
Abstract
The world has recently been plagued by a new coronavirus infection called SARS-CoV-2. This virus may lead to severe acute respiratory syndrome followed by multiple organ failure. SARS-CoV-2 has approximately 80–90% genetic similarity to SARS-CoV. Given the limited omics data available for host response to the viruses (more limited data for SARS-CoV-2), we attempted to unveil the crucial molecular mechanisms underlying the SARS-CoV-2 pathogenesis by comparing its regulatory network motifs with SARS-CoV. We also attempted to identify the non-shared crucial molecules and their functions to predict the specific mechanisms for each infection and the processes responsible for their different manifestations. Deciphering the crucial shared and non-shared mechanisms at the molecular level and signaling pathways underlying both diseases may help shed light on their pathogenesis and pave the way for other new drug repurposing against COVID-19. We constructed the GRNs for host response to SARS-CoV and SARS-CoV-2 pathogens (in vitro) and identified the significant 3-node regulatory motifs by analyzing them topologically and functionally. We attempted to identify the shared and non-shared regulatory elements and signaling pathways between their host responses. Interestingly, our findings indicated that NFKB1, JUN, STAT1, FOS, KLF4, and EGR1 were the critical shared TFs between motif-related subnetworks in both SARS and COVID-1, which are considered genes with specific functions in the immune response. Enrichment analysis revealed that the NOD-like receptor signaling, TNF signaling, and influenza A pathway were among the first significant pathways shared between SARS and COVID-19 up-regulated DEGs networks, and the term “metabolic pathways” (hsa01100) among the down-regulated DEGs networks. WEE1, PMAIP1, and TSC22D2 were identified as the top three hubs specific to SARS. However, MYPN, SPRY4, and APOL6 were the tops specific to COVID-19 in vitro. The term “Complement and coagulation cascades” pathway was identified as the first top non-shared pathway for COVID-19 and the MAPK signaling pathway for SARS. We used the identified crucial DEGs to construct a drug–gene interaction network to propose some drug candidates. Zinc chloride, Fostamatinib, Copper, Tirofiban, Tretinoin, and Levocarnitine were the six drugs with higher scores in our drug–gene network analysis.
Similar content being viewed by others
Introduction
COVID-19, SARS, and MERS are all frightening diseases caused by viral agents from a large family of viruses called coronaviruses (Munster et al. 2020). Although dozens of coronaviruses have been reported to infect various living organisms, including mammals, rodents, and birds, only seven have been able to infect humans. In 2020, Chen et al. listed six important pathogenic coronaviruses, including (HKU1, 229E, OC43, NL63, MERS, and SARS-CoV) (Chen et al. 2020). SARS-CoV-2 is considered the seventh member of the coronavirus family, which can infect humans and cause deadly pneumonia even in the young population (Paules et al. 2020). One of the predominant symptoms of patients can be severe acute respiratory syndrome (SARS), a respiratory condition with approximately 10% mortality (Organization 2003).
Analysis of the SARS-CoV-2 genome showed that this new virus shares about 80–90% sequence identity with the original SARS-CoV (Zhou et al. 2020). SARS-CoV is a positive-stranded ribonucleic acid (RNA) virus of the family of Coronaviridae (Guan et al. 2004; Peiris et al. 2004). Their genome is surrounded by a membrane that has various parts, such as the transmembrane (M) glycoprotein, the spike (S) glycoprotein, and the envelope (E) protein (Neuman et al. 2006). The S protein, an essential protein available on the SARS-CoV membrane, is crucial in penetrating host cells and initiating infection. The S protein interacts with the type 2 enzyme of humans converting angiotensin (ACE2) (Li et al. 2003). Thus, ACE2 is a functional SARS-CoV receptor in vivo (Imai et al. 2005; Kuba et al. 2005). Likewise, the SARS-CoV-2 spike S protein binds to its human receptor ACE2 (angiotensin-converting enzyme 2) through its receptor-binding domain (RBD), and ACE2 is recognized as a receptor in SARS-CoV-2 entry (Zhang et al. 2020).
Given their phylogenetic proximity, deciphering each of these two evolutionary cousins' molecular mechanisms will likely help us predict another viral pathology underlying mechanisms. However, more information is available on the SARS-CoV molecular mechanism than SARS-CoV-2; there are still many gaps in our knowledge of the host response mechanisms to SARS-CoV. For example, although some studies have tried approximating both illnesses’ mechanisms (Delgado-Chaves et al. 2020; Guzzi et al. 2020), no study has yet deciphered the regulatory motif elements in SARS-CoV host response GRN. Although both diseases were soon known for their pathogenesis in leading to acute respiratory distress, a recent single-cell sequencing study predicted that SARS-CoV-2 could harm most other organs in the body, such as the heart, kidney, and brain (Zaim et al. 2020). Their results proved that our understanding of the virus mechanisms has been insufficient over the past few months.
COVID-19 still kills some youth with no history of physical weakness daily and can potentially harm several vital organs, including the lung, brain, and digestive system (Renu et al. 2020). So far, no significantly effective drug has been discovered, perhaps due to its complex mechanism (Organization 2020). Unfortunately, many patients with severe forms of COVID-19 still die worldwide each day, and it highlights the importance of the immediate discovery of effective medications against the infection, and many research centers are working hard to find an effective drug against it. However, many common approaches to identifying disease mechanisms and drug design are reductionist in scope or at least time-consuming.
A relatively new and holistic study approach in biology, called systems biology, improved our understanding of various diseases' molecular mechanisms in the last twenty years. It has helped boost our knowledge of the regulatory interactions mediating many diseases (Dix et al. 2016; Sakata and Winzeler 2007). Various network analyses, including GRNs (gene regulatory networks), have been the main pivots in several studies (Noori et al. 2020; Sun et al. 2012). They have been used to decipher the mechanisms underlying drug effects or other treating interferences (Dehghan et al. 2022; Farahani et al. 2018; Khazaei‐Poul et al. 2022). Previous studies have shown that transcription factors and miRNAs are the main pillars of GRNs and play crucial roles in regulating their target genes (Zhou et al. 2007). It has been shown that TF-mediated transcriptional and posttranscriptional miRNA-mediated regulation is usually associated with each other in networks (Chen et al. 2011). GRNs have contributed to predicting the mechanisms underlying various diseases and even repurposing drugs (Zickenrott et al. 2016). New omics techniques have provided high-throughput transcript-omics data, paving the way to gain a more in-depth holistic insight into various processes mediated in disease pathogenesis. Shared crucial regulatory elements among some relevant diseases have recently been applied to heighten our perception and insights into the disease’s molecular mechanisms in more detail (Sun and Hu 2016). The new insights may clarify the crucial molecules that may presumably be applied to identify new prophylactic treatments, diagnosis methods, and even new drug designs.
Drug design methods use different in silico approaches to facilitate and accelerate the finding of proper medications. One common approach is applying docking methods and molecular dynamics. Several studies have attempted to introduce drug candidates against the crucial proteins of SARS-CoV-2 in a relatively short time after its emergence. SARS-CoV-2 3Clpro (Molavi et al. 2021), main protease (Singh et al. 2022a), helicase (Borgio et al. 2020), RdRp (RNA‐dependent RNA polymerase) (Singh et al. 2021), the non-structural and protein 15 (Singh et al. 2022b), have been targeted using these methods by several studies. They have also been applied in investigations on SARS-CoV-2 entry through the affinity of spike protein and its receptor (Aljindan et al. 2021). Another in silico approach to introducing drug candidates is applying drug-gene network analysis, and several studies have applied it to propose drug candidates against diseases (Dehghan et al. 2021; Huang et al. 2020).
In this study, we pursue three goals. First, we focus on analyzing the molecular mechanism of the host response to SARS-CoV. To do this, we first identify the molecular mechanisms of the SARS-CoV host respiratory system response by deciphering the SARS regulatory network motifs. As the second aim, we investigated the molecular relationships between SARS and COVID-19 regulatory networks, thereby identifying shared processes that are likely the crucial components of the molecular pathogenesis in both diseases. We also aimed to identify the non-shared crucial molecules and their functions to predict the specific mechanisms of each infection. It helps unravel the molecular mechanisms responsible for the two infections’ manifestations. For example, we identified six transcription factors as probably shared master regulators of SARS-CoV-2 and SARS-CoV pathogenesis, introduced in subsequent sections. Besides, we identified some crucial nodes in the SARS-specific networks, such as WEE1, PMAIP1, and TSC22D2. However, others were identified as crucial and specific to COVID-19 in vitro, like MYPN, SPRY4, and APOL6. Third, we aimed to repurpose new candidate drugs against COVID-19 or its symptoms based on our identified shared crucial molecules underlying both diseases’ pathogenesis. We utilize a drug–gene interaction network for the selected crucial genes to recommend new candidate drugs against COVID-19.
Methods
Overview of the study design
Initially, to retrieve DEGs (differentially expressed genes), three different microarray datasets were found for respiratory cell lines infected with SARS-CoV. For data related to COVID-19, the analyzed RNA-seq data from two different respiratory cell lines were used. The up- and down-regulated DEGs were filtered by P value and fold change (p value < 0.05 and |Log FC|> 0.5).
Several experimental databases were used to obtain four relationships between our DEGs, their regulating TFs, and miRNAs, and the TFs regulating the miRNAs to construct the GRNs (gene regulatory networks) for each disease. FANMOD software was used to identify the significant regulatory motifs for each network separately. Criteria of Z-score > 2.0 and p value < 0.05 were then applied to identify the significant motifs and motif-related subnetworks constructed using Cytoscape. The intersecting network between the motif-related subnetworks of each disease was constructed and analyzed to obtain the crucial nodes shared between the two diseases. The shared nodes with higher degree scores were identified. The non-shared nodes for each disease were also determined. We also constructed a drug–gene interaction network for the crucial shared DEGs between the two infections and the crucial DEGs specifically for COVID-19 using the dgidb database to propose possible candidate drugs and verified some using literature. The shared and non-shared pathways and gene ontology terms for the two infections were also identified and discussed as the possible mechanisms of the two diseases.
Expression data
COVID-19 data collection
Since COVID-19 is a relatively new infection, the related omics data are still insufficient in omics databanks. A recently published article regarding respiratory infections by Daniel Blanco-Melo et al. was utilized to extract the analyzed data related to COVID-19. In this study, two epithelial cell lines of the lungs' respiratory system (NHBE and A549) were applied as in vitro study models (Blanco-Melo et al. 2020b). A549 cells were infected at a multiplicity of infection (MOI) of 0.2 virus particles per cell for 24 h with SARS-CoV-2. However, NHBE cells were infected with SARS-CoV-2 at MOI of 2 for 24 h. Total RNA from infected and mock cells was extracted, and RNA-Seq libraries of poly-adenylated RNA were prepared using the TruSeq RNA Library Prep Kit v2 (Illumina). The cDNA libraries were sequenced using an Illumina NextSeq 500 platform (www.illumina.com). The raw data of each cell line were analyzed separately to obtain genes that were expressed differentially (DEGs), and then, up- and down DEGs were filtered. In this study, the criteria for filtering were p value < 0.05 and |Log FC|> 0.5. Our study’s results relate to respiratory epithelial cells, excluding immunological cells.
SARS data collection
Initially, to retrieve DEGs (differentially expressed gene data), we searched through NCBI Gene Expression Omnibus (GEO) and the European Array Express databank (Barrett et al. 2007). The SARS omics data was limited in databanks compared to data available for other diseases. A possible explanation might be that the SARS disease was controlled relatively soon and did not last long in populations compared with other diseases. Three different data sets were found and selected to be further analyzed (GSE33267, GSE37827, and GSE47960) (Aevermann et al. 2014; Mitchell et al. 2013; Sims et al. 2013b). In both GSE33267 and GSE37827, Calu-3 cells (a lung cancer cell line) were infected with icSARS-CoV (Mitchell et al. 2013). In GSE47960, HAE cells (human airway epithelium cell line) were infected by SARS-CoV (Sims et al. 2013b).
SARS data analysis of differentially expressed genes
The datasets relating to the 60 h early post-infection time-point, shared among the three SARS datasets, were analyzed. The three datasets were filtered, normalized, and analyzed (t Test) using the ge-Workbench_2.6.0 software. DEGs were filtered by criteria for P value and fold change (p value < 0.05 and |Log FC|> 0.5). We then selected the DEGs shared among the three dataset results using the online Venn diagram tool for the early time point (60 h post-infection) (http://bioinformatics.psb.ugent.be). We used the available raw data from the early 60-h post-infection time-point shared among the SARS datasets. We chose the 60 h post-infection data because first, it was available in all the three SARS-related datasets, and second, it had shown the most infection impact (among the shared time-points of the available datasets) and had the highest pick in the graph curves of the study by Sims et al. (Sims et al. 2013a). Note that, 60 and 24 h post-infection time-points are both considered as very early time-points in viral infection with SARS-CoV-2 among the three hypothetical stages and patients may show clinical manifestations even after 14 days post-infection and then continue in the first stage for five days on average (Molavi et al. 2021; Romagnoli et al. 2020).
Transcription factors and miRNAs regulating the DEGs
We used only the miRNAs whose interaction with their target gene was experimentally proven, so miRNAs regulating the DEGs were retrieved using two manually curated miRNA databases (miRTarBase and miRecords) (Chou et al. 2016; Xiao et al. 2009). To obtain transcription factors regulating genes, we used the TRRUST and TRANSFAC/JASPAR databases (http://jaspar.genereg.net) (Chekmenev et al. 2005; Han et al. 2018).
Identification of miRNAs regulating TFs
TFs were considered genes when identifying miRNA–TF interactions using the miRTarBase and the miRecords to find the regulatory relationships between miRNAs and TFs (Chou et al. 2016; Xiao et al. 2009).
Identification of TFs regulating miRNAs
The TransmiR database (http://www.cuilab.cn/transmir), an open-source database that identifies transcription factors regulating microRNAs, was used to find the regulatory relationship of TF-miRNA pairs (Tong et al. 2019; Wang et al. 2010).
Construction of gene regulatory networks (GRN) and network topological analysis
We combined the four types of relationships (miRNA͢Gene, TF͢Gene, miRNA͢TF, and TF͢miRNA) to construct a transcription factor-microRNA-target gene regulatory network for SARS and COVID-19 distinctly and visualized them using Cytoscape 3.8.0. The overall four regulatory networks for the up- and down-regulated DEGs in each disease were constructed separately.
The four regulatory networks were then analyzed topologically using the Cytoscape network analyzer tool (Shannon et al. 2003). Highly connected nodes were considered hubs (top 10% of nodes with the highest degree). Since bottleneck-ness is also a significant representative of essentiality in regulatory networks, we also identified the bottlenecks (Yu et al. 2007). The top 10% of nodes with the highest betweenness centrality were considered bottlenecks. The hub and bottlenecks were used to create the new subnetworks. The new intersection network was created using the shared nodes between hubs and bottlenecks (hub bottlenecks) in SARS GRN and COVID-19 GRNs for up- and down-regulated DEGs separately (Killcoyne et al. 2009).
Motif detection
FANMOD software was applied to identify the 3-node motifs. FANMOD is a tool for identifying motifs and uses recent algorithms to improve motif detection efficiency by some orders of magnitude compared to other software. It identifies motifs occurring significantly in the network more than detected in 1000 random networks using Z-score. We detected 3-node motifs based on Z-score > 2.0 and p value < 0.05 (Wernicke and Rasche 2006). The motifs with at least two colors in their edges (at least included two types of relationships) were selected as significant motifs in SARS and COVID-19 for up- and down-regulated genes separately. The nodes participating in the selected significant motifs were then retrieved using the Venn diagram. Further, they were visualized separately using Cytoscape, and a new regulatory subnetwork was constructed for each separate motif ID number (FANMOD motif ID number) in Cytoscape. The participating nodes that played dual roles (shared between the TF and DEG lists) were also identified.
Identification of motifs intersection network
The motif-related subnetworks obtained in the previous step were then merged to construct a new subnetwork in each disease (for up and down DEGs separately). The nodes shared among all the motif-related subnetworks were subsequently identified for each disease (for up and down DEGs separately) and introduced as essential molecules underlying the molecular mechanisms of host response in each viral infection independently. Besides, the non-shared nodes of the significant motifs for each disease were identified separately to predict the specific mechanisms for each infection.
Pathway and gene ontology functional analysis for hub-bottleneck genes of the union of motif-related subnetworks
Gene set enrichment analysis (GSEA) is a common approach for functional analysis and interpretation of gene expression data based on DEGs functional annotation. We used two online tools: the DAVID functional annotation tool (DAVID; http://david.abcc.ncifcrf.gov) and the STRING database (Huang et al. 2007).
Three types of gene sets were selected for further enrichment analysis. Firstly, genes related to the intersection between the two disease GRN networks were enriched. Secondly, genes related to the merged union of motif-related subnetworks in each of the four GRN networks were enriched, and finally, the non-shared nodes of the significant motifs were enriched separately. The non-shared nodes can probably help us predict the specific mechanisms for each infection. It will probably contribute to identifying the molecular mechanisms responsible for partially different manifestations of the two infections.
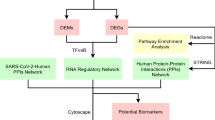
Following the construction of union subnetworks, we analyzed each subnetwork topologically to select the four subnetworks’ hub-bottleneck genes. The transcription factors were also selected if they were among the DEGs of the network disease. The four new gene sets were then enriched individually for the biochemical pathways of the Kyoto Encyclopedia of Genes and Genomes (KEGG) (KEGG; www.genome.ad.jp/KEGG) using both DAVID and STRING database, and the shared results between the two online tools were listed separately for each of the four sets of genes. The shared pathways and BPs terms were then sorted based on the p-value obtained from the STRING tool. Finally, we determined the shared and non-shared biochemical pathways and Gene Ontology terms (GOs) between SARS and COVID-19 (in vitro). A workflow representing data analysis, GRN construction, and further steps of the survey is depicted in Fig. 1.

This workflow represents the graphical overview of the study
Drug–gene interaction network construction
The crucial TF/genes identified exclusively related to COVID-19 and the crucial shared TF/genes between the two infections were selected to construct the drug–target interaction network for further drug screenings against COVID-19. We used the drug–gene interaction database (dgidb) to retrieve the medications with possible interactions with the selected genes (Cotto et al. 2018). Cytoscape (3.8.0) was used for network construction and visualization. The network was analyzed, and the drugs with higher degree scores (two or higher) were nominated to construct a new drug–gene sub-network. The new repurposed drug candidates were then validated using the ClinicalTrials.gov databank for COVID-19 clinical trials and discussed for COVID-19 or similar pathogens.
Results
We investigated the molecular regulatory mechanism of the host response to SARS-CoV-2 by comparing its regulatory network motifs with SARS-CoV. We aimed to investigate the molecular relationships between the SARS and COVID-19 regulatory networks. We identified the crucial shared and non-shared nodes between the networks of SARS and COVID-19 (in vitro). Finally, based on drug–gene interaction network analysis, we used the identified crucial molecules to repurpose drugs against COVID-19 or its manifestations.
COVID-19 and SARS data collection
The omics data for SARS-CoV-2 was available only for 24 h post-infection. We analyzed the SARS data for 60 h post-infection (the available shared early point data) to compare the two disease networks in the two available early time points. First, we filtered, normalized, and analyzed the raw data of three SARS-related datasets using ge-Workbench software (p value < 0.05 and |Log FC|> 0.5). We searched for the shared DEGs between the three data analysis results and identified them as DEGs related to the SARS-CoV host response in vitro for the time points. We identified 329 up-regulated and 228 down-regulated DEGs as SARS-related DEGs in 60 h. (Data are shown in Supplementary Figure S1 and Supplementary Table S1C.) For two COVID-19 datasets, we identified 572 up-regulated and 291 down-regulated genes as the COVID-19-related DEGs. (Data are presented in Supplementary Table S1D.) We searched for the shared DEGs among the two diseases. We also searched for the crucial shared DEGs between COVID-19 and DEGs related to SARS-related networks of 60 h. The results were related to respiratory epithelial cells, excluding immunological cells.
Transcription factors–miRNAs–DEGs relationships
Supplementary Tables S2, S3, S24, and S25 contain the regulatory elements of up-regulated and down-regulated SARS and COVID-19-related DEGs. miRecords and miRTarBase were searched to retrieve miRNAs that regulate the DEGs. Finally, 1512 microRNAs were retrieved for the up-regulated SARS DEGs, and 1406 miRNAs were obtained for the down-regulated DEGs. Moreover, 1569 miRNAs were extracted for the up-regulated COVID-19 DEGs and 1365 miRNAs for the down-regulated COVID-19 DEGs.
Transcription factors that regulate the DEGs were obtained from the TRRUST and TRANSFAC/JASPAR databases. Finally, 306 TFs were extracted for the SARS up-regulated DEGs and 211 TFs for the SARS down-regulated DEGs. Table S25 shows 319 and 242 transcription factors regulating the up- and down-regulated COVID-19 DEGs (Supplementary Table S3).
The subsequent regulatory relationships were the miRNAs regulating TFs. We detected 1493 unique miRNAs suppressing the TFs related to the SARS down-regulated DEGs and 1575 single miRNAs regulating the TFs related to the SARS up-regulated DEGs (Table S1D). TFs were considered genes, and two miRNA databases (miRTarBase and miRecord) were searched for TF-suppressing miRNAs in COVID-19. Table S25 shows that 1604 and 1526 microRNAs were obtained for the TFs related to the COVID-19 up- and down-regulated DEGs, respectively.
To construct the interaction of TFs regulating the miRNAs (TF → miRNA), we used the TransmiR database. We finally found 357 TFs regulating the miRNAs related to the SARS up- and down-regulated DEGs. The obtained miRNAs linked to the COVID-19 DEGs were also searched in the TransmiR database. Finally, 357 TFs were retrieved for the miRNAs regarding the up-regulated DEGs and 355 for the down-regulated (Tables S3 and S24).
Construction of gene regulatory networks and the intersection between SARS and COVID-19
Four separate regulatory networks of the TF-miRNA target gene were constructed by combining the four relationships (miRNA → gene, TF → gene, miRNA → TF, and TF → miRNA interactions) using the Cytoscape software for SARS and COVID-19 up- and down-regulated DEGs separately. A total of 2747 nodes and 24,903 edges for the SARS up-regulated DEGs network, and 2494 nodes and 18,138 edges for the SARS down-regulated DEGs regulatory network were identified. The COVID-19 up-regulated DEGs regulatory networks included 2755 nodes and 31,376 edges, and the down-regulated network included 2374 nodes and 19,163 edges.
Shared hub bottlenecks between SARS and COVID-19
To identify the shared hubs and bottlenecks between the SARS and COVID-19 regulatory networks, we first constructed four new subnetworks separately; each included the top 10% union of the hubs and the top 10% of the bottlenecks of its corresponding network (For COVID-19 and SARS up- and down-regulated separately). The four new subnetworks were then merged in Cytoscape (pairwise) using the intersectional merging analysis. Two new networks were created as intersection subnetworks between SARS and COVID-19 (for up and down networks separately). As visualized in Figure S2, the intersection between the two SARS and COVID-19 sub-networks of the top 10% hubs and bottlenecks was constructed (related to up-regulated DEGs). The new intersection subnetworks are included 138 nodes and 1344 edges, containing 64 TFs and 43 miRNAs, and 19 genes. Some crucial nodes played dual roles in the subnetwork as both Gene and TF, including STAT2, FOSL2, TNF, NFKB1, FOS, JUN, FOSL1, STAT1, IL6, PRDM1, KLF6, and HIF1A. Figure S3 demonstrates a similar comparison between the two subnetworks related to the down-regulated DEGs between the two diseases. It included 137 nodes and 936 edges, 52 miRNAs, 78 TFs, and some genes, including PPARGC1A, SPTLC3, TSC22D3, and VAV3. The nodes with the dual role (gene/TF) in this intersection were ZNF148 and TCF4.
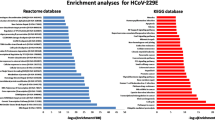
Top shared biochemical pathways and GOs between the two diseases
Shared hub-bottleneck nodes in SARS and COVID-19 regulatory networks were used to determine the functional enrichments. The NOD-like receptor signaling pathway was the top shared pathway between SARS and COVID-19 up-regulated DEGs (Table S4 and Table 1). Besides, as shown in Table S5 and Table 2, defense response, response to other organisms, and innate immune response were the top three shared biological processes between SARS and COVID-19 up-regulated DEGs. However, it is apparent from Table 2 that the most significant biological process shared between SARS and COVID-19 in down-regulated DEGs was energy homeostasis. As shown in Table 1, the most significant pathway between SARS and COVID-19 in down-regulated DEGs was the term “Metabolic pathways” (hsa01100).
Motif detection
To decipher the regulatory 3-node motifs, the data of the four mentioned types of the relationships (miRNA͢Gene, TF͢Gene, miRNA͢TF, and TF͢miRNA) were incorporated together. They were inserted as an input file to the FANMOD software (TSV format) for each of the four regulatory networks separately (SARS up, SARS down, COVID up, COVID down) (Wernicke and Rasche 2006). FANMOD software parameters were set to detect the significant 3-node size motifs. The FANMOD detected 13 significant motifs in the SARS up-regulated DEGs network and 12 in the SARS down-regulated DEGs (Supplementary Table S6A). Supplementary Table S6B shows nine significant motifs in the COVID-19 up-regulated DEGs network, and 12 significant motifs in the COVID-19 down-regulated DEGs were detected.
Motifs detected for the SARS DEGs
The significant motifs with edges with at least two colors were selected to construct the new motif-related sub-networks of the SARS up-regulated DEGs. The selected motifs identification codes are available in Table S24, and the motif nodes are depicted in Supplementary Table S7. The motifs with the same identity number were merged and considered unique motifs (Table S24). For example, we combined the subnetworks of the two motifs with the same identity number of 78. In this motif, 57 TFs and 91 unique microRNAs participated. We checked to see which of these TFs were among our related DEGs. Interestingly, we found that some TFs played a role as a gene and were available among the related DEGs; therefore, they were also considered dual TF/DEGs in the SARS network, including NFKB1, FOS, JUN, STAT1, EGR1, PRDM1, and KLF4. More detailed explanations of the other SARS Motif-related nodes are represented in Supplementary text S1.
Motifs detected for COVID-19 DEGs
As represented in Supplementary Table S8, nine motifs were identified as significant by FANMOD in the COVID-19 up-regulated DEGs networks and 12 motifs in the down-regulated. The selected motifs (with edges with at least two colors) and the number of participating nodes in each motif-related subnetwork are introduced in Table S25. In brief, three motif-related subnetworks were selected in the COVID-19 up-regulated DEG networks. For example, motif No.14 had 76 TFs and 300 microRNAs, of which HIF1A, RELB, FOSL1, STAT1, and PRDM1 were considered TF/Gene. More detailed explanations of the other COVID-19 Motif-related nodes are represented in Supplementary text S1.
Motifs intersection networks
The nodes participating in regulatory motifs are essential in disease molecular biology. Therefore, we attempted to identify the shared nodes among the selected motifs separately in each of the four networks. We first made a separate subnetwork for each motif to construct the intersectional subnetworks. We then merged them (the Cytoscape command) to create intersectional subnetworks. These intersections for SARS are shown in Fig. 2 for up-regulated DEGs, in which FOS, JUN, NFKB1, STAT1, KLF4, and EGR1 considered TF/DEGs, and Figure S4 depicts the junction between the significant motifs in down-regulated DEGs in SARS.

The intersection among motif-related subnetworks of SARS up-regulated DEGs network. The V-shape nodes show only TFs; circle nodes represent miRNAs; diamond nodes depict only genes. The regulatory nodes modulate the shared DEGs between all five significant motif-related subnetworks of the SARS up-regulated DEGs network. All motifs are represented by their identification motif number in the FANMOD software encyclopedia. Z-scores (> 2) and P values (< 0.05) are reported for each motif
Furthermore, the top ten shared nodes between the selected motifs related to SARS up- and down-regulated DEGs are shown in Table 3. The shared nodes between the selected motifs related to COVID-19 up-regulated DEGs are shown in Fig. 3. Some nodes matched to this intersection played dual roles as TF and DEGs, including STAT1, NFKB1, HIF1A, and PRDM1.

The intersection among motif-related subnetworks of COVID-19 up-regulated DEGs network. The V-shape nodes show only TFs; circle nodes represent miRNAs; diamond nodes depict TF/Genes (Dual role). The regulatory nodes modulate the shared DEGs between all three (three motif IDs) significant motif-related subnetworks of the COVID-19 up-regulated DEGs network. All motifs are represented by their identification motif number in the FANMOD software encyclopedia. Z-scores (> 2) and P values (< 0.05) are reported for each motif
Moreover, FOS, KLF4, EGR1, JUN, and ATF3 were the other shared gene/TFs between motif-related subnetworks. (They had dual regulatory roles). Moreover, the top ten shared nodes between the selected motifs related to COVID-19 up- and down-regulated DEGs are demonstrated in Table 4. All crucial nodes in SARS and COVID-19 are shown in Tables S1A and S1B.
We also identified 414 genes in COVID-19 exclusively and 192 nodes in SARS as up-regulated DEGs of the significant motif-related subnetworks for each disease (Supplementary Table S9). As shown in Table S9, 63 nodes in COVID-19 and 50 nodes in SARS were considered motifs down-regulated DEGs in each disease separately.
Topological analysis of hub-bottleneck genes of union motif networks
To obtain hub bottlenecks in the SARS and COVID-19 up- and down-regulated DEG networks, we merged the motif subnetworks in each of the four networks separately and created four new union subnetworks. Then, we analyzed each of these four subnetworks to identify their hub bottlenecks (the shared nodes between the top 10% of values of the degree and betweenness centrality), introduced in Supplementary Tables S10 and S11. The top five hubs and bottlenecks for each of the four networks are represented in Tables 3 and 4. Fifty-three nodes were considered the hub bottlenecks in the COVID-19 and 192 SARS up-regulated DEGs subnetwork and 166 nodes in COVID-19 and also 156 nodes in SARS as down-regulated nodes.
Overall, as represented in Tables S1A and S1B, we identified that NFKB1, JUN, STAT1, FOS, KLF4, and EGR1 were the critical shared TFs between motif-related subnetworks in both SARS and COVID-19. The crucial TF/DEGs shared among the two infections included NFKB1 and STAT1. Besides JUN, FOS, KLF4, and EGR1 were the shared TF/genes between motif-related subnetworks in SARS and COVID-19. Moreover, NFKB1, JUN, STAT1, and FOS also were identified as the shared hub bottlenecks. WEE1, PMAIP1, and TSC22D2 were identified as the top three DEGs with the highest degree scores specific to SARS. Besides, MYPN, SPRY4, and APOL6 were the crucial nodes specific to COVID-19 in vitro.
Functional analysis results of hub bottlenecks of union motif networks
The four lists of genes were prepared according to the method section details to perform the functional enrichment analysis. We then used DAVID and STRING tools for enrichment analysis and identified their shared pathways and GOs.
The top results of enrichment analysis for SARS-related biochemical pathways are available in Table 6, and other significant results are listed in Table S14 (based on p value). The most significant pathway term for the top 10% hub bottlenecks of the SARS network motifs (related to up-regulated DEGs) appeared to be apoptosis. The most significant biological process term (BP) was regulating the macromolecule metabolic process. (Top BP results for BPs are listed in Table 5 and others in Table S13.) As demonstrated in Table No.5 and Table S15, the top BP related to the SARS down-regulated DEGs subnetwork was the lipid metabolic process. The two top significant pathways related to its down-regulated subnetwork appeared to be the term “Metabolic pathways and Carbon metabolism” (Table 6).
The nodes exclusively for each disease were enriched separately to obtain the BPs and pathways not shared between the two diseases, and the BPs and pathways for each disease were identified. Afterward, we determined the shared and non-shared BPs and pathways using the Venn diagram. We reported the non-shared BPs and pathways specific to each disease separately (Supplementary Tables S16–S19).
As available in Tables S16–17, the most significant BP. for COVID-19 up-regulated DEGs was the pattern recognition receptor signaling (GO: 0002221), and the most significant pathway was complement and coagulation cascades. Furthermore, the most significant BP in SARS was the regulation of response to the stimulus, and the first pathway was the MAPK signaling pathway.
The top enrichment analysis results for COVID-19-related gene ontology terms are available in Table 7, and other significant results are listed in Table S20. The top GO term (in COVID-19 up-regulated DEGs subnetwork) was transcription by RNA polymerase II. Noteworthy, the Hepatitis B pathway had the first rank among the COVID-19 up-regulated DEGs subnetwork results (Table S21).
The top three GOs are energy homeostasis, regulation of the developmental process, and multicellular organismal homeostasis (Tables S22 and 7). Glucagon signaling was the most significant signaling pathway in COVID-19 down-regulated DEGs network (Tables S23 and 8).
Drug–target interaction network results
As available in Supplementary Table S12, 51 unique nodes were considered the selected genes, and 83 unique medicines were nominated as the related drugs to the selected genes in the drug–target interaction network. Among the 83 drugs, 31 with higher degrees than two (based on the results between two and nine) were selected to create a new sub-network, as visualized in Fig. 4. The new repurposed drug candidates were then validated using the ClinicalTrials.gov databank for COVID-19 clinical trials and discussed for COVID-19 or similar pathogens. As shown in Table 9, five drugs were previously registered in ClinicalTrials.gov to be evaluated as possible treatments for COVID-19 and were found as possible logical choices for investigation against COVID-19 treatment by other researchers.

The drug–gene interaction network. Green ovals show the crucial identified Genes, and diamonds depict our repurposed medications. The medicines previously registered for clinical trials against COVID-19 are distinct, using violet color
Discussion
Regulatory networks are comprised of a set of recurrence regulatory patterns named motifs. They are re-selected based on their biological functions in evolution. They are found in various organisms, showing that they are the basic building blocks of biological networks (Alon 2007). These repeating circuit elements usually appear much higher frequency than randomized networks (Milo et al. 2004). Here, we have investigated these regulatory motifs to decipher the host response complex mechanism to COVID-19 and SARS-CoV pathogenesis (in vitro). The present study is the first system biology regulatory network study for COVID-19 (in vitro) that has attempted to decipher the regulatory network motifs to identify the molecular mechanism of COVID-19 pathogenesis. We have finally applied the identified targets in repurposing new drug candidates against the deadly COVID-19, hoping our recommended medications appear effective in further in vitro and in vivo investigations.
This study compared the motif-related subnetworks of differentially expressed genes between respiratory cells treated with SARS-CoV-2 and SARS-CoV. We revealed the shared crucial nodes, molecular mechanisms, and signaling pathways underlying both viral infections. Our findings indicated that NFKB1 and STAT1 were shared DEG/TFs between motif-related subnetworks in SARS and COVID-19 up-regulated networks. They also were identified as the shared hub bottlenecks. Enrichment analysis revealed that the NOD-like receptor signaling pathway was the first significant pathway shared between SARS and COVID-19 up-regulated DEGs networks. However, the term “metabolic pathways” (hsa01100) was identified as the most significant pathway term shared among the down-regulated DEGs networks. The infected cell almost always experiences metabolic changes when viruses infect it. Deepak Sumbria et al. recently reviewed several examples, including how viruses can affect the metabolism of fatty acids, glutaminolysis, and energy (Sumbria et al. 2021). Some viruses only infect the already metabolically active cells. Their infection usually downregulates some metabolic processes. It happens, for instance, with HIV, which prefers to infect cells in an activated metabolic state (Datta et al. 2016; Sumbria et al. 2021).
The SARS-CoV-2 infection has also been linked to metabolic changes. During COVID-19, this metabolic dysregulation affects multiple organs. Therefore, its early detection may serve as a prognosis marker (Datta et al. 2016).
WEE1, PMAIP1, and TSC22D2 were identified as the top three DEGs with the highest degree scores specific to SARS. The "Complement and coagulation cascades" pathway was identified as the first top non-shared pathway for COVID-19, and the MAPK signaling pathway was identified as the first for SARS. Here, we discuss and verify that some of our identified crucial molecules and pathways proposed in this network model study are previously experimentally proven in other similar viral pathologies and recommend them for further experimental studies for COVID-19.
NF-κB1 (Nuclear Factor Kappa B Subunit 1) was shared between the two diseases and identified as a node with dual roles (gene/TF) that were up-regulated in both SARS and COVID-19 in vitro. The NF-κB1 plays a role in some immune diseases and is associated with some immunodeficiency diseases based on the GeneCards databank (Stelzer et al. 2016). The regulation of NF-κB is linked to inflammatory and autoimmune diseases and some viral infections. The NF-κB has been reported in many animal cell types. It is an essential intermediate in various biochemical pathways related to cell life, such as apoptosis, cellular responses to cytokines, and viral antigens.
Regarding apoptosis regulation, NF-κB acts pro-apoptotic and antiapoptotic, depending on cell type. However, it serves more as an apoptosis blocker. Some studies have suggested that finding more in-depth insight into the NF-κB mechanism might help develop suitable therapeutics for some immune diseases (Baichwal and Baeuerle 1997; Barkett and Gilmore 1999; Gilmore 2006). NF-κB has been proposed as an essential signaling hub. It integrates the intracellular and intercellular signaling pathways, therefore facilitating the host response without inflammatory injuries (Quinton and Mizgerd 2011).
Some studies have reported the activation of NF-κB1 in lung alveolar epithelial cells in patients infected with other respiratory pathogens (Quinton et al. 2007; Xu et al. 2008) In line with this, Frieman et al. revealed that the SARS-CoV papain-like protease (PLP) could block the NF-KB signaling pathway and affect the host immune system. Indeed, viral PLP antagonizes the host IFN induction and NF-κB signaling pathways. Using the PLP, the virus regulates the host's innate immune responses against itself (Frieman et al. 2009). Thus, SARS-CoV probably uses PLP as a strategy against NF-κB signaling and IFN secretion to overcome the host immune response. Moreover, an in silico docking screening study has recently attempted to identify some candidate drugs against SARS-CoV-2 PLP to overcome the COVID-19 dilemma (Kumar et al. 2020). These findings confirm that NF-κB1 is a crucial regulatory element in the host immune response to SARS-CoV-2, regulates apoptosis and cell life, and targets its viral antagonists (like PLP). It may help to develop suitable medicines against SARS-CoV-2.
STAT1 (The signal transducer and activator of transcription 1) was another shared crucial gene. It mediates cellular responses to different cytokines and growth factors, including cytokine KITLG/SCF and interferons (IFNs). Liang; et al. reported that STAT1 activation plays a vital role in IFNα signaling for the NK cell cytolysis function (Liang et al. 2003). The role of STAT1 in various viral infections, including the Ebola virus (EBOV)(Reid et al. 2006), Hepatitis C virus (HCV) (Lin et al. 2005), Nipah virus (NiV) (Rodriguez et al. 2002), and influenza A (Melen et al. 2003), has been investigated in several studies. They reported that the expression of some viral proteins suppresses type-I interferon signaling by reducing phosphorylated STAT1, and also, some of them could block the nuclear accumulation of STAT1 (Mu et al. 2020). A study on knocked-out mice (STAT−/−) showed that lack of STAT might contribute to wound healing loss and fibrosis induction. It led to a fatal condition after SARS-CoV infection (Frieman et al. 2010). Lung injuries such as fibrosis are considered a severe health problem in patients with severe COVID-19. These findings suggest that STAT1 plays a crucial role in the immune response to various pathogens; however, it could also intermediate in other aspects of the COVID-19 pathogenesis, such as lung fibrosis. These findings support the hypothesis that the STAT1 gene is likely a critical mediating element in the SARS-CoV-2 pathogenesis. It may affect the severity of COVID-19 symptoms and the body’s response. However, further experimental studies are required to accurately determine the details of its molecular response in the body against SARS-CoV-2.
Interestingly, Lei et al. recently uncovered that SARS-CoV-2 initiated an unusual type-I IFN response in vitro. The expressions of IFN-β and ISG56 were scarcely initiated early amid viral disease, whereas they surged at late time points. The postponed antiviral response could give a window for viral replication. The lack of an on-time and sufficient antiviral response could be pivotal to the COVID-19 pathogenesis. The finding of Lei et al. also seems consonant with our hypothesis since the data used in this study were related to an early time-point of infection for COVID-19 (Lei et al. 2020). Blanco-Melo et al. also compared SARS-CoV-2 with other respiratory infections. It appeared that SARS-CoV-2 disease drives a lower antiviral transcriptional response stamped by low IFN-I and IFN-III protein levels and lifted chemokine expression, clarifying the COVID-19 pro-inflammatory state in line with this speculation (Blanco-Melo et al. 2020a).
Our findings indicated that FOS, KLF4, EGR1, and JUN were four shared Gene/TFs between motif-related subnetworks in SARS and COVID-19. (They had dual regulatory roles). FOS and JUN also were identified as the shared hub bottlenecks. These four genes were as DEGs up-regulated in SARS-infected samples but not in COVID-19. However, they were predicted as TFs in our network with a crucial regulatory role in both disease networks.
Our results predicted FOS as a critical shared TF between the networks. The FOS gene family consists of four members: FOS, FOSB, FOSL1, and FOSL2. These genes encode leucine zipper proteins that can dimerize with JUN family proteins, forming the AP-1 transcription factor complex regulating TGF-beta-mediated signaling (Zhang et al. 1998). It has been reported that the expression of the N protein in SARS-CoV activates the AP-1 signaling pathway (He et al. 2003). Another study confirmed that augmentation of AP-1 due to SARS-CoV 3b protein causes pro-inflammatory cytokines and is responsible for the cytokine storm during SARS-CoV (Varshney and Lal 2011). FOS was predicted as an essential TF shared between the two diseases’ up-regulated networks. It can be concluded that FOS probably plays a role as an intermediate in the COVID-19 pathogenesis by activating the AP-1 signaling pathway, ultimately leading to the cytokine storm in severe COVID-19 cases. Therefore, using it as a target in the drug–gene interaction network seems reasonable in the path to candidate medications.
We also reported KLF4 (Kruppel-like factor 4) as another essential shared TF between the networks. KLF4 is a transcription factor that mediates the proliferation, epithelial cells’ differentiation, and apoptosis (Zhang et al. 2010). It also modifies the JAK–STAT3 signaling in some progenitor cells (McConnell and Yang 2010; Qin et al. 2013). Luo et al. indicated that KLF4 negatively regulates cellular antiviral response and inhibits virus-triggered type I IFN signaling. In this study, KLF4 was reported to bind to the IFNB promoter. Therefore, it inhibited the recruitment of IRF3 to the promoter of the IFNB gene.
They also showed that the knockdown of KLF4 increases the induction of IFNB1, thus reducing viral replication (Luo et al. 2016). KLF4 appeared to be overexpressed in our estimation following SARS-CoV entry and has a critical regulatory role in the SARS-CoV-2 GRN. It could be hypothesized that the KLF4 inhibits the IFNB gene promoter and indirectly contributes to further viral replication in SARS and probably in COVID-19. Therefore, it plays a crucial role in the host response to SARS-CoV and SARS-CoV-2 pathogenesis. Further empirical investigations are required to confirm these assumptions.
EGR1 (early growth response 1) was another critical shared TF between the up-regulated networks of SARS and COVID-19. SARS-CoV PLpro has been reported to up-regulate EGR1, which affects the activation of the TGF-β1 promoter. Therefore, it induces the TGF-β1-mediated pro-fibrotic responses in SARS-CoV pathogenesis (Li et al. 2016). Nishi et al. showed that EGR1 could activate the basal transcriptional activity of the EGFR promoter (Nishi et al. 2002). The excessive activation of EGFR signaling leads to increased fibrosis after SARS-CoV infection (Venkataraman et al. 2017). Another study reported that EGR1 is a gene of the pro-coagulation pathway affected by SARS-CoV infection in vitro. The study used a microarray technique. Its findings were confirmed by reverse transcription-quantitative PCR and immunoassays for some encoded proteins (Tang et al. 2005). This finding would probably explain the critical regulatory role of EGR1 in thrombolytic problems in a subset of COVID-19 patients. The EGFR1 is probably mediated in at least two ways in the molecular pathogenesis of COVID-19. The first way is through coagulopathy, and the other is to induce fibrosis.
JUN, another shared node between COVID-19 and SARS networks, is a transcription factor that plays a critical role in the CCR5 signaling pathway in macrophages (based on the GeneCard data for JUN). The CCR5 raises macrophage activation and survival during parainfluenza and influenza infection in mice by activating MAPKs and PI3K signaling (Tyner et al. 2005). MAPKs (mitogen-activated protein kinases) are well-known as signal transducers that respond to extracellular stimulation by cytokines, viral infections, growth factors, and stress. They play crucial roles in regulating cell differentiation, proliferation, survival, and apoptosis (Garrington and Johnson 1999; Kyriakis and Avruch 2001; Whitmarsh and Davis 2000). In vitro and in vivo studies regarding SARS have clarified that activation of MAPKs is essential in regulating cytokine expression (Mizutani 2007). Cytokine storm and excessive cytokine secretion are severely life-threatening in patients with SARS and COVID-19 (Henderson et al. 2020; Huang et al. 2005). In vitro study by Mizutani et al. demonstrated that SARS-CoV infection induced the p38 MAPK signaling pathway in permissive Vero E6 cells (Mizutani et al. 2004). Therefore, targeting p38 MAPK or its downstream molecules could be a drug target. These findings also confirm that JUN molecular functions are related to the host response to the SARS-CoV-2 pathogenesis. It mediates macrophage activation and survival and probably affects the cytokine storm in COVID-19 patients. These findings further support our result in JUN emerging as an essential molecule at the intersection of networks among hundreds of other regulatory nodes.
HIF1A and PRDM1 were identified as two crucial genes exclusively in COVID-19 (up-regulated) in this study. HIF1A (hypoxia-inducible factor 1 subunit alpha) is a master transcriptional regulator in the adaptive response to hypoxia. It can activate genes capable of increasing oxygen delivery or facilitating metabolic adaptation to hypoxia (Li et al. 2009; Masson et al. 2001). Philip et al. have investigated the role of HIF1A in a fatal chronic lung disease called idiopathic pulmonary fibrosis (IPF). They have shown that HIF1A leads to pulmonary fibrosis.
Furthermore, they demonstrated that hypoxia, through HIF1A, leads to pulmonary fibrosis by up-regulating the ADORA2B receptors on AAMs (alternatively activated macrophages) and producing pro-fibrotic mediators (Philip et al. 2017). Since the lungs are the main target organ in SARS-CoV-2 pathogenesis, HIF1A probably mediates pulmonary events in severe COVID-19. It may also be a critical regulatory element influencing the host response to SARS-CoV-2 and needs further investigation.
PRDM1 (positive regulatory domain I), the other up-regulated crucial transcription factor in COVID-19, plays roles in different adaptive and innate tissue-resident T cell types. It also drives B-cell differentiation into terminal IG-secreting plasma cells (Shapiro-Shelef and Calame 2005). Besides, it mediates the Ag presentation by MHC class I. An evolutionary study has identified the PRDM1 as a conserved critical regulator with a unique role in Ag presentation by MHC class I (Doody et al. 2007). These findings may explain that different aspects of host immune response in COVID-19 patients are related to PRDM1.
In addition to identifying crucial nodes, we also recognized the shared biochemical pathways between the two diseases in this study. The NOD-like receptor (NLR) signaling pathway was a shared significant pathway between SARS and COVID-19 up-regulated DEGs networks. NLR signaling plays an essential role in host antiviral immunity pro-inflammatory mediators (Allen et al. 2011; Schneider et al. 2012). These (nucleotide-binding oligomerization domain-like) receptors are intracellular sensors for PAMPs (pathogen-associated molecular patterns). Each mediates in various signaling pathways, including NF-kB and MAPK, IFN type I response, autophagy, and the generation of reactive oxygen species (ROS) (Lei et al. 2013; Moore et al. 2008; Tattoli et al. 2007). Animal studies have shown that the NLRP3 inflammasome is a vital component of the host immune response to viral infections (Allen et al. 2009).
The top shared biochemical pathway between the two diseases' down-regulated DEG networks was the “metabolic pathways” term. Metabolism is a major factor that regulates the function and differentiation of immune cells and influences the immune response process (Pearce and Pearce 2013; van der Windt et al. 2012; Wang and Green 2012). Our enrichment analysis results related to down-regulated DEG networks revealed that the Glucagon signaling pathway, one of the metabolic-related signaling, was the most significant in the COVID-19 network. These findings suggest that metabolic studies likely need more attention to discover the molecular aspects of host response in the pathogenesis of SARS-CoV and SARS-CoV-2.
VAV3 and SPTLC3 were the two other shared crucial nodes participating in down-regulated DEG motif subnetworks. VAV3 is one of the GEFs (guanine nucleotide exchange factors) that activates some pathways leading to transcriptional changes. Vav3 is reported to participate in B cell antigen receptor (BCR) signaling. In this way, VAV3 regulates different B cell receptor responses (Inabe et al. 2002). In this area, Zhivaki et al. have explained that Neonates Regulatory B Cells (nBreg) are infected by Respiratory Syncytial Virus (RSV) via the BCR. RSV-treated nBreg cells secreted anti-inflammatory cytokines (IL-10) in response to HRSV. Still, nBreg cells could not secret cytokine IL-10 when treated with other viruses, including coronavirus 229E, influenza A (IAV), and HIV (Zhivaki et al. 2017). These findings highlight the importance of VAV3 in different possible cell responses to various viruses via BCR. Therefore, our results indicate that SARS-CoV and SARS-CoV-2 entry to some cells may also be affected by VAV3, affecting BCR signaling in host response. Our analysis was performed only on respiratory epithelial cells, not immune cells like macrophages. Considering that VAV3 has appeared as a crucial node in our results, we propose that its related immune signaling had better be experimentally investigated in further studies for COVID-19.
SPTLC3 (Serine Palmitoyltransferase Long Chain Base Subunit 3) was another down-regulated. It participates in the metabolism of some lipids and lipoproteins, and Ziv et al. investigated the role of Serine Palmitoyltransferase in a marine viral infection. They showed that Serine Palmitoyltransferase is required for the viral infection. It activates sphingolipid biosynthesis. They explained that the EhV virus utilizes the host sphingolipid to produce some virus-specific lipids. These unique sphingolipids were vital for the life cycle of the EhV virus. Sphingolipids have a role in viral assembly and infectivity. They might have an evolutionary role in the pathogenesis of other viral infections, such as HIV and HCV (Ziv et al. 2016). SARS-CoV-2 is a positive-sense single-stranded RNA virus similar to HIV and HCV. SPTLC3 possibly mediates in SARS-CoV-2 pathogenesis through its effect on sphingolipid metabolism. The sphingolipid is crucial for viral components. The host cell probably has down-regulated SPTLC3 to reduce the viral access to the sphingolipid.
Our study identified some shared crucial molecules responsible for the immune response to COVID-19 and SARS in vitro (Table S1A and S1B) and crucial molecules specific in response to COVID-19 and SARS. (Supplementary Table S9).
We have represented the overview of our crucial identified molecules and pathways in an integrative diagram in Fig. 5. Among several critical immune system players, our study found some responsible for response against SARS-CoV-2 and SARS_CoV pathogens; some were identified as a specific response in one disease. SARS-CoV and SARS-CoV-2 pathogens are sensed by the mammalian Toll-like and NOD-like receptors. It induces pro-inflammatory cytokines (TGF-b and type I interferons). They are induced through three pathways (NFkB, MAPK (c-Jun, c-Fos), and IRF). The pro-inflammatory cytokines act via the JAK-STAT (STAT1, KLF4) pathway. We found that some of the crucial identified TFs are related to these pathways and can be considered the leading players in the immune response to SARS-CoV-2 and SARS-CoV pathogens. We also found that some biochemical pathways, including Complement and coagulation cascade and Phagosome, and crucial molecules such as WEE1, PMAIP1, and TSC22D2 were related to SARS in vitro. Besides MYPN, SPRY4 and APOL6 were the top crucial nodes specific to COVID-19 in vitro.

The integrative diagram summarizes the crucial molecules and mechanisms
This study constructed a gene–drug interaction network and applied network analysis to propose several candidate drugs (Table S12). We applied the identified shared genes and crucial genes specific to COVID-19 to candidate new drugs interacting with a higher number of the crucial genes. The Drugs with higher degree scores than two (based on the results between two and nine) were selected to create a new subnetwork (Fig. 4). Six previously FDA-approved drugs interacting with COVID-19 target genes included Fostamatinib, Tretinoin, Tirofiban, Zinc chloride, copper, and Levocarnitine. They were previously registered in ClinicalTrials.gov to be evaluated as possible treatments for COVID-19 patients. They were found as possible logical choices for investigation against COVID-19 treatment by other researchers. Their clinical trial registration codes are represented in Table 9. Herein, we have briefly verified them, representing them proposed by other investigations against COVID-19.
Treatment with Fostamatinib might be beneficial. It guards against COVID-19 patients’ neutrophil extracellular traps (Strich et al. 2021). Thirty synergistic drug combinations, including Tretinoin and Fostamatinib, were suggested by Yang Liu et al. using a complementary exposure model to treat COVID-19 (Liu et al. 2021). Hypercoagulability may be life-threatening in patients with severe COVID-19. Tirofiban enhanced oxygenation in patients with elevated D-dimers who had already received antiplatelet therapy.
There are more elderly victims of COVID-19. A combination of micronutrients, especially zinc, and copper, can boost the immunity of aged people. They have a significant role in promoting public health (Chandra 1997). An ideal zinc level may increase host resistance to COVID-19 (Razzaque 2020).
Poor outcomes have been recorded in COVID-19 individuals with inadequate zinc. Hypozincemia has been linked to COVID-19 exacerbation (Jothimani et al. 2020; Yasui et al. 2020). Interestingly, low zinc levels have been linked to immune cell malfunction. Zinc's immunomodulatory effects as a COVID-19 preventive tool and supportive strategy have been proposed (Mossink 2020; Tayyib et al. 2020). Zinc supplementation is also indicated as a COVID-19 adjunct therapy. It has even been recommended with high doses for treatment investigations against COVID-19; however, achieving its therapeutic potential is considered challenging (Chinni et al. 2021).
Copper is also suggested as a possible adjunct therapy for COVID-19 patients who are critically ill (Fooladi et al. 2020). Copper and N-acetylcysteine (NAC) may have a role in a combination of prospective antiviral therapies against SARS-CoV-2, according to Andreou et al. Their findings supported the combination. They suggested that copper, NAC, colchicine, NO, and Remdesivir/EIDD-2801 be studied as a potential SARS-COV-2 treatment scheme (Andreou et al. 2020). Overall, reports about some of the proposed drugs verify the rationale behind the network model and show that our other proposed medications are worth further experimentally investigating against SARS-CoV-2 in vitro and in vivo. Besides, since our data was related to the early time points of the infection with SARS-CoV-2, it probably is better to first investigate the candidate medications in the early stages of the disease.
Conclusion
Designing suitable therapeutic drugs demands elucidation of the mechanisms underlying the disease. System biology approaches have recently contributed to predicting a faster and more in-depth insight into diseases' molecular pathology. This study provides an exciting opportunity to advance our knowledge of the molecular mechanism of COVID-19 and drug repurposing through its gene regulatory network. Since SARS-CoV-2 is a new virus, in vivo omics data accessible for it are limited. This study uses the network analysis of in vitro data to fill the knowledge gap in vivo.
Comparing the motif-related subnetworks of DEGs between respiratory cells treated with SARS-CoV-2 and SARS-CoV revealed the shared molecular mechanisms and signaling pathways underlying both viral infections. NLRs signaling pathway was the first pivotal pathway between SARS and COVID-19 up-regulated DEGs networks. NLR signaling plays an essential role in pro-inflammatory mediators of host antiviral immunity. NLRs are sensors (intracellular) for PAMPs and mediate in several immune-related pathways, including NF-kB, MAPK, IFN type I response, and ROS generation. Some are vital components of the host’s immune response to viral infections. The “metabolic pathways signaling” term was also the most significant pathway term for the down-regulated DEGs network. The metabolism is an essential factor that regulates immune cell function and differentiation and influences the immune response process. We suggest that studies related to metabolic pathways signaling will likely need more attention in the route to discover the molecular aspects of the host response to SARS-CoV and SARS-CoV-2.
The motif-related subnetworks helped candidate six transcription factors as probably shared critical regulators of SARS-CoV-2 and SARS-CoV pathogenesis, including NFKB1, JUN, STAT1, FOS, KLF4, and EGR1. Besides, the top three non-shared biochemical pathways enriched specifically for COVID-19 included Complement- coagulation cascade, Staphylococcus aureus infection, and Phagosome. Some top crucial nodes in the network specific to SARS were WEE1, PMAIP1, and TSC22D2. Besides, MYPN, SPRY4, and APOL6 are the crucial nodes specific to COVID-19 in vitro. Our findings suggest that they probably play roles in the molecular regulatory mechanisms mediating the host response to SARS-CoV-2.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article or its supplementary materials.
Abbreviations
- GRN:
-
Gene regulatory network
- DEGs:
-
Differentially expressed genes
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene ontology
- BP:
-
Biological process
- SARS-CoV:
-
Severe acute respiratory syndrome coronavirus
- TF:
-
Transcription factor
- ACE2:
-
Angiotensin-converting enzyme 2
References
Aevermann BD, Pickett BE, Kumar S, Klem EB, Agnihothram S, Askovich PS, Bankhead A 3rd, Bolles M, Carter V, Chang J, Clauss TR, Dash P, Diercks AH, Eisfeld AJ, Ellis A, Fan S, Ferris MT, Gralinski LE, Green RR, Gritsenko MA, Hatta M, Heegel RA, Jacobs JM, Jeng S, Josset L, Kaiser SM, Kelly S, Law GL, Li C, Li J, Long C, Luna ML, Matzke M, McDermott J, Menachery V, Metz TO, Mitchell H, Monroe ME, Navarro G, Neumann G, Podyminogin RL, Purvine SO, Rosenberger CM, Sanders CJ, Schepmoes AA, Shukla AK, Sims A, Sova P, Tam VC, Tchitchek N, Thomas PG, Tilton SC, Totura A, Wang J, Webb-Robertson BJ, Wen J, Weiss JM, Yang F, Yount B, Zhang Q, McWeeney S, Smith RD, Waters KM, Kawaoka Y, Baric R, Aderem A, Katze MG, Scheuermann RH (2014) A comprehensive collection of systems biology data characterizing the host response to viral infection. Sci Data 1:140033
Aljindan RY, Al-Subaie AM, Al-Ohali AI, Kamaraj B (2021) Investigation of nonsynonymous mutations in the spike protein of SARS-CoV-2 and its interaction with the ACE2 receptor by molecular docking and MM/GBSA approach. Comput Biol Med 135:104654
Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity 30:556–565
Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, Ranjan P, Monroe KM, Pickles RJ, Sambhara S, Ting JP (2011) NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity 34:854–865
Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev Genet 8:450–461
Andreou A, Trantza S, Filippou D, Sipsas N, Tsiodras S (2020) COVID-19: The potential role of copper and N-acetylcysteine (NAC) in a combination of candidate antiviral treatments against SARS-CoV-2. In Vivo 34:1567–1588
Baichwal VR, Baeuerle PA (1997) Apoptosis: activate NF-κB or die? Curr Biol 7:R94–R96
Barkett M, Gilmore TD (1999) Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18:6910–6924
Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R (2007) NCBI GEO: mining tens of millions of expression profiles–database and tools update. Nucleic Acids Res 35:D760-765
Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Møller R, Panis M, Sachs D, Albrecht RA (2020a) SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems. BioRxiv
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, TenOever BR (2020b) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell
Borgio JF, Alsuwat HS, Al Otaibi WM, Ibrahim AM, Almandil N, Al Asoom LI, Salahuddin M, Kamaraj B, AbdulAzeez S (2020) State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch Med Sc 16
Chandra RK (1997) Nutrition and the immune system: an introduction. Am J Clin Nutr 66:460S-463S
Chekmenev DS, Haid C, Kel AE (2005) P-Match: transcription factor binding site search by combining patterns and weight matrices. Nucleic Acids Res 33:W432-437
Chen CY, Chen ST, Fuh CS, Juan HF, Huang HC (2011) Coregulation of transcription factors and microRNAs in human transcriptional regulatory network. BMC Bioinform 12(Suppl 1):S41
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92:418–423
Chinni V, El-Khoury J, Perera M, Bellomo R, Jones D, Bolton D, Ischia J, Patel O (2021) Zinc supplementation as an adjunct therapy for COVID-19: challenges and opportunities. Br J Clin Pharmacol 87:3737–3746
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, Tsai TR, Ho SY, Jian TY, Wu HY, Chen PR, Lin NC, Huang HT, Yang TL, Pai CY, Tai CS, Chen WL, Huang CY, Liu CC, Weng SL, Liao KW, Hsu WL, Huang HD (2016) miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 44:D239-247
Cotto KC, Wagner AH, Feng YY, Kiwala S, Coffman AC, Spies G, Wollam A, Spies NC, Griffith OL, Griffith M (2018) DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res 46:D1068–D1073
Datta PK, Deshmane S, Khalili K, Merali S, Gordon JC, Fecchio C, Barrero CA (2016) Glutamate metabolism in HIV-1 infected macrophages: role of HIV-1 Vpr. Cell Cycle 15:2288–2298
Dehghan Z, Mohammadi-Yeganeh S, Sameni M, Mirmotalebisohi SA, Zali H, Salehi M (2021) Repurposing new drug candidates and identifying crucial molecules underlying PCOS pathogenesis based on bioinformatics analysis. DARU J Pharmaceutical Sci 29:353–366
Dehghan Z, Mirmotalebisohi SA, Sameni M, Bazgiri M, Zali H (2022) A motif-based network analysis of regulatory patterns in doxorubicin effects on treating breast cancer, a systems biology study. Avicenna J Med Biotechnol 14:137
Delgado-Chaves FM, Gómez-Vela F, Divina F, García-Torres M, Rodriguez-Baena DS (2020) Computational analysis of the global effects of Ly6E in the immune response to coronavirus infection using gene networks. Genes 11:831
Dix A, Vlaic S, Guthke R, Linde J (2016) Use of systems biology to decipher host-pathogen interaction networks and predict biomarkers. Clin Microbiol Infect 22:600–606
Doody GM, Stephenson S, McManamy C, Tooze RM (2007) PRDM1/BLIMP-1 modulates IFN-gamma-dependent control of the MHC class I antigen-processing and peptide-loading pathway. J Immunol 179:7614–7623
Farahani M, Rezaei-Tavirani M, Zali H, Arefi Oskouie A, Omidi M, Lashay A (2018) Deciphering the transcription factor-microRNA-target gene regulatory network associated with graphene oxide cytotoxicity. Nanotoxicology 12:1014–1026
Fooladi S, Matin S, Mahmoodpoor A (2020) Copper as a potential adjunct therapy for critically ill COVID-19 patients. Clin Nut ESPEN 40:90–91
Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS (2009) Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol 83:6689–6705
Frieman MB, Chen J, Morrison TE, Whitmore A, Funkhouser W, Ward JM, Lamirande EW, Roberts A, Heise M, Subbarao K, Baric RS (2010) SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog 6:e1000849
Garrington TP, Johnson GL (1999) Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol 11:211–218
Gilmore TD (2006) Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25:6680–6684
Guan Y, Peiris JS, Zheng B, Poon LL, Chan KH, Zeng FY, Chan CW, Chan MN, Chen JD, Chow KY, Hon CC, Hui KH, Li J, Li VY, Wang Y, Leung SW, Yuen KY, Leung FC (2004) Molecular epidemiology of the novel coronavirus that causes severe acute respiratory syndrome. Lancet 363:99–104
Guzzi PH, Mercatelli D, Ceraolo C, Giorgi FM (2020) Master regulator analysis of the SARS-CoV-2/human interactome. J Clin Med 9:982
Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, Lee S, Kang B, Jeong D, Kim Y, Jeon HN, Jung H, Nam S, Chung M, Kim JH, Lee I (2018) TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res 46:D380–D386
He R, Leeson A, Andonov A, Li Y, Bastien N, Cao J, Osiowy C, Dobie F, Cutts T, Ballantine M, Li X (2003) Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem Biophys Res Commun 311:870–876
Henderson LA, Canna SW, Schulert GS, Volpi S, Lee PY, Kernan KF, Caricchio R, Mahmud S, Hazen MM, Halyabar O, Hoyt KJ, Han J, Grom AA, Gattorno M, Ravelli A, De Benedetti F, Behrens EM, Cron RQ, Nigrovic PA (2020) On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheumatol 72:1059–1063
Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, Lei HY (2005) An interferon-gamma-related cytokine storm in SARS patients. J Med Virol 75:185–194
Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA (2007) The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 8:R183
Huang Q, Zhai S-J, Liao X-W, Liu Y-C, Yin S-H (2020) Gene expression, network analysis, and drug discovery of neurofibromatosis type 2-associated vestibular schwannomas based on bioinformatics analysis. J Oncol 2020:1–9
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116
Inabe K, Ishiai M, Scharenberg AM, Freshney N, Downward J, Kurosaki T (2002) Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J Exp Med 195:189–200
Jothimani D, Kailasam E, Danielraj S, Nallathambi B, Ramachandran H, Sekar P, Manoharan S, Ramani V, Narasimhan G, Kaliamoorthy I (2020) COVID-19: Poor outcomes in patients with zinc deficiency. Int J Infect Dis 100:343–349
Khazaei‐Poul Y, Mirmotalebisohi SA, Zali H, Molavi Z, Mohammadi‐Yeganeh S (2022) Identification of miR‐3182 and miR‐3143 target genes involved in the cell cycle as a novel approach in TNBC treatment: a systems biology approach. Chem Biol Drug Design
Killcoyne S, Carter GW, Smith J, Boyle J (2009) Cytoscape: a community-based framework for network modeling. Methods Mol Biol 563:219–239
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11:875–879
Kumar Y, Singh H, Patel CN (2020) In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. journal of infection and public health
Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81:807–869
Lei Y, Wen H, Ting JP (2013) The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy 9:432–433
Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L (2020) Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11:1–12
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454
Li Y, Lim S, Hoffman D, Aspenstrom P, Federoff HJ, Rempe DA (2009) HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J Cell Biol 185:1065–1081
Li SW, Wang CY, Jou YJ, Yang TC, Huang SH, Wan L, Lin YJ, Lin CW (2016) SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-beta1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep 6:25754
Liang S, Wei H, Sun R, Tian Z (2003) IFNα regulates NK cell cytotoxicity through STAT1 pathway. Cytokine 23:190–199
Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, Chung RT (2005) Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology 128:1034–1041
Liu D-Y, Liu J-C, Liang S, Meng X-H, Greenbaum J, Xiao H-M, Tan L-J, Deng H-W (2021) Drug repurposing for COVID-19 treatment by integrating network pharmacology and transcriptomics. Pharmaceutics 13:545
Luo WW, Lian H, Zhong B, Shu HB, Li S (2016) Kruppel-like factor 4 negatively regulates cellular antiviral immune response. Cell Mol Immunol 13:65–72
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J 20:5197–5206
McConnell BB, Yang VW (2010) Mammalian Kruppel-like factors in health and diseases. Physiol Rev 90:1337–1381
Melen K, Fagerlund R, Franke J, Kohler M, Kinnunen L, Julkunen I (2003) Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J Biol Chem 278:28193–28200
Milo R, Itzkovitz S, Kashtan N, Levitt R, Shen-Orr S, Ayzenshtat I, Sheffer M, Alon U (2004) Superfamilies of evolved and designed networks. Science 303:1538–1542
Mitchell HD, Eisfeld AJ, Sims AC, McDermott JE, Matzke MM, Webb-Robertson BJ, Tilton SC, Tchitchek N, Josset L, Li C, Ellis AL, Chang JH, Heegel RA, Luna ML, Schepmoes AA, Shukla AK, Metz TO, Neumann G, Benecke AG, Smith RD, Baric RS, Kawaoka Y, Katze MG, Waters KM (2013) A network integration approach to predict conserved regulators related to pathogenicity of influenza and SARS-CoV respiratory viruses. PLoS ONE 8:e69374
Mizutani T (2007) Signal transduction in SARS-CoV-infected cells. Ann N Y Acad Sci 1102:86–95
Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S (2004) Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem Biophys Res Commun 319:1228–1234
Molavi Z, Razi S, Mirmotalebisohi SA, Adibi A, Sameni M, Karami F, Niazi V, Niknam Z, Aliashrafi M, Taheri M (2021) Identification of FDA approved drugs against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) and 3-chymotrypsin-like protease (3CLpro), drug repurposing approach. Biomed Pharmacotherapy 138:111544
Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, Ting JP (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451:573–577
Mossink JP (2020) Zinc as nutritional intervention and prevention measure for COVID-19 disease. BMJ Nutr Prev Health 3:111–117
Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, Jin L, Deng F, Qiu Y, Zhou X (2020) SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov 6:65
Munster VJ, Koopmans M, van Doremalen N, van Riel D, de Wit E (2020) A novel coronavirus emerging in China–key questions for impact assessment. N Engl J Med 382:692–694
Neuman BW, Adair BD, Yoshioka C, Quispe JD, Orca G, Kuhn P, Milligan RA, Yeager M, Buchmeier MJ (2006) Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J Virol 80:7918–7928
Nishi H, Nishi KH, Johnson AC (2002) Early growth response-1 gene mediates up-regulation of epidermal growth factor receptor expression during hypoxia. Cancer Res 62:827–834
Noori E, Kazemi B, Bandehpour M, Zali H, Khalesi B, Khalili S (2020) Deciphering crucial genes in coeliac disease by bioinformatics analysis. Autoimmunity 53:102–113
Organization WH (2003) Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. http://www.who.int/csr/sars/country/table2004_04_21/en/index.html
Organization WH (2020) Clinical management of severe acute respiratory infection (SARI) when COVID-19 disease is suspected: interim guidance, 13 March 2020. World Health Organization
Paules CI, Marston HD, Fauci AS (2020) Coronavirus infections-more than just the common cold. JAMA
Pearce EL, Pearce EJ (2013) Metabolic pathways in immune cell activation and quiescence. Immunity 38:633–643
Peiris JS, Guan Y, Yuen KY (2004) Severe acute respiratory syndrome. Nat Med 10:S88-97
Philip K, Mills TW, Davies J, Chen NY, Karmouty-Quintana H, Luo F, Molina JG, Amione-Guerra J, Sinha N, Guha A, Eltzschig HK, Blackburn MR (2017) HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J 31:4745–4758
Qin S, Zou Y, Zhang CL (2013) Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat Commun 4:2633
Quinton LJ, Mizgerd JP (2011) NF-kappaB and STAT3 signaling hubs for lung innate immunity. Cell Tissue Res 343:153–165
Quinton LJ, Jones MR, Simms BT, Kogan MS, Robson BE, Skerrett SJ, Mizgerd JP (2007) Functions and regulation of NF-kappaB RelA during pneumococcal pneumonia. J Immunol 178:1896–1903
Razzaque MS (2020) COVID-19 pandemic: can maintaining optimal zinc balance enhance host resistance? Tohoku J Exp Med 251:175–181
Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF (2006) Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol 80:5156–5167
Renu K, Prasanna PL, Valsala Gopalakrishnan A (2020) Coronaviruses pathogenesis, comorbidities and multi-organ damage—a review. Life Sci 255:117839
Rodriguez JJ, Parisien JP, Horvath CM (2002) Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol 76:11476–11483
Romagnoli S, Peris A, De Gaudio AR, Geppetti P (2020) SARS-CoV-2 and COVID-19: from the bench to the bedside. Physiol Rev 100:1455–1466
Sakata T, Winzeler EA (2007) Genomics, systems biology and drug development for infectious diseases. Mol Biosyst 3:841–848
Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, Davis BK, Allen IC, Holl EK, Ye Z, Rahman AH, Conti BJ, Eitas TK, Koller BH, Ting JP (2012) The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol 13:823–831
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Shapiro-Shelef M, Calame K (2005) Regulation of plasma-cell development. Nat Rev Immunol 5:230–242
Sims AC, Tilton SC, Menachery VD, Gralinski LE, Schäfer A, Matzke MM, Webb-Robertson B-JM, Chang J, Luna ML, Long CE (2013a) Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J Virol 87:3885–3902
Sims AC, Tilton SC, Menachery VD, Gralinski LE, Schafer A, Matzke MM, Webb-Robertson BJ, Chang J, Luna ML, Long CE, Shukla AK, Bankhead AR 3rd, Burkett SE, Zornetzer G, Tseng CT, Metz TO, Pickles R, McWeeney S, Smith RD, Katze MG, Waters KM, Baric RS (2013b) Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J Virol 87:3885–3902
Singh R, Bhardwaj VK, Purohit R (2021) Potential of turmeric-derived compounds against RNA-dependent RNA polymerase of SARS-CoV-2: An in-silico approach. Comput Biol Med 139:104965
Singh R, Bhardwaj VK, Das P, Bhattacherjee D, Zyryanov GV, Purohit R (2022a) Benchmarking the ability of novel compounds to inhibit SARS-CoV-2 main protease using steered molecular dynamics simulations. Comput Biol Med 146:105572
Singh R, Bhardwaj VK, Purohit R (2022b) Inhibition of non-structural protein 15 of SARS‐CoV‐2 by golden spice: a computational insight. Cell Biochem Funct
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, Kaplan S, Dahary D, Warshawsky D, Guan-Golan Y, Kohn A, Rappaport N, Safran M, Lancet D (2016) The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform 54:1–33
Strich JR, Ramos-Benitez MJ, Randazzo D, Stein SR, Babyak A, Davey RT, Suffredini AF, Childs RW, Chertow DS (2021) Fostamatinib inhibits neutrophils extracellular traps induced by COVID-19 patient plasma: a potential therapeutic. J Infect Dis 223:981–984
Sumbria D, Berber E, Mathayan M, Rouse BT (2021) Virus infections and host metabolism—can we manage the interactions? Front Immunol 11:594963
Sun YV, Hu YJ (2016) Integrative Analysis of multi-omics data for discovery and functional studies of complex human diseases. Adv Genet 93:147–190
Sun J, Gong X, Purow B, Zhao Z (2012) Uncovering MicroRNA and transcription factor mediated regulatory networks in glioblastoma. PLoS Comput Biol 8:e1002488
Tang BS, Chan KH, Cheng VC, Woo PC, Lau SK, Lam CC, Chan TL, Wu AK, Hung IF, Leung SY, Yuen KY (2005) Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. J Virol 79:6180–6193
Tattoli I, Travassos LH, Carneiro LA, Magalhaes JG, Girardin SE (2007) The Nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol 29:289–301
Tayyib NA, Ramaiah P, Alsolami FJ, Alshmemri MS (2020) Immunomodulatory effects of zinc as a supportive strategies for COVID-19. J Pharmaceutical Res Int 14–22
Tong Z, Cui Q, Wang J, Zhou Y (2019) TransmiR v2.0: an updated transcription factor-microRNA regulation database. Nucleic Acids Res 47:D253–D258
Tyner JW, Uchida O, Kajiwara N, Kim EY, Patel AC, O’Sullivan MP, Walter MJ, Schwendener RA, Cook DN, Danoff TM, Holtzman MJ (2005) CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med 11:1180–1187
van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL (2012) Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36:68–78
Varshney B, Lal SK (2011) SARS-CoV accessory protein 3b induces AP-1 transcriptional activity through activation of JNK and ERK pathways. Biochemistry 50:5419–5425
Venkataraman T, Coleman CM, Frieman MB (2017) Overactive epidermal growth factor receptor signaling leads to increased fibrosis after severe acute respiratory syndrome coronavirus infection. J Virol 91
Viecca M, Radovanovic D, Forleo GB, Santus P (2020) Enhanced platelet inhibition treatment improves hypoxemia in patients with severe COVID-19 and hypercoagulability. A case control, proof of concept study. Pharmacological Res 158:104950
Wang R, Green DR (2012) Metabolic checkpoints in activated T cells. Nat Immunol 13:907–915
Wang J, Lu M, Qiu C, Cui Q (2010) TransmiR: a transcription factor-microRNA regulation database. Nucleic Acids Res 38:D119-122
Wernicke S, Rasche F (2006) FANMOD: a tool for fast network motif detection. Bioinformatics 22:1152–1153
Whitmarsh AJ, Davis RJ (2000) A central control for cell growth. Nature 403:255–256
Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T (2009) miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 37:D105-110
Xu F, Xu Z, Zhang R, Wu Z, Lim JH, Koga T, Li JD, Shen H (2008) Nontypeable Haemophilus influenzae induces COX-2 and PGE2 expression in lung epithelial cells via activation of p38 MAPK and NF-kappa B. Respir Res 9:16
Yasui Y, Yasui H, Suzuki K, Saitou T, Yamamoto Y, Ishizaka T, Nishida K, Yoshihara S, Gohma I, Ogawa Y (2020) Analysis of the predictive factors for a critical illness of COVID-19 during treatment-relationship between serum zinc level and critical illness of COVID-19. Int J Infect Dis 100:230–236
Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M (2007) The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol 3:e59
Zaim S, Chong JH, Sankaranarayanan V, Harky A (2020) COVID-19 and multiorgan response. Curr Probl Cardiol 45:100618
Zhang Y, Feng XH, Derynck R (1998) Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 394:909–913
Zhang P, Andrianakos R, Yang Y, Liu C, Lu W (2010) Kruppel-like factor 4 (Klf4) prevents embryonic stem (ES) cell differentiation by regulating Nanog gene expression. J Biol Chem 285:9180–9189
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 46:586–590
Zhivaki D, Lemoine S, Lim A, Morva A, Vidalain PO, Schandene L, Casartelli N, Rameix-Welti MA, Herve PL, Deriaud E, Beitz B, Ripaux-Lefevre M, Miatello J, Lemercier B, Lorin V, Descamps D, Fix J, Eleouet JF, Riffault S, Schwartz O, Porcheray F, Mascart F, Mouquet H, Zhang X, Tissieres P, Lo-Man R (2017) Respiratory syncytial virus infects regulatory B cells in human neonates via chemokine receptor CX3CR1 and promotes lung disease severity. Immunity 46:301–314
Zhou Y, Ferguson J, Chang JT, Kluger Y (2007) Inter- and intra-combinatorial regulation by transcription factors and microRNAs. BMC Genomics 8:396
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273
Zickenrott S, Angarica VE, Upadhyaya BB, del Sol A (2016) Prediction of disease-gene-drug relationships following a differential network analysis. Cell Death Dis 7:e2040
Ziv C, Malitsky S, Othman A, Ben-Dor S, Wei Y, Zheng S, Aharoni A, Hornemann T, Vardi A (2016) Viral serine palmitoyltransferase induces metabolic switch in sphingolipid biosynthesis and is required for infection of a marine alga. Proc Natl Acad Sci USA 113:E1907-1916
Acknowledgements
This study is related to project NO 1399/60914 from the Student Research Committee, Shahid Beheshti University of Medical Sciences, Tehran, Iran. We also appreciate the Student Research Committee and Research & Technology Chancellor at Shahid Beheshti University of Medical Sciences for their financial support of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sameni, M., Mirmotalebisohi, S.A., Dehghan, Z. et al. Deciphering molecular mechanisms of SARS-CoV-2 pathogenesis and drug repurposing through GRN motifs: a comprehensive systems biology study. 3 Biotech 13, 117 (2023). https://doi.org/10.1007/s13205-023-03518-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03518-x