticut soil. Phycologia 4:145–148
Meehan CJ, Beiko RG (2014) A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol Evol 6:703–713
Mello A, Murat C, Bonfante P (2006) Truffles: much more than a prized and local fungal delicacy. FEMS Microbiol Lett 260:1–8
Mello A, Ding G-C, Piceno YM, Napoli C, Tom LM, DeSantis TZ, Andersen GL, Smalla K, Bonfante P (2013) Truffle Brûlés have an impact on the diversity of soil bacterial communities. PLoS One 8:e61945
Milanez GD, Masangkay FR, Thomas RC, Ordona MOGO, Bernales GQ, Corpuz VCM, Fortes HSV, Garcia CMS, Nicolas LC, Nissapatorn V (2017) Molecular identification of Vermamoeba vermiformis from freshwater fish in lake Taal, Philippines. Exp Parasitol 183:201–206
Mohd Nor MN, Sabaratnam V, Tan GYA (2017) Devosia elaeis sp. nov., isolated from oil palm rhizospheric soil. Int J Syst Evol Microbiol 67:851–855
Mouchacca J (1973) Deux Alternatria des sols arides d’Égypte: A. Chlamydosporum sp. nov. et A. Phragmospora van Emden. Mycopathol Mycol Appl 50:217–225
Mylnikov AP, Karpov SA (2004) Review of the diversity and taxonomy of cercomonads. Protistology 3:201–217
Nagel M, Andreesen JR (1991) Bacillus niacini sp. nov., a nicotinate-metabolizing mesophile isolated from soil. Int J Syst Bacteriol 41:134–139
Nakagawa Y, Sakane T, Yokota A (1996) Emendation of the genus Planococcus and transfer of Flavobacterium okeanokoites Zobell and Upham 1944 to the genus Planococcus as Planococcus okeanokoites comb. nov. Int J Syst Bacteriol 46:866–870
Navarrete AA, Venturini AM, Meyer KM, Klein AM, Tiedje JM, Bohannan BJM, Nüsslein K, Tsai SM, Rodrigues JLM (2015) Differential response of Acidobacteria subgroups to forest-to-pasture conversion and their biogeographic patterns in the Western Brazilian Amazon. Front Microbiol 6:1443. https://doi.org/10.3389/fmicb.2015.01443
Nawrocki EP, Eddy SR (2013) Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29(22):2933–2935
O’Sullivan NJ, Teasdale MD, Mattiangeli V, Maixner F, Pinhasi R, Bradley DG, Zink A (2016) A whole mitochondria analysis of the Tyrolean Iceman’s leather provides insights into the animal sources of Copper Age clothing. Sci Rep 6:31279. https://doi.org/10.1038/srep31279
Ogórek R, Dyląg M, Kozak B (2016) Dark stains on rock surfaces in Driny Cave (Little Carpathian Mountains, Slovakia). Extremophiles 20:641–652
Orsi WD, Smith JM, Liu S, Liu Z, Sakamoto CM, Wilken S, Poirier C, Richards TA, Keeling PJ, Worden AZ, Santoro AE (2016) Diverse, uncultivated bacteria and archaea underlying the cycling of dissolved protein in the ocean. ISME J 10:2158–2173
Otlewska A, Adamiak J, Stryszewska T, Kańka S, Gutarowska B (2017) Factors determining the biodiversity of halophilic microorganisms on historic masonry buildings. Microbes Environ 32:164–173
Patterson DJ (1989) Stramenopiles: chromophytes from a protistan perspective. In: Green JC, Leadbeater BSC, Diver WL (eds) The chromophyte algae: problems and perspectives. Clarendon. ISBN 0198577133
Pitt J. and Hocking A (2009) Fungi and food spoilage. Blackie Academic and Professional, London. https://doi.org/10.1007/978-0-387-92207-2
Quaedvlieg W, Binder M, Groenewald JZ, Summerell BA, Carnegie AJ, Burgess TI, Crous PW (2014) Introducing the consolidated species concept to resolve species in the Teratosphaeriaceae. Persoonia 33:1–40
Ragan MA, Goggin CL, Cawthorn RJ, Cerenius L, Jamieson AV, Plourde SM, Rand TG, Söderhäll K, Gutell RR (1996) A novel clade of protistan parasites near the animal-fungal divergence. PNAS 93:11907–11912
Reddy GS, Garcia-Pichel F (2007) Sphingomonas mucosissima sp. nov. and Sphingomonas desiccabilis sp. nov., from biological soil crusts in the Colorado Plateau, USA. Int J Syst Evol Microbiol 57:1028–1034
Rey T, Dumas B (2017) Plenty is no plague: Streptomyces symbiosis with crops. Trends Plant Sci 22:30–37
Riedel T, Spring S, Fiebig A, Petersen J, Göker M, Klenk HP (2014) Genome sequence of the pink to light reddish-pigmented Rubellimicrobium mesophilum type strain (DSM 19309(T)), a representative of the Roseobacter group isolated from soil, and emended description of the species. Stand Genomic Sci 9:902–913
Rosenberg E (2014) The family Prevotellaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes. Springer, Berlin
Ruuskanen MO, St. Pierre KA, St. Louis VL, Aris-Brosou S, Poulain AJ (2018) Physicochemical drivers of microbial community structure in sediments of Lake Hazen, Nunavut, Canada. Front Microbiol 9:1138. https://doi.org/10.3389/fmicb.2018.01138
Ryan MP, Pembroke JT (2018) Brevundimonas spp: emerging global opportunistic pathogens. Virulence 9:480–493
Saitoh S, Suzuki T, Nishimura Y (1998) Proposal of Craurococcus roseus gen. nov., sp. nov. and Paracraurococcus ruber gen. nov., sp. nov., novel aerobic bacteriochlorophyll a-containing bacteria from soil. Int J Syst Bacteriol 48:1043–1047
Samson RA, Peterson SW, Frisvad JC, Varga J (2011) New species in Aspergillus section Terrei. Stud Mycol 69:39–55
Schmitt S, Deines P, Behnam F, Wagner M, Taylor MW (2011) Chloroflexi bacteria are more diverse, abundant, and similar in high than in low microbial abundance sponge. FEMS Microbiol Ecol 78:497–510
Schommer NN, Gallo RL (2013) Structure and function of the human skin microbiome. Trends Microbiol 21:660–668
Selvakumar G, Kundu S, Joshi P, Nazim S, Gupta AD, Gupta HS (2010) Growth promotion of wheat seedlings by Exiguobacterium acetylicum 1P (MTCC 8707) a cold tolerant bacterial strain from the Uttarakhand Himalayas. Indian J Microbiol 50:50–56
Singleton DR, Furlong MA, Peacock AD, White DC, Coleman DC, Whitman WB (2003) Solirubrobacter pauli gen. nov., sp. nov., a mesophilic bacterium within the Rubrobacteridae related to common soil clones. Int J Syst Evol Microbiol 53:485–490
Sivapalan A, Metussin R, Harndan F, Zain RM (1998) Fungi associated with postharvest fruit rots of Durio graveolens and D. kutejensis in Brunei Darussalam. Australas Plant Pathol 27:274–277
Song W, Wang X, Gu J, Zhang S, Yin Y, Li Y, Qian X, Sun W (2017) Effects of different swine manure to wheat straw ratios on antibiotic resistance genes and the microbial community structure during anaerobic digestion. Bioresour Technol 231:1–8
Sterflinger K, Piñar G (2013) Microbial deterioration of cultural heritage and works of art—tilting at windmills? Appl Microbiol Biotechnol 97:9637–9646
Stirnberg A, Djamei A (2016) Characterization of ApB73, a virulence factor important for colonization of Zea mays by the smut Ustilago maydis. Mol Plant Pathol 17:1467–1479
Sun JQ, Wang XY, Wang LJ, Xu L, Liu M, Wu XL (2016) Saccharibacillus deserti sp. nov., isolated from desert soil. Int J Syst Evol Microbiol 66:623–627
Teasdale MD, Fiddyment S, Vnouček J, Mattiangeli V, Speller C, Binois A, Carver M, Dand C, Newfield TP, Webb CC, Bradley DG, Collins MJ (2017) The York Gospels: a 1000-year biological palimpsest. R Soc Open Sci 4:170988
Urich T, Lanzén A, Qi J, Huson DH, Schleper C, Schuster SC (2008) Simultaneous assessment of soil microbial community structure and function through analysis of the metatranscriptome. PLoS One 3(6):e2527
Urzì C, Salamone P, Schumann P, Rohde M, Stackebrandt E (2004) Blastococcus saxobsidens sp. nov., and emended descriptions of the genus Blastococcus Ahrens and Moll 1970 and Blastococcus aggregatus Ahrens and Moll 1970. Int J Syst Evol Microbiol 54:253–259
Velegraki A, Cafarchia C, Gaitanis G, Iatta R, Boekhout T (2015) Malassezia infections in humans and animals: pathophysiology, detection, and treatment. PLoS Pathog 11(1):e1004523
Venkateswaran K, Kempf M, Chen F, Satomi M, Nicholson W, Kern R (2003) Bacillus nealsonii sp. nov., isolated from a spacecraft-assembly facility, whose spores are gamma-radiation resistant. Int J Syst Evol Microbiol 53:165–172
Vetriani C, Crespo-Medina M, Antunes A (2014) The family Salinisphaeraceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes. Springer, Berlin, Heidelberg ISBN: 978-3-642-38921-4
Vinokurova NG, Boĭchenko LV, Arinbasarov MU (2003) Formation of alkaloids from Penicillium species fungi during growth on wheat kernels. Prikl Biokhim Mikrobiol 39:457–460
Visagie CM, Hirooka Y, Tanney JB, Whitfield E, Mwange K, Meijer M, Amend AS, Seifert KA, Samson RA (2014) Aspergillus, Penicillium and Talaromyces isolated from house dust samples collected around the world. Stud Mycol 78:63–139
Wang X, Xue Y, Yuan S, Zhou C, Ma Y (2008) Salinicoccus halodurans sp. nov., a moderate halophile from saline soil in China. Int J Syst Evol Microbiol 58:1537–1541
Wang K, Zhang L, Liu Y, Pan Y, Meng L, Xu T, Zhang C, Liu H, Hong S, Huang H, Jiang J (2015) Kocuria dechangensis sp. nov., an actinobacterium isolated from saline and alkaline soils. Int J Syst Evol Microbiol 65:3024–3030
Wang XW, Houbraken J, Groenewald JZ, Meijer M, Andersen B, Nielsen KF, Crous PW, Samson RA (2016) Diversity and taxonomy of Chaetomium and chaetomium-like fungi from indoor environments. Stud Mycol 84:145–224
Wang X, Jiang WK, Cui MD, Yang ZG, Yu X, Hu G, Zhang H, Hong Q (2017) Nocardioides agrisoli sp. nov., isolated from farmland soil. Int J Syst Evol Microbiol 67:3722–3727
Wang WC, Yan FF, Hu JY, Amen OA, Cheng HW (2018) Supplementation of Bacillus subtilis-based probiotic reduces heat stress-related behaviors and inflammatory response in broiler chickens. J Anim Sci 96:1654–1666
Weon HY, Kim BY, Kim MK, Yoo SH, Kwon SW, Go SJ, Stackebrandt E (2007) Lysobacter niabensis sp. nov. and Lysobacter niastensis sp. nov., isolated from greenhouse soils in Korea. Int J Syst Evol Microbiol 57:548–551
Wexler HM (2007) Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20:593–621
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR protocols: a guide to methods and applications. Academic, New York, pp 315–322
Wilson MC, Mori T, Rückert C, Uria AR, Helf MJ, Takada K, Gernert C, Steffens UA, Heycke N, Schmitt S, Rinke C, Helfrich EJ, Brachmann AO, Gurgui C, Wakimoto T, Kracht M, Crüsemann M, Hentschel U, Abe I, Matsunaga S, Kalinowski J, Takeyama H, Piel J (2014) An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506:58–62
Xi L, Zhang L, Ruan J, Huang Y (2012) Description of Verrucosispora qiuiae sp. nov., isolated from mangrove swamp sediment, and emended description of the genus Verrucosispora. Int J Syst Evol Microbiol 62:1564–1569
Yadav S, Kaushik R, Saxena AK, Arora DK (2011) Diversity and phylogeny of plant growth-promoting bacilli from moderately acidic soil. J Basic Microbiol 51:98–106
Yamamoto M, Ishii A, Nogi Y, Inoue A, Ito M (2006) Isolation and characterization of novel denitrifying alkalithermophiles, AT-1 and AT-2. Extremophiles 10:421–426
Yi TH, Han CK, Srinivasan S, Lee KJ, Kim MK (2010) Sphingomonas humi sp. nov., isolated from soil. J Microbiol 48:165–169
Yoon JH, Lee KC, Weiss N, Kang KH, Park YH (2003) Jeotgalicoccus halotolerans gen. nov., sp. nov. and Jeotgalicoccus psychrophilus sp. nov., isolated from the traditional Korean fermented seafood jeotgal. Int J Syst Evol Microbiol 53:595–602
Zalar P, de Hoog GS, Schroers HJ, Crous PW, Groenewald JZ, Gunde-Cimerman N (2007) Phylogeny and ecology of the ubiquitous saprobe Cladosporium sphaerospermum, with descriptions of seven new species from hypersaline environments. Stud Mycol 58:157–183
Zgurskaya HI, Evtushenko LI, Akimov VN, Voyevoda HV, Dobrovolskaya TG, Lysak LV, Kalakoutskii LV (1992) Emended description of the genus Agromyces and description of Agromyces cerinus subsp. cerinus sp. nov., subsp. nov., Agromyces cerinus subsp. nitratus sp. nov., subsp. nov., Agromyces fucosus subsp. fucosus sp. nov., subsp. nov., and Agromyces fucosus subsp. hippuratus sp. nov., subsp. nov. Int J Syst Bacteriol 42:635–641
Zhang YQ, Li WJ, Zhang KY, Tian XP, Jiang Y, Xu LH, Jiang CL, Lai R (2006) Massilia dura sp. nov., Massilia albidiflava sp. nov., Massilia plicata sp. nov. and Massilia lutea sp. nov., isolated from soils in China. Int J Syst Evol Microbiol 56:459–463
Zhang DC, Liu HC, Xin YH, Yu Y, Zhou PJ, Zhou YG (2009) Planomicrobium glaciei sp. nov., a psychrotolerant bacterium isolated from a glacier. Int J Syst Evol Microbiol 59:1387–1390
Zhang Q, Sun J, Liu S, Wei Q (2013a) Manure refinement affects apple rhizosphere bacterial community structure: a study in sandy soil. PLoS One 8(10):e76937
Zhang X, Yao Q, Cai Z, Xie X, Zhu H (2013b) Isolation and identification of myxobacteria from saline-alkaline soils in Xinjiang, China. PLoS One 8(8):e70466
Zhang ZY, Gao XH, Zhang YJ, Jia M, Lu XJ, Ma YC, Tian F, Xie Q, Tang SK (2015) Skermanella rubra sp. nov., a bacterium isolated from the desert of Xinjiang, China. Antonie Van Leeuwenhoek 108:627–632
Zhang T, Wang NF, Zhang YQ, Liu HY, Yu LY (2016) Diversity and distribution of aquatic fungal communities in the Ny-Ålesund region, Svalbard (High Arctic): aquatic fungi in the Arctic. Microb Ecol 71:543–554
Zhao J, Guo L, Sun P, Han C, Bai L, Liu C, Li Y, Xiang W, Wang X (2015) Actinomadura jiaoheensis sp. nov. and Actinomadura sporangiiformans sp. nov., two novel actinomycetes isolated from muddy soil and emended description of the genus Actinomadura. Antonie Van Leeuwenhoek 108:1331–1339
Zhou N, Zhang Y, Liu F, Cai L (2016) Halophilic and thermotolerant Gymnoascus species from several special environments, China. Mycologia 108:179–191
Zhu WY, Zhao LX, Zhao GZ, Duan XW, Qin S, Li J, Xu LH, Li WJ (2012) Plantactinospora endophytica sp. nov., an actinomycete isolated from Camptotheca acuminata Decne., reclassification of Actinaurispora siamensis as Plantactinospora siamensis comb. nov. and emended descriptions of the genus Plantactinospora and Plantactinospora mayteni. Int J Syst Evol Microbiol 62:2435–2442
Acknowledgements
The authors wish to thank Dr. Georg Plattner from the KHM (Vienna) for the sampling facilities and for the support.
Funding
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU). This study was funded by the Federal Criminal Police Office and the Federal Monuments Office, Vienna, Austria.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 35 kb)
Supplementary Figure 1
(PPTX 183 kb)
Supplementary Figure 2
(PPTX 55 kb)
Supplementary Figure 3
(HTML 688 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Piñar, G., Poyntner, C., Tafer, H. et al. A time travel story: metagenomic analyses decipher the unknown geographical shift and the storage history of possibly smuggled antique marble statues. Ann Microbiol 69, 1001–1021 (2019). https://doi.org/10.1007/s13213-019-1446-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13213-019-1446-3