
Abstract
Vaccines are used as one of the major weapons for the eradication of pandemic. However, the rise of different variants of the SARS-CoV-2 virus is creating doubts regarding the end of the pandemic. Hence, there is an urgent need to develop more drug candidates which can be useful for the treatment of COVID-19. In the present research for the scientific hypothesis, emphasis was given on the direct antiviral therapy available for the treatment of COVID-19. In lieu of this, the available molecular targets which include Severe Acute Respiratory Syndrome Chymotrypsin-like Protease (SARS-3CLpro), Papain-Like Cysteine Protease (PLpro), and RNA-Dependent RNA Polymerase (RdRp) were explored. As per the current scientific reports and literature, among all the available molecular targets, RNA-Dependent RNA Polymerase (RdRp) was found to be a crucial molecular target for the treatment of COVID-19. Most of the inhibitors which are reported against this target consisted of the free amine group and carbonyl group which might be playing an important role in the binding interaction with the RdRp protein. Among all the reported RdRp inhibitors, remdesivir, favipiravir, and molnupiravir were found to be the most promising drugs against COVID-19. Overall, the structural features of this RNA-Dependent RNA Polymerase (RdRp) inhibitors proved the importance of pyrrolo-triazine and pyrimidine scaffolds. Previous computational models of these drug molecules indicated that substitution with the polar functional group, hydrogen bond donor, and electronegative atoms on these scaffolds may increase the activity against the RdRp protein. Hence, in line with the proposed hypothesis, in the present research work for the evaluation of the hypothesis, new molecules were designed from the pyrrolo-triazine and pyrimidine scaffolds. Further, molecular docking and MD simulation studies were performed with these designed molecules. All these designed molecules (DM-1, DM-2, and DM-3) showed the results as per the proposed hypothesis. Among all the designed molecules, DM-1 showed promising results against the RdRp protein of SARS-CoV-2. In the future, these structural features can be used for the development of new RdRp inhibitors with improved activity. Also, in the future lead compound DM-1 can be explored against the RdRp protein for the treatment of COVID-19.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
COVID-19
WHO declared COVID-19 as a Global pandemic last year, which infected around 289 million people around the world until December 2021 and nearly 5 million people died due to this infection (https://covid19.who.int/). It is considered as one of the biggest pandemics after the Spanish flu, affecting most of the population during 1918–1920. One of the biggest problems associated with COVID-19 is the high transmission rate from person to person through droplets or breath (Liu et al. 2020). Sometimes, asymptomatic patients are also carriers of the virus which make it difficult to contain the virus (Liu et al. 2020). Symptoms of COVID-19 patients vary from person to person, but the most common symptoms include fever, cough, fatigue, shortness of breath, and loss of smell and taste (Liang et al. 2020; Amin et al. 2021a; Tian et al. 2021a; Ma et al. 2021). Depending upon the immunity of an individual, severity varies from person to person. In severe cases, COVID-19 patients developed the complications like pneumonia, respiratory distress, blood clots, and organ-failure that consequently resulted in death (Liang et al. 2020; Amin et al. 2021a; Tian et al. 2021a; Ma et al. 2021). Through the continuous efforts of the scientific community, several vaccines have been developed. However, the mutation in the virus is causing breakthrough infections in fully vaccinated people, which is a cause of concern for the world.
Available WHO-approved vaccines and their limitations
To date, 10 vaccines have been approved by the WHO shown in Table 1 which include Pfizer-BioNTech-BNT162b2 (RNA-based Vaccine), Moderna-mRNA-1273-(RNA based Vaccine), Johnson & Johnson’s Janssen Ad26.COV2.S-(Non Replicating Viral Vector-based vaccine), AstraZeneca/Oxford-AZD1222-(Non-Replicating Viral Vector-based vaccine), Serum Institute of India Covishield (Oxford/AstraZeneca formulation-(Non-Replicating Viral Vector- based vaccine), SinoPharm Beijing-BBIBP-CorV (Vero Cells based vaccine), Sinovac (CoronaVac vaccine), Novavax (NVX-CoV2373-protein subunit), Serum Institute of India COVOVAX (Novavax formulation-Protein Subunit) and Bharat Biotech Covaxin (https://covid19.who.int/). Vaccine discovery played an important role in the mitigation of COVID-19 in most parts of the world but unexpected mutation in the SARS-CoV-2 virus is still a big challenge for the scientific community (Ma et al. 2021; Plante et al. 2021; Chen et al. 2020a; Manuel and Timothy 2020; Tao et al. 2021; Harvey et al. 2021). Recently, the Omicron variant is haunting the world for the next outbreak as it caused some breakthrough infections in fully vaccinated people (https://covid19.who.int/). However, the deaths reported due to the Omicron variant are fewer, and it seems to be less lethal compared to the delta variant. It is well understood that the virus will continue to mutate which consequently may turn into a lethal variant in the future that may break the immunity developed by the vaccines. Hence, there are many uncertainties in the scientific community regarding the end of the current pandemic. Therefore, there is an urgent need to discover the effective SARS-CoV-2 inhibitors which may help to eradicate the pandemic from the world.
General features of SARS-CoV-2 virus and possible molecular targets
Coronavirus belongs to the family Coronaviridae that also has a subfamily Coronavirinae which contains four genera Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus. SARS-CoV-2 mainly belongs to the beta corona virus family (Fu et al. 2021) which comprises of the other two deadlier viruses, i.e., Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and Middle East Respiratory Syndrome Coronavirus (MERS-CoV). These viruses also created the havoc in the past. SARS-CoV-2 is a single-stranded RNA that has a genome of 27–32 kb which is larger than any other virus. It consists of four different types of structural protein, i.e., Spike Protein (S), Envelope Protein (E), Membrane Protein (M), Nucleocapsid Protein (N), and sixteen nonstructural proteins (NSP 1–16) (Wang et al. 2020). Among all the proteins, spike protein (S) plays an important role in the entry into the host cell through Angiotensin Converting Enzyme (ACE-2) as shown in Fig. 1 (Luan et al. 2020; Hussain et al. 2020). The Receptor Binding Domain (RBD) of the spike glycoprotein mediates the binding to the ACE-2 receptor; therefore, both ACE-2 and RBD are attractive targets for inhibition of the entry of the SARS-CoV-2 virus in the host cells. The advantage of targeting these receptors leads to the prevention of the spread of the infection in the body in the early stages. After the entry of the virus in the host cells, two viral polyproteins are produced with the aid of the host cell’s ribosomes (Ghanbari et al. 2020). These polyproteins get converted into RNA-Dependent RNA Polymerase (RdRp) protein in the presence of the viral protease enzymes such as Severe Acute Respiratory Syndrome Chymotrypsin-like Protease (SARS-3CLpro), and Papain-Like Cysteine Protease (PLpro) as shown in Fig. 1 (Mody et al. 2021; Rut et al. 2020; Gao et al. 2020). This non-structural RdRp protein is encoded with the viral genome which further produces the subgenomic mRNA as shown in Fig. 1. This mRNA further produces the viral structural protein through the translation process as shown in Fig. 1. This viral protein along with genome RNA gets converted into the new virion. This new virion gets transported outside the cell through exocytosis. Therefore, inhibition of these enzymatic targets, i.e., RBD-ACE2 fusion, RdRp, SARS-3CLpro, PLpro, plays a crucial role in ceasing the viral replication in the body.

Pathogenesis of COVID-19 along with the important molecular targets of the SARS-CoV-2
Introduction to available treatment for COVID-19
In the early days of the COVID-19 pandemic, the genome sequence of the five SARS-CoV-2 patients from Wuhan revealed a similarity with the SARS-CoV-2 virus that forms the basis of the preliminary treatment of COVID-19 (Wang et al. 2020; Rossi et al. 2020; Abdelrahman et al. 2020; Maldonado et al. 2021; Fani et al. 2020). During this pandemic, many strategies were used which included the use of antibodies, IFNs, inhibitors of viral and host proteases, and host-directed therapies. A few of them were used as an experiment during the current pandemic. But these strategies were found to be ineffective against the delta strain of the SARS-CoV-2 virus (Fu et al. 2021; de Wit et al. 2016). Similarly, a few other drugs were repurposed for the treatment of the COVID-19 which include favipiravir, remdesivir, hydroxychloroquine, lopinavir, interferon, etc., which were found to be not that effective for the prevention of the mortality rate (Hassanipour et al. 2021; Bansal et al. 2021; Axfors et al. 2021; Cao et al. 2020). During the pandemic, the delta strain created a catastrophic effect on COVID-19 patients by inducing a cytokine storm that led to a sharp rise in the number of patients hospitalized. To overcome this problem, a few other therapies were utilized like mono-clonal antibodies (bamlanivimab + etesevimab) (Dougan et al. 2021), Interleukin-1 Inhibitors (canakinumab) (Cremer et al. 2021), Interleukin-6-Inhibitors (sarilumab) which have been effectively used for the treatment of COVID-19. All these therapies were found to be useful for the treatment of the hyperactivated immune response rather than against the virus.
Overall, many efforts are being put in by the scientific community to eradicate the COVID-19 pandemic from the world. In lieu of this, in the present work, we have tried to collect the possible molecular targets for the SARS-CoV-2 virus and their structural features with respect to the binding domain. Additionally, we enumerated various SARS-CoV-2 inhibitors to date to find the common features of these inhibitors. Based on the structural information of the various receptors of the SARS-CoV-2 virus and reported SARS-CoV-2 inhibitors, we have proposed the required features of the molecules that can be effective against SARS-CoV-2.
Severe acute respiratory syndrome (SARS) chymotrypsin-like Protease (SARS-3CLpro) and its inhibitors
3CLpro is a major replicating enzyme present inside the SARS-CoV-2 virus. It is responsible for the replication of the SARS-CoV-2 virus inside the host cells. 3CLpro is responsible for the production of 16 non-structural proteins (NSPs). These NSPs are responsible for the replication, transcription, and virus recombination during the infection phase. Hence, inhibition of the 3CLpro would be one of the effective strategies for the antiviral therapy for COVID-19. The binding pocket of 3CLpro is highly conserved with amino acids like glutamine, leucine, and methionine. 3CLpro is responsible for the cleavage of SARS-CoV-2 polyprotein at the 11 sites which consequently results in the production of the NSPs. The 3CLpro protein comprises three domains: domain-I, domain-II and domain-III. Among these domains, domain III has prime importance as it is involved in the dimerization and formation of an active 3CLpro protease. The active site of the 3CLpro lies between domain-I and domain-II. Recent studies of 3CLpro indicated that it consisted of the His41 (part of domain-I) and Cys145 (part of domain-II) residues of S-glycoprotein which are the prime targets for the inhibition of the replication of the SARS-CoV-2 virus (Ferreira and Rabeh 2020).
Earlier research on this target showed some promising drug candidates as shown in Table 2 (Konwar and Sarma 2021; Chen et al. 2005; Ghosh et al. 2007; Wu et al. 2020). Most of the drugs are polyphenolic as shown in (Fig. 2; Konwar and Sarma 2021; Chen et al. 2005; Ghosh et al. 2007; Wu et al. 2020) indicating the importance of the hydroxyl group required for the binding affinity with the 3CLpro

Severe Acute Respiratory Syndrome (SARS) chymotrypsin-like protease (SARS-3CLpro) inhibitors
Papain-like cysteine protease (PLpro) and its inhibitors
Basically, the PLpro protein of SARS-CoV-2 consists of two domains, i.e., the N-terminal UBL domain and the C-terminal USP domain, where all the process of regulation takes place. PLpro of the SARS-CoV-2 virus is cysteine proteases which are responsible for protein maturation, dysregulating host inflammation response and impairing the host type I interferon antiviral immune responses. All these changes take place due to the interferon-stimulated gene (ISG15) modification. Therefore, the binding cleft of the PLpro protein which comprises ISG15 is considered a hotspot for antiviral therapy (Shin et al. 2020).
The recent protein structure of PLpro complexed with the GRL-0617 revealed that the side chains of the Glu269 and Tyr268 revealed the polar interaction with the co-crystalized ligand and is responsible for the stabilization of the protein–ligand complex. Carbonyl oxygen and the free amine group of the ligand played an important role in this polar binding interaction with the protein. Additionally, naphthalene of the inbound ligand showed binding interactions with the Tyr264 and Tyr268. Overall, this complex structure represented the importance of the carbonyl oxygen and hydrophobic ring like naphthalene for the appropriate binding interactions of any ligand with this target protein (Fu et al. 2021; Wu et al. 2020; Shin et al. 2020; Verma et al. 2021; Amin et al. 2021b; Mirza et al. 2020; Bhati 2020; Swaim et al. 2021; Rajpoot et al. 2021). Most of the PL-pro inhibitors shown in Fig. 3 (Fu et al. 2021; Wu et al. 2020; Shin et al. 2020; Verma et al. 2021; Amin et al. 2021b; Mirza et al. 2020; Bhati 2020; Swaim et al. 2021; Rajpoot et al. 2021) bears the free amine group along with the carbonyl group which might be playing an important role in the inhibition if the PLpro protein.

Papain-like cysteine protease (PLpro) inhibitors
Angiotensin converting enzyme (ACE2) and its inhibitors
ACE2 is one of the most attractive targets for the SARS-CoV-2 virus (South et al. 2020; Arno et al. 2020; Li et al. 2020; Lan et al. 2020). This receptor is usually present in the lower respiratory system of the human body; hence, this virus is responsible for more damage in the lower respiratory tract (South et al. 2020; Arno et al. 2020; Li et al. 2020; Lan et al. 2020). S-glycoprotein of the SARS-CoV-2 interacts with the ACE2 receptor which aids the entry of the virus inside the host cell and further replication takes place (Lan et al. 2020; Duan et al. 2020). This S-glycoprotein present in the spikes of SARS-CoV-2 virus is considered to be the protective envelope of the virus. This S-glycoprotein comprises two subunits, i.e., S1 and S2. The S1 subunit identifies the receptor in the host and attaches itself to the receptor. This attachment of the S1 subunit with the host receptor takes place between the Receptor Binding Domain (RBD) of the S1 subunit of the viral protein and the peptidase domain (PD) of the ACE2 receptor. On the other hand, S2 is responsible for the fusion process between the virus and host receptor. Once this S1-RBD attaches itself to the receptor of the host cell, host proteases cleave the S-glycoprotein which resulted in the shedding of the S1 subunit. The previous finding suggested that the amino acid residues of S1-RBD (Thr333-Gly526) interact with the amino acid residues of the ACE2 receptor (Ser19-Asp615) which is responsible for the entry of the virus in the host cells (Singh et al. 2021a).
In the initial period of the COVID-19 pandemic, antimalarial drugs like hydroxychloroquine and chloroquine phosphate were found to be effective and taken for prophylactic measure by healthcare professionals (Chen et al. 2020b). The administration of these antimalarial drugs is based on a possible hypothesis that these drugs may act as fusion inhibitors of the S-RBD and ACE2. Later on, the scientific community proved that this hypothesis was wrong. Also, the inhibition of ACE-2 may create other pharmacological problems like vasoconstriction, inflammation as ACE-2 is involved in the Renin Angiotensin Aldosterone System (RAAS) pathway and consequently reducing the harmful effect of vasoconstrictor, anti-inflammatory peptide, and angiotensin 2. Its inhibition may disturb the RASS pathway which may create other severe health issues (Behl et al. 2020).
Despite this, there are certain molecules as shown in Table 2 (McKee et al. 2020) sent for the clinical trials meant for ACE2 inhibition.
Non-structural proteins (NSP)
Apart from the structural protein, the virus also comprises non-structural proteins (NSPs), i.e., NSP1 to NSP10 and NSP12 to NSP16. These proteins are encoded by the genes and are located in the 5l -region of viral RNA. NSP1 is an N terminal product of the viral replicase and it comprises 180 amino acid residues. The major function of the NSP1 is to degrade the host mRNA (Yadav et al. 2021; Singh et al. 2022). NSP2 is also the N-terminal product of the viral protein which consists of 638 amino acids. It binds to prohibitin 1 and prohibitin 2. NSP3 is a papain-like proteinase which consists of the 1945 amino acid residues. This protein is responsible for the release of the NSP1, NSP2, and NSP3. NSP4 comprises 500 amino acids and is involved in the viral replication. NSP5 is also known as proteinase which consist of 306 amino acid residues. The major role of this protein involves cleaving the protein at multiple sites which consequently results in the formation of the intermediate non-structural protein. NSP6 is the Putative transmembrane domain that is responsible for the formation of ER-derived autophagosomes. NSP7 and NSP8 are two important non-structural proteins that play a vital role in the RdRp activity in the SARS-CoV-2. NSP7 comprises 83 amino acid residues. Mainly, it forms the complex with the NSP8 and NSP12 and induces the RNA polymerase activity of the NSP8. NSP8 is involved in the formation of the heterodimer with the NSP8 and 12. The major role of NSP 9 to 11 in SARS-CoV-2 is not yet clear. NSP12 and 13 are involved in viral replication. Both these proteins comprise 932 amino acid residues. NSP14 with the 527 amino acid residues is involved in the N7-guanine methyltransferase activity. Similarly, NSP15 which comprises of 346 amino acid residues is responsible for the Mn(2+)-dependent endoribonuclease activity.
Among all the non-structural proteins, NSP16 plays a vital role in the reproduction of the CoV RNA genome. This NSP16 consists of 301 amino acid residues, among which Phe6947, Asp6912, Leu6898, Cys6913, Met6929, Gly6871, Asp6897, Asn6899, Asp6928, Tyr6845, Asn6841, and Gly6871 are parts of the binding pocket of the NSP16 protein. Inhibition of the NSP16 would be advantageous for inhibiting the SARS-CoV-2 virus.
RNA-dependent RNA polymerase (RdRp) and its inhibitors
Until now, and since the pandemic, this target is one of the most important target meant for the treatment of the SARS-CoV-2 infection (Elfiky 2020; Gao et al. 2020; Pachetti et al. 2020; Aftab et al. 2020; Khater et al. 2021). RNA-Dependent RNA Polymerase is an enzyme responsible for RNA synthesis, and hence, it plays an important role in viral replication. In the current pandemic, many antiviral drugs like remdesivir and favipiravir were repurposed against the SARS-CoV-2 virus (Tian et al. 2021b). One of the interesting things observed in the RdRp inhibitor is that most of these inhibitors consisted of the free amine group along with the carbonyl group shown in Fig. 4 (Tian et al. 2021b). Few of these inhibitors showed binding interactions with the key amino acids of the RdRp protein as shown in Table 3 (Sada et al. 2020; Nimgampalle et al. 2021; Poustforoosh et al. 2021; Alexpandi et al. 2020). In addition to this synthetic molecule, lots of natural molecules are also screened against the RdRp protein as shown in Table 3. Examples of these natural molecules include 7-hydroxyaloin-B of Aloe vera; Isoquercetin and sonimocinolide of Azadirachta indica; which showed good binding affinity with the RdRp protein (Kushwaha et al. 2021). Interestingly, as per the Piplani et.al study (Piplani et al. 2021), digoxin showed a good binding affinity and interaction with the RdRp protein. It showed key binding interactions with the Ser682, Arg553, Arg624, and Cys622 of the RdRp protein. Similarly, Singh et.al reported curcumin and diacetylcurcumin as promising drug candidates as an RdRp inhibitor for the mitigation of COVID-19 (Singh et al. 2021b). During the pandemic, the plants which were explored more were Swertia chirayita, Tinospora cordifolia, and Withania somnifera. Koulgi et.al in their study (Koulgi et al. 2021) reported important phytoconstituents of these plants which include swertiapuniside, cordifolide A, sitoindoside IX, and amarogentin. Glycosidic moiety of these phytoconstituents formed the key binding interactions with the polar and charged amino acids of the RdRp protein which includes Arg553, Arg555, Asp618, Asp760, Asp761, Glu811, and Ser814. Similarly, Singh et.al (Singh et al. 2021c) showed the importance of polyphenolic compounds for the treatment of COVID-19. He reported a few polyphenolic phytoconstituents which include Epigallocatechin Gallate (EGCG), theaflavin (TF1), theaflavin-3’-O-gallate (TF2a), theaflavin-3’-gallate (TF2b), theaflavin 3,3'-digallate (TF3), hesperidin, quercetagetin, and myricetin. These phytoconstituents revealed excellent binding affinity with the RdRp protein.

RNA-dependent RNA polymerase (RdRp) inhibitors
RdRp protein also known as Non-Structural Protein 12 (NSP12) consists of two subunits NSP7 and NSP8 which have an important role in the replication of the SARS-CoV-2 virus (Kirchdoerfer and Ward 2019; Velthuis et al. 2012; Sun 2020; Bhalla et al. 2021). A schematic representation of this RdRp protein (PDB ID:7D4F) is shown in Fig. 5. In addition to this, RdRp protein consists of a few other important domains. These include the polymerase domain (Ser367 to Phe920), and nidovirus-specific N-terminal expansion domain (NiRAN domain) which consists of the amino acids from Asp60 to Arg249. RdRp protein also consists of an interface domain that joins the polymerase and NiRAN domains together. This interface domain consists of amino acids ranging from Ala250 to Arg365. NiRAN domain is responsible for the SARS-CoV-2 viral replication (Venkataraman et al. 2018; Bhardwaj et al. 2021a; Yin et al. 2021).

Schematic representation of RdRp protein (PDB ID:7D4F) with its important structural domains, where orange and green color helix represent the NSP8 protein, violet-red color helix represents RdRp protein and purple color helix represents NSP7 protein
Apart from this, RdRp also has a few other domains which include the palm domain (Tyr582-Pro620 and Tyr680-Gln815), the finger domain (Leu366-Ala581 and Lys621-Gly679), and the thumb domain. The palm domain of the RdRp protein consists of the catalytic aspartate and RNA Recognizing motif which are responsible for the catalysis. The finger domain is responsible for the proper geometry of the active site of the RdRp protein. The thumb domain is required for nucleic acid synthesis. The N-terminal hairpin which has an amino acid residue from Asp29 to Lys50 is responsible for the stability of the RdRp structure.
Among all the antivirals acting against the RdRp, the remdesivir monophosphate was found to be the most effective. A recent electron microscopical study of a complex of remdesivir monophosphate and RdRp revealed the importance of the pyrrolo-triazine scaffold of the remdesivir (Bravo et al. 2021). RdRp protein consequently delays the replication of the viral protein. However, this interaction is not enough to completely stop the viral replication. Hence, there is the scope for suitable structural modification of remdesivir which could lead to the discovery of a powerful RdRp inhibitor. In addition to this, remdesivir showed many side effects such as anemia, infusion site reactions, increased liver enzymes, cutaneous rash, kidney injuries, and hypotension. Hence, its use has to be restricted in critically comorbid patients (Nabati and Parsaee 2022). Similar to the remdesivir, one more drug was found to be effective against the SARS-CoV-2, i.e., molnupiravir showed crucial binding interaction with the RdRp protein (Yip et al. 2022).
Hypothesis
Currently, there are various therapies available against SARS-CoV-2 which include both natural and synthetic sources that act against the above-mentioned targets of the SARS-CoV-2 virus. Singh et.al. in their studies reported various bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. In their study, they found theaflavin as a potential lead candidate as an NSP-16 inhibitor where theaflavin forms a hydrogen bond with the key amino acids, i.e., Asp6928, Asp6897, Lys6844, Asp6931, Met6929. This study proved the importance of polyphenolic compounds in the treatment of SARS-CoV-2. Hence, during the designing of NSP16 inhibitors, the incorporation of the hydroxyl (OH) group in the structure can play a crucial role in NSP-16 inhibition (Singh et al. 2022).
Similarly, Singh et.al. in their other in silico study reported Dicaffeoylquinic acid and Diacetylcurcumin as potential inhibitors of the Receptor Binding Domain (RBD)-S protein of the SARS-CoV-2 virus which is mainly involved in the fusion process with the ACE-2 receptor of the host cells. Dicaffeoylquinic acid is mainly involved in the amino acid interactions with the Lys417, Gln493, Tyr489, Phe456, Tyr473, and Glu484 of S-RBD. Inhibition of these residues prevents the entry of the virus into the host cell as these amino acid residues are involved in the interaction with the ACE-2 protein of the host cell. Here also, the hydroxyl group (OH) played an important role in the binding interaction with the target protein (Singh et al. 2021a).
Bhardwaj et.al in their study reported the acridinedione analogs as potential SARS-CoV-2 main protease inhibitors (Mpro or 3CLpro). In their study, they found these analogues better than saquinavir, which is a well-known antiviral drug. This study proved the importance of the carbonyl group (C=O) which can play a crucial role in the designing of 3CLpro inhibitors (Bhardwaj et al. 2021b).
Among all the molecular targets available for the inhibition of the SARS-CoV-2 virus, the RNA-Dependent RNA Polymerase (RdRp) has evolved as an important and crucial target for the treatment of COVID-19. Drugs like remdesivir, favipiravir and molnupiravir were used against these targets, and they have shown some promising outcomes during this pandemic.
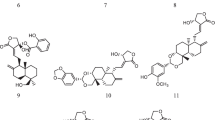
Remdesivir is one of the broad-spectrum antivirals, invented in the year 2014–2016 used for the treatment of the Ebola pandemic spread in West Africa. It consisted of the pyrrolo-triazine, a carboxylic ester, a nitrile scaffold, a phosphoramidate ester, and aromatic amine which is considered as a prodrug. Further, this prodrug is converted into the active metabolite GS-441524 which acts as an ATP competitive inhibitor. The research study carried out by Nguyen et.al of the remdesivir–RdRp complex indicated that –NH2 of pyrrolo-triazine of remdesivir active metabolite is an essential part of the binding interactions with the crucial amino acid of A chain which includes Thr677 as shown in Fig. 6A. The hydroxyl (OH) group of the furan ring of the remdesivir indicated the binding interactions with the Asp757, Asn688, and Ser756 as Fig. 6A (Nguyen et al. 2020; Khan et al. 2021).

Similar to remdesivir, favipiravir was discovered for the treatment of the influenza virus as a prodrug. It consists of pyrazine carboximide as a core scaffold which is converted into triphosphate which is an active form of the drug that consequently exerts its biological effect against the RdRp. As per the study conducted by Sada et.al., after the conversion into triphosphate, the phosphate group (P=O) plays an important role in the binding interactions with the crucial amino acids of the RdRp protein which includes binding interactions with the Ser602, Thr600, Thr607, Asn611, Ser679, and Arg 473 as shown in Fig. 6B (Sada et al. 2020).
Molnupiravir is another category of prodrug which exerts RdRp inhibitory activity after the conversion into triphosphate. It consists of a pyrimidine scaffold where the hydroxyl group showed important binding interaction with the Arg555 and Lys545 as shown in Fig. 6C (Wang et al. 2021) and is one of the important parameter for the inhibition of the RdRp. In addition to this, the nitrogen of the pyrimidine ring indicated the important binding interaction with the target protein.
Overall, the summary of all the scientific studies on RdRp inhibitors, to date, indicated the importance of the triazine and pyrimidine scaffolds as shown in Fig. 7A–F. The important features of these drugs include the electronegative nitrogen atom of the aromatic ring system. In addition to this, free amine and hydroxyl groups also play an important role in the binding process with the RdRp protein.

A Amino, hydroxyl, phosphate and carbonyl groups of pyrrolo-triazine of remdesivir. B Amino, fluorine and carbonyl groups of pyrimidine scaffold of favipiravir and C amino, hydroxyl and carbonyl groups of pyrimidine scaffold of molnupiravir are essential part biological activity against the RdRp protein. D Essential features of the pyrrolo-triazine scaffold of remdesivir required for the binding affinity with the RdRp protein. E Essential features of the pyrimidine scaffold of favipiravir required for the binding affinity with the RdRp protein. F Essential features of the pyrimidine scaffold of molnupiravir required for the binding affinity with the RdRp protein
In line with these facts, we have proposed the designing strategy as shown in Fig. 7A–F for the novel RdRp inhibitors which can be used against the SARS-CoV-2. Many docking-based analysis of the remdesivir–RdRp complex suggested that free amine group of triazine–pyrrole complex is surrounded by the charged and polar amino acids as shown in Fig. 6A. This computational model indicated the importance of polar functional groups at this position of the scaffold as shown in Fig. 7A. Therefore, this position of the scaffold can be explored for substitution with the various electronegative atoms and hydrogen bond donors groups as shown in Fig. 7D. Similarly, remdesivir acts as a prodrug which get converted to monophosphate and then consequently exerts its effect against the RdRp. This conversion takes place at the pyrrole ring of the remdesivir. Therefore, this position is favorable for the electronegative atoms and the polar functional groups as shown in Fig. 7D. If the intention is to design the prodrug, then this pyrrole ring should not be modified as monophosphate attaches itself to the pyrrole ring of the remdesivir and get converted into active metabolite.
Similar to the remdesivir, previous research related to the favipiravir–RdRp complex indicated that the amide functional group of the favipiravir is surrounded by the polar and charged amino acids as shown in Fig. 6B. Therefore, this position of the pyrimidine scaffold of the favipiravir is favorable to the polar functional groups as shown in Fig. 7B and E. Further to this, –NH of the pyrimidine scaffold as shown in Fig. 7E is important for the conversion of the favipiravir into the favipiravir phosphate which is an active form of the drug. This position of the scaffold is favorable for the polar functional group as the previous research findings revealed that this position is surrounded by the polar and charged amino acids.
Recently, new drug molecule like molnupiravir showed some excellent results in the clinical trials. It also consists of the pyrimidine scaffold as shown in Fig. 7C and F. Previous computational model of this molecule in complex with the RdRp as shown in Fig. 6C indicated that it is also surrounded by the polar and charged amino acids. Hence, the modification with the polar functional groups on the pyrimidine scaffold may give favorable results against the RdRp protein.
In a nutshell, our hypothesis suggested that these scaffolds along with the required substitutions can be explored for the development of novel RdRp inhibitors which can helpful in the treatment of COVID-19.
Evaluations of hypothesis
Overall, in this work, we enumerated the important targets of SARS-CoV-2 along with their inhibitors. Additionally, we proposed important chemical scaffolds and groups which are responsible for SARS-CoV-2 inhibition. Among all the molecular targets, RdRp is one of the choices for the development of the new lead compound which can be used for the treatment of COVID-19. Hence, more emphasis was given on the RdRp protein. In lieu of this, to prove the importance of our suggested hypothesis, we have designed a few molecules against the RdRp protein and their molecular docking and dynamics study was performed.
Design and molecular docking study
As suggested in the hypothesis, triazine and pyrimidine scaffolds were found to be promising scaffolds for the designing of RdRp inhibitors. In line with this, based upon the available data and a proposed hypothesis, the modification was carried out in the triazine and pyrimidine scaffolds of remdesivir, favipiravir, and molnupiravir, which resulted in the Designed Molecule-1 (DM-1), Designed Molecule-2 (DM-2), Designed Molecule-3 (DM-3), respectively, as shown in Table 4. While designing the molecules against the RdRp protein, modifications carried out at the free amine groups of the standard drugs showed that it may be beneficial for the reduction in the toxicity which may occur due to the free amine groups.
Further docking study of these designed molecules along with the standard drugs which include remdesivir, favipiravir, and molnupiravir was carried out with the aid of AutoDock Vina as shown in Fig. 8A–F. All the standard drugs and designed molecules were converted into the pdbqt file with the aid of Open Babel software. RdRp protein (PDB ID: 7D4F) was taken from the protein data bank. Further, the protein was prepared by adding polar hydrogens and Kollman Charges. A Grid box was generated surrounding the binding pocket of the RdRp protein and converted into the pdbqt file. After, the completion of the molecular docking study, the docking interactions were analyzed with the help of Pymol and a web-based protein–ligand interaction profiler.

Molecular Docking Study was carried out against the RdRp Protein (PDB ID:7D4F) for the evaluation of the hypothesis, where the designed molecules showed similar binding interactions as that of the standard drug molnupiravir. A Remdesivir showed hydrogen bond interactions (solid blue color lines), hydrophobic interactions (green color dotted lines), salt bridge (yellow color dotted line) with the RdRp protein. B Favipiravir showed hydrogen bond interactions (solid blue color lines) with the RdRp protein. C Molnupiravir showed hydrogen bond interactions (solid blue color lines) and hydrophobic interactions (green color dotted lines) with the RdRp protein. D DM-1 showed hydrogen bond interactions (solid blue color lines), π-cation interaction (orange color dotted lines) and hydrophobic interactions (green color dotted lines) with the RdRp protein. E DM-2 showed hydrogen bond interactions (solid blue color lines) and hydrophobic interactions (green color dotted lines) with the RdRp protein. F DM-3 showed hydrogen bond interactions (solid blue color lines) and hydrophobic interactions (green color dotted lines) with the RdRp protein
The DM-1 designed from the triazine scaffold of the remdesivir showed a similar docking score of − 7.9 kcal/Mol compared to the remdesivir docking score of − 7.8 kcal/Mol. Also, DM-1 showed binding interactions with the Pro461A and Val315A which is similar to remdesivir. It validates the effectiveness of the designed molecule against the RdRp protein.
The DM-2 which is designed from the pyrimidine scaffold of the favipiravir showed an excellent docking score of − 7.1 kcal/Mol compared to the favipiravir docking score of − 5.8 kcal/Mol. Also, this DM-2 indicated the binding interaction with the Pro677A and Asn628A which is similar to the binding interactions of the favipiravir that validates the proper binding orientation of the DM-2.
The DM-3 is also designed from the pyrimidine scaffold of molnupiravir indicated a docking score of − 7.1 kcal/Mol which is good as compared to the molnupiravir which showed a docking score of − 6.7 kcal/Mol. Further, DM-3 showed binding interactions with the Pro461A and Pro677A similar to molnupiravir which validated the potency of the DM-3.
Molecular dynamics simulation study
Further, for the support of the docking analysis, an MD simulation study was performed with the aid of the DESMOND module of DE Shaw Research. Solvation of the docked complex was carried out with the help implicit solvent model in a cubic box of 10 Å spacing. The neutralization of the solvated system was carried out with the help of the counter ions. The salt concentration was maintained at 0.15 M. All the systems were set up for 50 ns at 300 K temperature and 1.013 atmospheric pressure (Pandey et al. 2021). As DM-1 showed good binding affinity, i.e., − 7.9 kcal/mol as compared to the standard drug, it was selected for this analysis. DM-1 which is present in the binding cavity of the RdRp protein taken for the MD simulation study for 50 ns is shown in Fig. 9A. After the completion of the MD simulation study, the RMSD value protein–ligand complex revealed that both the protein and ligand achieved their equilibrium at 10 ns and remain stabilized for the remaining 40 ns as shown in Fig. 9A. The RMSD value for the receptor remains within the 3.2 Å. Interestingly, the lower RMSD value of docked molecule (DM-1) within the 1.2 Å shown in pink color in Fig. 9A validates the stability of the complex and proved that the docked ligand does not diffuse away from its actual binding pocket.

A RMSD plot of RdRp protein with the Lead compound (DM-1) during 50 ns MD simulation time. B RMSF profiles of RdRp protein with the Lead compound (DM-1) during 50 ns MD simulation
Further, for accessing the local changes in the protein chains during the MD simulation study, root mean square fluctuation (RMSF) was determined as shown in Fig. 9B. It indicated that no major conformational changes occurred in the RdRp protein. After docking, the RMSF value remained within the 2 Å indicated in green bars as shown in Fig. 9B. It validated the hypothesis proving that the docked complex of RdRp protein remained more stable without major fluctuation.
Similar to the protein, ligand RMSD value as shown in Fig. 10A remains within the 1.2 Å indicating the conformation of the docked ligand (DM-1) does not change so much with reference to the parent conformation of the ligand (DM-2). Similar, to the RMSD, the RMSF value of DM-1 lies within the 1.5 Å as shown in Fig. 10B which proved that each atom of the ligand does not deviate so much from its original position. The ligand torsion profile during the simulation is shown in Fig. 11A–F which consists of the radial plots which indicate the flexible position of the docked ligand (DM-1) as shown in Fig. 11G. This flexible point of a ligand can further be considered for the modifications to get the novel RdRp inhibitors. In the case of DM-1, dots are more scattered in Figs. 11C and E. Figure 11C corresponds to the 5th position of the triazine scaffold as shown in Fig. 11G with the same color coding indicating that this position is flexible for further modification to get the best binding fit in the RdRp protein. Figure 11E corresponds to the hydroxyl group of the tetrahydrothiophene ring shown in Fig. 11G with the same color coding. This position can be modified further to get the best binding mode of the ligand in the binding pocket of the RdRp protein.

A RMSD and B RMSF plot of the docked ligand DM-1

Ligand torsion profile of compound DM-1, where A radial plot corresponds to the carbonyl group of the phenyl ring of DM-1 indicated in the cyan color. B Radial plot corresponds to the (-CONH) group of DM-1 indicated in light green color. C Radial plot corresponds to the 5th position of the triazine scaffold indicated in the peach color. D Radial plot corresponds to the 9th position of the triazine scaffold indicated in orange color. E Radial plot corresponds to -OH group of the tetrahydrothiophene of the DM-1 ligand indicated in the purple color. F Radial plot corresponds to -OH group of the tetrahydrothiophene of the DM-1 ligand indicated in the dark green color. G Docked ligand (DM-1) with different color codes corresponds to the radial plots
Conclusion
In this work, efforts were made to explore the available treatments and protective vaccines for COVID-19. Overall, the current scenario indicated that the available vaccines played an important role in slowing down the pandemic in the world, although certain mutations in the virus are creating hindrances in the vaccine effectiveness. The rise of different strains across the world is creating challenges for the scientific community. Based on the previously published literature, it is well understood that the RdRp is one of the important molecular targets for drug development against SARS-CoV-2. All the published data to date related to the RdRp protein revealed some key amino acids of the binding pocket of the RdRp which consisted a combination of polar amino acids along with some hydrophobic amino acids. Based on this, we have proposed various hypotheses for designing novel potent RdRp inhibitors which will be beneficial for the treatment of SARS-CoV-2. All the given hypotheses were evaluated by the molecular docking-assisted molecular dynamics simulation study.
Overall, as per the evaluation of the hypothesis, designed analogues showed a good binding affinity with the target protein RdRp compared to the standard drugs which validate the accuracy of the proposed hypothesis. Among all the designed molecules, DM-1 showed promising results in the MD simulation-assisted molecular docking study against the RdRp protein. In the future, this lead compound can be explored for the inhibition of the RdRp protein which will be beneficial for the treatment of SARS-CoV-2.
Availability of data and material (Code availability)
All data analyzed during this study are included in this published article. Also, the data analyzed during the current study are available from the corresponding author on reasonable request as per the journal guidelines.
References
Abdelrahman Z, Li M, Wang X (2020) Comparative review of SARS-CoV-2, SARS-CoV, MERS-CoV, and influenza a respiratory viruses. Front Immunol 11:552909. https://doi.org/10.3389/fimmu.2020.552909
Aftab SO, Ghouri MZ, Masood MU, Haider Z, Khan Z, Ahmad A, Munawar N (2020) Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J Transl Med 18:275. https://doi.org/10.1186/s12967-020-02439-0
Alexpandi R, De Mesquita JF, Pandian SK, Ravi AV (2020) Quinolines-based SARS-CoV-2 3CLpro and RdRp inhibitors and Spike-RBD-ACE2 inhibitor for drug-repurposing against COVID-19: an in silico analysis. Front Microbiol 11:1796. https://doi.org/10.3389/fmicb.2020.01796
Amin SA, Banerjee S, Gayen S, Jha T (2021a) Protease targeted COVID-19 drug discovery: what we have learned from the Past SARS-CoV inhibitors? Eur J Med Chem 215:113294. https://doi.org/10.1016/j.ejmech.2021.113294
Amin SKA, Banerjee S, Ghosh K, Gayen S, Jha T (2021b) Protease targeted COVID 19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg Med Chem 29:115860. https://doi.org/10.1016/j.bmc.2020.115860
Arno RB, Amaal EA, Wim T, Jan LH, Gerjan JN, Sanne JG, Marieke CB, Gerard D, Adriaan AV, Albert DO, Peter H, Douwe JM, Harry G (2020) Angiotensin-converting Enzyme 2 ACE2 SARS-CoV-2 and the pathophysiology corona virus disease (COVID-19). J Pathol 251:228. https://doi.org/10.1002/path.5471
Axfors C, Schmitt AM, Janiaud P, van Hooft J, Abd-Elsalam S, Abdo EF, Abella BS, Akram J, Amaravadi RK, Angus DC, Arabi YM, Azhar S, Baden LR, Baker AW, Belkhir L, Benfield T, Berrevoets MAH, Chen CP, Chen TC, Cheng SH, Cheng CY, Chung WS, Cohen YZ, Cowan LN, Dalgard O, de Almeida-e-Val FF, de Lacerda MVG, de Melo GC, Derde L, Dubee V, Elfakir A, Gordon AC, Hernandez-Cardenas CM, Hills T, Hoepelman AIM, Huang YW, Igau B, Jin R, Jurado-Camacho F, Khan KS, Kremsner PG, Kreuels B, Kuo CY, Le T, Lin YC, Lin WP, Lin TH, Lyngbakken MN, McArthur C, McVerry BJ, Meza-Meneses P, Monteiro WM, Morpeth SC, Mulligan MA, MJ, Murthy S, Naggie S, Narayanasamy S, Nichol A, Novack LA, O’Brien SM, Okeke NL, Perez L Perez-Padilla R, Perrin L, Remigio-Luna A, Rivera-Martinez NE, Rockhold FW, Rodriguez-Llamazares S, Rolfe R, Rosa R, Røsjø H, Sampaio VS, Seto TB, Shahzad M, Soliman, S, Stout JE, Thirion-Romero I, Troxel AB, Tseng TY, Turner NA, Ulrich RJ, Walsh SR, Webb SA, Weehuizen JM, Velinova M, Wong HL, Wrenn R, Zampieri, FG, Zhong W, Moher D, Goodman SN, Ioannidis JPA, Hemkens LG (2021) Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat Commun 12:2349. https://doi.org/10.1038/s41467-021-22446-z
Bansal V, Mahapure KS, Bhurwal A, Gupta I, Hassanain S, Makadia J, Madas N, Armaly P, Singh R, Mehra I, O’Horo JC, Kashyap R (2021) Mortality benefit of remdesivir in COVID-19: a systematic review and meta-analysis. Front Med 7:606429. https://doi.org/10.3389/fmed.2020.606429
Behl T, Kaur I, Bungau S, Kumar A, Uddin MS, Kumar C, Pal G, Shrivastava SK, Zengin G, Arora S (2020) The dual impact of ACE2 in COVID-19 and ironical actions in geriatrics and pediatrics with possible therapeutic solutions. Life Sci 257:118075. https://doi.org/10.1016/j.lfs.2020.118075
Bhalla V, Blish CA, South AM (2021) A historical perspective on ACE2 in the COVID-19 era. J Hum Hypertens 35:935. https://doi.org/10.1038/s41371-020-00459-3
Bhardwaj VK, Singh R, Sharma J, Rajendran V, Purohit R, Kumar S (2021a) Bioactive molecules of tea as potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2. Front Med 8:684020. https://doi.org/10.3389/fmed.2021.684020
Bhardwaj VK, Singh R, Das P, Purohit R (2021b) Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Comput Biol Med 128:104117. https://doi.org/10.1016/j.compbiomed.2020.104117
Bhati S (2020) Structure-based drug designing of naphthalene based SARS-CoV PLpro inhibitors for the treatment of COVID-19. Heliyon. https://doi.org/10.1016/j.heliyon.2020.e05558
Bravo JPK, Dangerfield TL, Taylor DW, Johnson KA (2021) Remdesivir is a delayed translocation inhibitor of SARS-CoV-2 replication. Mol Cell 81(7):1548-1552.e4. https://doi.org/10.1016/j.molcel.2021.01.035
Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, Ruan L, Song B, Cai Y, Wei M, Li X, Xia J, Chen N, Xiang J, Yu T, Bai T, Xie X, Zhang L, Li C, Yuan Y, Chen H, Li H, Huang H, Tu S, Gong F, Liu Y, Wei Y, Dong C, Zhou F, Gu X, Xu J, Liu Z, Zhang Y, Li H, Shang L, Wang K, Li K, Zhou X, Dong X, Qu Z, Lu S, Hu X, Ruan S, Luo S, Wu J, Peng L, Cheng F, Pan L, Zou J, Jia C, Wang J, Liu X, Wang S, Wu X, Ge Q, He J, Zhan H, Qiu F, Guo L, Huang C, Jaki T, Hayden FG, Horby PW, Zhang D, Wang CA (2020) Trial of lopinavir-ritonavir in adults hospitalized with severe COVID-19. N Engl J Med 382:1787. https://doi.org/10.1056/NEJMoa2001282
Chen CN, Lin CPC, Huang KK, Chen WC, Hsieh HP, Liang PH, Hsu JT (2005) A Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3,3’-digallate (TF3). Evid Based Complement Alternat Med 2:209. https://doi.org/10.1093/ecam/neh081
Chen J, Wang R, Wang M, Wei GW (2020a) Mutations strengthened SARS-CoV-2 infectivity. J Mol Biol 432:5212. https://doi.org/10.1016/j.jmb.2020.07.009
Chen L, Chen H, Dong S, Huang W, Chen L, Wei Y, Shi L, Li J, Zhu F, Zhu Z, Wang Y, Lv X, Yu X, Li H, Wei W, Zhang K, Zhu L, Qu C, Hong J, Hu C, Dong J, Qi R, Lu D, Wang H, Peng S, Hao G (2020b) The effects of chloroquine and hydroxychloroquine on ACE2-related coronavirus pathology and the cardiovascular system: an evidence-based review. Function. https://doi.org/10.1093/function/zqaa012
Cremer PC, Sheng CC, Sahoo D, Dugar S, Prada RA, Wang TKM, Hassan OKA, Hernandez-Montfort J, Wolinsky DA, Culver DA, Rajendram P, Duggal A, Brennan DM, Wolski KE, Lincoff AM, Nissen SE, Menon V (2021) Double-blind randomized proof-of-concept trial of canakinumab in patients with COVID-19 associated cardiac injury and heightened inflammation. Eur Heart J. https://doi.org/10.1093/ehjopen/oeab002
de Wit E, FalzaranoMunster DVJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523. https://doi.org/10.1038/nrmicro.2016.81
Dougan M, Nirula A, Azizad M, Mocherla B, Gottlieb RL, Chen P, Hebert C, Perry R, Boscia J, Heller B, Morris J, Crystal C, Igbinadolor A, Huhn G, Cardona J, Shawa I, Kumar P, Adams AC, Van Naarden J, Custer KL, Durante M, Oakley G, Schade AE, Holzer TR, Ebert PJ, Higgs RE, Kallewaard NL, Sabo J, Patel DR, Dabora MC, Klekotka P, Shen L, Skovronsky DM (2021) Bamlanivimab plus etesevimab in mild or moderate COVID-19. N Engl J Med 385:1382. https://doi.org/10.1056/NEJMoa2102685
Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, Wang H (2020) The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike-based vaccine immunogens. Front Immunol 11:576622. https://doi.org/10.3389/fimmu.2020.576622
Elfiky AA (2020) SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: an in silico perspective. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1761882
Fani M, Teimoori A, Ghafari S (2020) Comparison of the COVID-2019 (SARS-CoV-2) pathogenesis with SARS-CoV and MERS-CoV infections. Future Virol 15:317. https://doi.org/10.2217/fvl-2020-0050
Ferreira JC, Rabeh WM (2020) Biochemical and biophysical characterization of the main protease, 3-chymotrypsin-like protease (3CLpro) from the novel coronavirus SARS-CoV 2. Sci Rep 10(1):22200. https://doi.org/10.1038/s41598-020-79357-0
Fu Z, Huang B, Tang J, Liu S, Liu M, Ye Y, Liu Z, Xiong Y, Zhu W, Cao D, Li J, Niu X, Zhou H, Zhao YJ, Zhang G, Huang H (2021) The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun 12:488. https://doi.org/10.1038/s41467-020-20718-8
Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J, Zheng L, Zhang Y, Wang H, Zhu Y, Zhu C, Hu T, Hua T, Zhang B, Yang X, Li J, Yang H, Liu Z, Xu W, Guddat LW, Wang Q, Lou Z, Rao Z (2020a) Structure of the RNA-dependent RNA polymerase from virus. Science 368:779. https://doi.org/10.1126/science.abb7498
Ghanbari R, Teimoori A, Sadeghi A, Mohamadkhani A, Rezasoltani S, Asadi E, Jouyban A, Sumner SC (2020) Existing antiviral options against SARS-CoV-2 replication in COVID-19 patients. Future Microbiol 15(18):1758. https://doi.org/10.2217/fmb-2020-0120
Ghosh AK, Xi K, Grum Tokars V, Xu X, Ratia K, Fu W, Houser KV, Baker SC, Johnson ME, Mesecar AD (2007) Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett 17:5876. https://doi.org/10.1016/j.bmcl.2007.08.031
Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, Ludden C, Reeve R, Rambaut A, Genomics UK (COG-UK) Consortium, Peacock SJ, Robertson DL (2021) SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 19:409. https://doi.org/10.1038/s41579-021-00573-0
Hassanipour S, Arab-Zozani M, Amani B, Heidarzad F, Fathalipour M, Martinez-de-Hoyo R (2021) The efficacy and safety of favipiravir in treatment of COVID-19: a systematic review and meta-analysis of clinical trials. Sci Rep 11:11022. https://doi.org/10.1038/s41598-021-90551-6
https://covid19.who.int/. Assessed 2nd Jan 2022
Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, Aziz B (2020) Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol 92:15801. https://doi.org/10.1002/jmv.25832
Khan FI, Kang T, Ali H, Lai D (2021) Remdesivir strongly binds to RNA-dependent RNA polymerase, membrane protein, and main protease of SARS-CoV-2: indication from molecular modeling and simulations. Front Pharmacol 12:710778. https://doi.org/10.3389/fphar.2021.710778
Khater S, Kumar P, Dasgupta N, Das G, Ray S, Prakash A (2021) Combining SARS-CoV-2 proofreading exonuclease and RNA-dependent RNA polymerase inhibitors as a strategy to combat COVID-19: a high-throughput in silico screening. Front Microbiol 12:647693. https://doi.org/10.3389/fmicb.2021.647693
Kirchdoerfer RN, Ward AB (2019) Structure of the SARS-CoV Nsp12 polymerase bound to Nsp7 and Nsp8 Co-factors. Nat Commun 10:2342. https://doi.org/10.1038/s41467-019-10280-3
Konwar M, Sarma D (2021) Advances in developing small molecule SARS 3CLpro inhibitors as potential remedy for corona virus infection. Tetrahedron 77:131761. https://doi.org/10.1016/j.tet.2020.131761
Koulgi S, Jani V, Uppuladinne VN, Uonavane U, Joshi R (2021) Natural plant products as potential inhibitors of RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus-2. PLoS ONE 16(5):e0251801. https://doi.org/10.1371/journal.pone.0251801
Kushwaha PP, Singh AK, Bansal T, Yadav A, Prajapati KS, Shuaib M, Kumar S (2021) Identification of natural inhibitors against SARS-CoV-2 drugable targets using molecular docking, molecular dynamics simulation, and MM-PBSA approach. Front Cell Infect Microbiol 11:730288. https://doi.org/10.3389/fcimb.2021.730288
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215. https://doi.org/10.1038/s41586-020-2180-5
Li G, He X, Zhang L, Ran Q, Wang J, Xiong A, Wu D, Chen F, Sun J, Chang C (2020) Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J Autoimmun 112:102463. https://doi.org/10.1016/j.jaut.2020.102463
Liang C, Tian L, Liu Y, Hui N, Qiao G, Li H, Shi Z, Tang Y, Zhang D, Xie X, Zhao X (2020) A Promising Antiviral Candidate Drug for the COVID-19 Pandemic: A Mini-Review of Remdesivir. Eur J Med Chem 201:112527. https://doi.org/10.1016/j.ejmech.2020.112527
Liu Y, Liang C, Xin L, Ren X, Tian L, Ju X, Li H, Wang Y, Zhao Q, Liu H, Cao W, Xie X, Zhang D, Wang Y, Jian Y (2020) The Development of Coronavirus 3C-Like Protease (3CLpro) Inhibitors from 2010 to 2020. Eur J Med Chem 206:112711. https://doi.org/10.1016/j.ejmech.2020.112711
Luan J, Lu Y, Jin X, Zhang L (2020) Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem Biophys Res Commun 526:165. https://doi.org/10.1016/j.bbrc.2020.03.047
Ma L, Liu H, Liu X, Yuan X, Xu C, Wang F, Lin J, Xu R, Zhang D (2021) Screening S protein – ACE2 blockers from natural products: strategies and advances in the discovery of potential inhibitors of COVID-19. Eur J Med Chem 226:113857. https://doi.org/10.1016/j.ejmech.2021.113857
Maldonado LL, Bertelli AM, Kamenetzky L (2021) Molecular features similarities between SARS-CoV-2, SARS, MERS and key human genes could favour the viral infections and trigger collateral effects. Sci Rep 11:4108. https://doi.org/10.1038/s41598-021-83595-1
Manuel BF, Timothy C (2020) SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract 74:13525. https://doi.org/10.1111/ijcp.13525
McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C (2020) Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res 157:104859. https://doi.org/10.1016/j.phrs.2020.104859
Mirza MU, Ahmad S, Abdullah I, Froeyen M (2020) Identification of novel human USP2 inhibitor and its putative role in treatment of COVID-19 by inhibiting SARS-CoV-2 papain-like (PLpro) protease. Comput Biol Chem 89:107376. https://doi.org/10.1016/j.compbiolchem.2020.107376
Mody V, Ho J, Wills S, Mawri A, Lawson L, Ebert MC, Fortin GM, Rayalam S, Taval S (2021) Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun Biol 4:93. https://doi.org/10.1038/s42003-020-01577-x
Nabati M, Parsaee H (2022) Potential cardiotoxic effects of remdesivir on cardiovascular system: a literature review. Cardiovasc Toxicol 22(3):268. https://doi.org/10.1007/s12012-021-09703-9
Nguyen HL, Thai NQ, Truong DT, Li MS (2020) Remdesivir strongly binds to both RNA-dependent RNA polymerase and main protease of SARS-CoV-2: evidence from molecular simulations. J Phys Chem B 124:11337. https://doi.org/10.1021/acs.jpcb.0c07312
Nimgampalle M, Devanathan V, Saxena A (2021) Screening of chloroquine, hydroxychloroquine and its derivatives for their binding affinity to multiple SARS-CoV-2 protein drug targets. J Biomol Struct Dyn 39(14):4949. https://doi.org/10.1080/07391102.2020.1782265
Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, Masciovecchio C, Angeletti S, Ciccozzi M, Gallo RC, Zella D, Ippodrino R (2020) Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med 18:179. https://doi.org/10.1186/s12967-020-02344-6
Pandey K, Lokhande KB, Venkateswara K, Nagar S, Dake M (2021) In silico exploration of phytoconstituents from phyllanthus emblica and aegle marmelos as potential therapeutics against SARS-CoV-2 RdRp. Bioinform Biol Insights 15:1–13. https://doi.org/10.1177/11779322211027403
Piplani S, Singh PK, Winkler DA, Petrovsky N (2021) Computationally repurposed drugs and natural products against RNA dependent RNA polymerase as potential COVID-19 therapies. Mol Biomed 2(1):28. https://doi.org/10.1186/s43556-021-00050-3
Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, Zhang X, Muruato AE, Zou J, Fontes-Garfias CR, Mirchandani D, Scharton D, Bilello JP, Ku Z, An Z, Kalveram B, Freiberg AN, Menachery VD, Xie X, Plante KS, Weaver SC, Shi PY (2021) Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592:116. https://doi.org/10.1038/s41586-020-2895-3
Poustforoosh A, Hashemipour H, Tüzün B, Pardakhty A, Mehrabani M, Nematollahi MH (2021) Evaluation of potential anti-RNA-dependent RNA polymerase (RdRP) drugs against the newly emerged model of COVID-19 RdRP using computational methods. Biophys Chem 272:106564. https://doi.org/10.1016/j.bpc.2021.106564
Rajpoot S, Alagumuthu M, Baig MS (2021) Dual targeting of 3CLpro and PLpro of SARS-CoV-2: a novel structure-based design approach to treat COVID-19. Curr Res Struct Biol 3:9. https://doi.org/10.1016/j.crstbi.2020.12.001
Rossi GA, Sacco O, Mancino E, Cristiani L, Midulla F (2020) Differences and similarities between SARS-CoV and SARS-CoV-2: spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection 48:665. https://doi.org/10.1007/s15010-020-01486-5
Rut W, Lv Z, Zmudzinski M, Patchett S, Nayak D, Snipas SJ, Oualid F, Huang TT, Bekes M, Drag M, Olsen SK (2020) Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti– drug design. Sci Adv. https://doi.org/10.1126/sciadv.abd4596
Sada M, Saraya T, Ishii H, Okayama K, Hayashi Y, Tsugawa T, Nishina A, Murakami K, Kuroda M, Ryo A, Kimura H (2020) Detailed molecular interactions of favipiravir with SARS-CoV-2, SARS-CoV, MERS-CoV, and influenza virus polymerases in silico. Microorganisms 8(10):1610. https://doi.org/10.3390/microorganisms8101610
Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van heden van Noort GJ, Ovaa H, Müller S, Knobeloch K-P, Rajalingam K, Schulman BA, Cinatl J, Dikic I (2020) Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587(7835):657. https://doi.org/10.1038/s41586-020-2601-5
Singh R, Bhardwaj VK, Sharma J, Kumar D, Purohit R (2021a) Identification of potential plant bioactive as SARS-CoV-2 Spike protein and human ACE2 fusion inhibitors. Comput Biol Med 136:104631. https://doi.org/10.1016/j.compbiomed.2021.104631
Singh R, Bhardwaj VK, Purohit R (2021b) Potential of turmeric-derived compounds against RNA-dependent RNA polymerase of SARS-CoV-2: An in-silico approach. Comput Biol Med 139:104965. https://doi.org/10.1016/j.compbiomed.2021.104965
Singh S, Sk MF, Sonawane A, Kar P, Sadhukhan S (2021c) Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: An in-silico analysis. J Biomol Struct Dyn 39(16):6249. https://doi.org/10.1080/07391102.2020.1796810
Singh R, Bhardwaj VK, Sharma J, Purohit R, Kumar S (2022) In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. J Tradit Complement Med 12(1):35. https://doi.org/10.1016/j.jtcme.2021.05.005
South AM, Diz DI, Chappell MC (2020) COVID-19, ACE2, and the cardiovascular consequences. Am J Physiol Heart Circ 318:1084–1090. https://doi.org/10.1152/ajpheart.00217.2020
Sun D (2020) Remdesivir for treatment of COVID-19: combination of pulmonary and IV administration may offer additional benefit. AAPS J 22:77. https://doi.org/10.1208/s12248-020-00459-8
Swaim CD, Dwivedi V, Perng YC et al (2021) 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro. iScience 24:103213. https://doi.org/10.1016/j.isci.2021.103213
Tao K, Tzou PL, Nouhin J, Gupta RK, de Oliveira T, Kosakovsky Pond SL, Fera D, Shafer RW (2021) The biological and clinical significance of emerging SARS-CoV-2 variants. Nat Rev Genet 22:757. https://doi.org/10.1038/s41576-021-00408-x
te Velthuis AJW, van den Worm SHE, Snijder EJ (2012) The SARS-coronavirus Nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res 40:1747. https://doi.org/10.1093/nar/gkr893
Tian L, Qiang T, Liang C, Ren X, Jia M, Zhang J, Li J, Wan M, Yu Wen X, Li H, Cao W, Liu H (2021a) RNA-dependent RNA polymerase (RdRp) inhibitors: the current landscape and repurposing for the COVID-19 pandemic. Eur J Med Chem 213:113201. https://doi.org/10.1016/j.ejmech.2021.113201
Tian L, Qiang T, Liang C, Ren X, Jia M, Zhang J, Li J, Wan M, Yu Wen X, Li H, Cao W, Liu H (2021b) RNA-Dependent RNA polymerase (RdRp) inhibitors: the current landscape and repurposing for the COVID-19 pandemic. Eur J Med Chem 213:113201. https://doi.org/10.1016/j.ejmech.2021.113201
Venkataraman S, Prasad B, Selvarajan R (2018) RNA dependent RNA polymerases: insights from structure. Funct Evol Viruses 10(2):76. https://doi.org/10.3390/v10020076
Verma D, Mitra D, Paul M et al (2021) Potential inhibitors of SARS-CoV-2 (COVID 19) proteases PLpro and Mpro/ 3CLpro: molecular docking and simulation studies of three pertinent medicinal plant natural components. Curr Res Pharmacol Drug Discov 2:100038. https://doi.org/10.1016/j.crphar.2021.100038
Wang MY, Zhao R, Gao LJ, Gao XF, Wang DP, Cao JM (2020) SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front Cell Infect Microbiol 10:587269. https://doi.org/10.3389/fcimb.2020.587269
Wang Y, Li P, Solanki K, Li Y, Ma Z, Maikel PP, Baig MS, Pan Q (2021) Viral polymerase binding and broad spectrum antiviral activity of molnupiravir against human seasonal coronavirus. Virol J 564:33. https://doi.org/10.1016/j.virol.2021.09.009
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 10:766. https://doi.org/10.1016/j.apsb.2020.02.008
Yadav R, Chaudhary JK, Jain N, Chaudhary PK, Khanra S, Dhamija P, Sharma A, Kumar A, Handu S (2021) Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells 10(4):821. https://doi.org/10.3390/cells10040821
Yin W, Luan X, Li Z, Zhou Z, Wang Q, Gao M, Wang X, Zhou F, Shi J, You E, Liu M, Wang Q, Jiang Y, Jiang H, Xiao G, Zhang L, Yu X, Zhang S, Eric HX (2021) Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat Struct Mol Biol 28(3):319. https://doi.org/10.1038/s41594-021-00570-0
Yip AJW, Low ZY, Chow VTK, Lal SK (2022) Repurposing molnupiravir for COVID-19: the mechanisms of antiviral activity. Viruses 14(6):1345. https://doi.org/10.3390/v14061345
Funding
The authors declare that no funds and grants were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent statement/ethical approval
Not required.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chaube, U., Patel, B.D. & Bhatt, H.G. A hypothesis on designing strategy of effective RdRp inhibitors for the treatment of SARS-CoV-2. 3 Biotech 13, 12 (2023). https://doi.org/10.1007/s13205-022-03430-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-022-03430-w