
Abstract
Omicron, a variant of concern (VOC) of SARS-CoV-2, emerged in South Africa in November 2021. Omicron has been continuously acquiring a series of new mutations, especially in the spike (S) protein that led to high infectivity and transmissibility. Peptides targeting the receptor-binding domain (RBD) of the spike protein by which omicron and its variants attach to the host receptor, angiotensin-converting enzyme (ACE2) can block the viral infection at the first step. This study aims to identify antiviral peptides from the Antiviral peptide database (AVPdb) and HIV-inhibitory peptide database (HIPdb) against the RBD of omicron by using a molecular docking approach. The lead RBD binder peptides obtained through molecular docking were screened for allergenicity and physicochemical criteria (isoelectric point (pI) and net charge) required for peptide-based drugs. The binding affinity of the best five peptide inhibitors with the RBD of omicron was validated further by molecular dynamics (MD) simulation. Our result introduces five antiviral peptides, including AVP1056, AVP1059, AVP1225, AVP1801, and HIP755, that may effectively hinder omicron-host interactions. It is worth mentioning that all the three major sub-variants of omicron, BA.1 (B.1.1.529.1), BA.2 (B.1.1.529.2), and BA.3 (B.1.1.529.3), exhibits conserved ACE-2 interacting residues. Hence, the screened antiviral peptides with similar affinity can also interrupt the RBD-mediated invasion of different major sub-variants of omicron. Altogether, these peptides can be considered in the peptide-based therapeutics development for omicron treatment after further experimentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since December 2019, the world has been battling the SARS-CoV-2 pandemic. The constant rate at which new Variants of Concern (VOC) are emerging, clasping more risky mutations than previous variants, suggests that it will be a long battle before it is over (World Health Organization (WHO), https://www.who.int/en/activities/tracking-SARS-CoV-2-variants). The VOC omicron was reported as the variant with the highest number of mutations that led to higher transmissibility and some extent of resistance to immunity induced by various vaccines (Variant; Kumar et al. 2022). Omicron is represented by three sublineages, including BA.1, BA.2, and BA.3 (Dyer 2021). Omicron variant BA.1 is highly divergent from the Wuhan-Hu-1, the ancestral strain, with some deletions and more than 30 mutations (e.g., 69–70del, T95I, G142D/143–145del, K417N, T478K, N501Y, N655Y, N679K, and P681H) (Chen and Wei 2022). BA.2 has 32 mutations in common with BA.1 but 28 distinct mutations. The RBD of BA.2 has four unique mutations, and 12 mutations are shared with BA.1 (Chen and Wei 2022). The BA.2 sublineage has been reported with a higher replication rate and enhanced fusogenicity than BA.1. All the variants of SARS-CoV-2 employ the surface-bound spike protein (S) to interact with the angiotensin-converting enzyme 2 (ACE2) receptor of the host cell for attachment, fusion, and entry (Lan et al. 2020; Wang et al. 2020c). S protein is divided into two subunits, the S1 subunit, essential for binding receptors, and the S2 subunit, essential for fusing the cell membranes. S1 subunit has the receptor-binding domain (RBD) that interacts with the ACE2 receptors for viral entry into the host cell (Wang et al. 2020a). Notably, the BA.3 variant has a recombinant S protein of BA.1 and BA.2 variants (Das et al. 2022; Li et al. 2022), with 15 mutations in the RBD domain like BA.1 and BA.2 (Chen and Wei 2022).
As the protein–protein interaction between RBD of S1 and hACE-2 is the first and most crucial step for SARS-CoV-2 entry into the host cell (Sharifkashani et al. 2020), this interaction site has been considered a promising target for therapeutic interventions for blocking the SARS-CoV-2 infection. Molecules targeting this viral entry site need not be cell-penetrating hence may have minimum detrimental effects. Several chemical and natural compounds and small molecules are employed to target the RBD and hACE-2 interaction (Bruno et al. 2013). Still, peptides and peptide-based inhibitors represent the most attractive therapeutic alternative as peptides are more specific, efficient, and can be better tolerated than small molecule drugs (Unciti-Broceta et al. 2013; Brauer et al. 2013). Moreover, peptides can disrupt the protein–protein interactions more effectively compared to small molecules as they can efficiently bind the interface-binding region due to their larger surface area (Kaspar and Reichert 2013). Peptide inhibitors from various resources have also been reported to be effective against SARS-CoV-2 during the COVID-19 pandemic (Schütz et al. 2020). The peptide inhibitors specifically designed against SARS-CoV2 RBD are shown to neutralize the binding affinity of the virus and prevent the virus from entering the human cell (Baig et al. 2020; Han and Král 2020). As the molecular docking method provide the opportunity to investigate biomolecular interactions at the atomic level in a time and cost-effective manner (Moradi et al. 2019; Shahlaei et al. 2021), in this study, we screened 759 antiviral peptides from the Antiviral peptide database (AVPdb) and HIV-inhibitory peptide database (HIPdb) by using HDOCK docking program against the RBD of the S protein of omicron. These two databases consist of a wide range of experimentally verified natural and modified peptide sequences (Qureshi et al. 2014). As the amino acid residues selected in the docking site of RBD are conserved in omicron and its three sublineages (Supplementary Fig. 2), the peptides with higher docking scores are predicted to bind the RBD of omicron and its variants effectively.
The allergenicity and physiochemical properties of 177 peptides with a higher docking score were further analyzed. This study demonstrates that 18 peptides are non-allergenic and bear suitable druggable properties. The five best peptides were further analyzed with molecular dynamics (MD) simulation to obtain high-affinity binders for the RBD of S protein of omicron and its variants. Therefore, these peptides identified herein are worthy of further in vitro and in vivo experimental investigation to develop peptide-based therapeutics of omicron and its variants.
Materials and methods
Selection of efficient peptide libraries for screening
To begin, we chose the two most diverse and experimentally confirmed peptide libraries for our investigation, the AVPdb (https://crdd.osdd.net/servers/avpdb/) and the (HIV-Inhibitory peptide database) (HIPdb) (https://crdd.osdd.net/servers/hipdb/). AVPdb and HIPdb are manually curated databases that include antiviral agents experimentally validated for antiviral activity. As per the AVPdb database, the peptides target 60 medically significant viruses (Qureshi et al. 2013). HIPdb, on the other hand, contains peptides tested on 35 different cell lines that can potentially halt the virus from infecting the host cells (Qureshi et al. 2014). Out of 2683 antiviral peptides, we have selected 759 peptides based on the availability of information on their known inhibitory biological activity.
Retrieval of protein sequence and preparation for docking
As the crystallographic structure of the SARS-CoV-2 omicron variant spike protein was not determined at the initial time of this study, a three-dimensional model of omicron spike protein RBD was developed using homology modeling. This computational approach matured into an essential technique in structural biology, significantly contributing to narrowing the gap between known protein sequences and experimentally determined structures (Waterhouse et al. 2018). For homology modeling, we retrieved the amino acid sequence of omicron spike protein from the NCBI database (https://www.ncbi.nlm.nih.gov/). The obtained protein sequence was modeled into a 3D protein structure using the SWISS-MODEL server (https://swissmodel.expasy.org/), which is regarded as the pioneer of automated comparative modeling and the most frequently used server for 3D protein structure modeling (Schwede et al. 2003). The interacting amino acid residues of omicron RBD with hACE2 were identified by sequence alignment with wild-type SARS-CoV-2-RBD (Supplementary Fig. 2).
The modeled structure was validated by examining the quality of the model in terms of global model quality estimation (GMQE), QMEAN score, and conformational orientation through the structure assessment tool and Ramachandran plot. Generally, the GMQE scores range between 0 and 1, with the higher the score indicating higher reliability of the modeled structure. In the case of QMEAN, a score lower than 4.0 shows reliability (Patel et al. 2019). Furthermore, to get the best conformational state in terms of the lowest energy of the modeled protein, we performed a MD simulation for 10 ns using GROMACS 2020.5 before molecular docking.
Molecular docking of selected peptides with spike RBD
With the lowest energy state modeled RBD, we performed molecular screening of 759 peptides using the high ambiguity-driven protein–protein docking HDOCK server (https://hdock.phys.hust.edu.cn/) (Yan et al. 2017b). We execute site-specific docking in which amino acid residues of RBD that are involved in the protein–protein interaction with the hACE-2 receptor (N414, S443, Y446, Y450, L452, F453, A472, F483, N484, Y103, R490, S493, R495, T497, Y498, G499, and H502) (Wang et al. 2020c) were given as input, enabling peptides to dock at the hACE2 binding site of the RBD. HDOCK enables information-driven, flexible docking for complex biomolecular modeling. This HDOCK server contains binding interface information from PDB as well as user-supplied biological data such as residue constraints and molecule size/shape information collected from small-angle X-ray scattering (SAXS) to perform flexible docking. It uses an intrinsic scoring function to determine protein–protein interactions (Yan et al. 2020b). Besides this, it is a user-friendly, highly integrated, and fast result-producing server, which utilizes a hybrid docking strategy, i.e., template-based and template-free method, which strengthens its prediction accuracy over other docking servers (Yan et al. 2017a).
Physiochemical property and allergenicity toxicity assessment of peptides
The physicochemical properties, including isoelectric point (pI), and net charge at physiological pH of the lead peptides, were analyzed. It was found that peptide/protein solubility is directly related to their pI. Avoiding peptide aggregation will allow for its efficient interaction with viral receptors. pI between 11 and 13 avoids self-aggregation of peptides in biological conditions (Giangaspero et al. 2001). Highly cationic peptides, ranging from +2 to +9, directly correlate with their attraction to negatively charged membranes, whereas a lower net charge reduces interaction between ligand and Receptor (Tossi et al. 1994; Gurtovenko and Vattulainen 2005). PepCalc, an online tool developed by Innovagen AB (https://pepcalc.com/), calculated the isoelectric peptide point (pI) and the net charge. (https://pepcalc.com/). We have further assessed the allergenicity of peptides using the AllerTop v2.0 online tool. (https://www.ddg-pharmfac.net/AllerTOP/method.html). Using amino acid E-descriptors and k-nearest neighbor (kNN) machine-learning approaches, the tool has been demonstrated to predict allergenicity accurately and reliably (Dimitrov et al. 2014).
Molecular dynamics (MD) simulation
The molecular dynamic simulation (MDS) was carried out for the top five successful protein-peptide complexes (AVP1056-RBD, AVP1059-RBD, AVP1225-RBD, AVP1801-RBD, and HIP755-RBD) using GROMACS 2020.5 (Adewole et al. 2022). A 100 ns of MDS was conducted for each complex at 1.0 bar pressure and 300 K temperature by placing each complex in a periodic cubic box (1.2-nm edge). All the simulated systems were set at pH 7.5. For all complexes, the topology files were prepared using the pdb2gmx program (Kumar and Ramanathan 2015). A CHARMM27 all-atom force field (Huang and MacKerell 2013) and TIP3P water model (Mark and Nilsson 2001) were employed in this MDS study. The added water molecules for AVP1056, AVP1059, AVP1225, AVP1801, and HIP755 complex systems were 17,801, 14,787, 15,726, 14,770, and 15,297, respectively. Each of the systems was neutralized by using Na+ or Cl− ions. For instance, the AVP1056, AVP1059, AVP1225, AVP1801, and HIP755 complexes were neutralized by adding 8Cl, 9Cl, 6Cl, 6Cl, and 6Cl ions respectively to the system. The steepest descent algorithm (Banavath et al. 2014) was being used for the energy minimization of the complexes with 50,000 steps. Following this, pre-equilibration was done by NVT and NPT for 1 ns each to attain constant pressure (1 bar) and temperature (300 K). Long-range electrostatics were calculated using Particle Mesh Ewald Method (Wang et al. 2010). The final MD production run was carried out for 100 ns for each complex (Kumari and Subbarao 2021). The MDS trajectories of each complex were analyzed through the root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and hydrogen bond interactions parameters to study their conformational changes and stability in the system. Further, the result analysis was done through XMGrace (GRaphing, Advanced Computation, and Exploration of data) software (Cowan and Grosdidier 2000).
Results
Homology modeling, assessment, and validation of Spike RBD
In the absence of crystallographic spike protein of omicron, we developed a homology model of omicron’s RBD using its amino acid sequence, retrieved from the NCBI database. Omicron RBD’s interacting amino acid residues with hACE2 were identified by sequence alignment with wild-type SARS-CoV-2-RBD (Lan et al. 2020) (Supplementary Fig. 2). The homology model was created using the SWISS-MODEL server. Validation of structure was done based on Ramachandran plot analysis. The RBD was modeled on template 7e8m.1.A (RBD of SARS-CoV-2) with a resolution of 2.09 Å and an overall sequence identity of 93.08%. The GMQE and QMEAN scores of the modeled RBD were 0.81 and −0.45, suggesting the best quality model. Ramachandran plot reveals 97.66% of RBD amino acid residues lie in the favored region (Supplementary Fig. 1). Before molecular docking, we ran a MD simulation for 10 ns to find the optimal conformational state in terms of the lowest energy (−927,143.56 kJ/mole) of the modeled protein. The original numbering of amino acid residues of the omicron RBD crucial for interaction with ACE2 receptors after homology modeling is changed from N414 to N31, S443 to S60, Y446 to Y63, Y450 to Y67, L452 to L69, F453 to F70, A472 to A89, F483 to F100, N484 to N101, Y103 to Y486, R490 to R107, S493 to S110, R495 to R112, T497 to T114, Y498 to Y115, G499 to G116, and H502 to H119, respectively.
Molecular docking of peptides against spike RBD
A molecular docking approach reveals the binding affinity of the individual ligand to a specific target protein structure to assist rational drug design (Jones and Willett 1995). Understanding the interaction dynamics and possible binding mechanisms will facilitate the discovery of strong protein inhibitors (Liu et al. 2018; Patel et al. 2019). Using the HDOCK server, we evaluated the binding affinities, interactions between the query peptide sequences from AVPdb and HIPdb, and the amino acid residues of omicron RBD crucial for interaction with hACE2.
The RBD of the virus interacts mainly with the α-1 helix of host ACE-2 (Yan et al. 2020a). By introducing mutations in the non-interacting residues of the α-1 helix, Panda et al. (2021) designed peptides that showed a better binding affinity for SARS-CoV2 RBD than hACE-2’s α-1 helix. Among all his designed mutated peptides, P13 exhibited the highest affinity for the SARS-CoV2 RBD (Panda et al. 2021). Our study considered P13 (IDWQFWFHYDKWDHEWEDEWYQSS) as a reference peptide for the molecular docking analysis. One hundred seventy-seven peptides from the AVPdb and HIPdb have shown better docking scores than reference peptides indicating a higher affinity for omicron spike protein RBDs. The docking scores of the best eighteen peptides after filtering through physiochemical and allergenic parameters (explained in detail in the following section) are illustrated in Fig. 1a. The molecular docking of the best five peptides is further performed using PATCHDOCK docking program. The PATCHDOCK docking scores are shown in supplementary Table 1. The docking scores of AVP1059 and AVP1801 were found more than the control, however, the docking scores of AVP1056, HIP755, and AVP1225 are slightly lower than the control peptide. This visual difference is in the docking score is seen due to the different algorithm used by these two free web servers. The PATCHDOCK server, uses geometric hashing and pose-clustering matching techniques to detect the best-docked peptide (Duhovny et al. 2002). Whereas, the HDOCK web server uses a template-based hybrid docking algorithm to generates the docking scores (Yan et al. 2020b).

Graphical illustration of docking Score of 18 antiviral peptides and their control
Hydrogen bonds (H-bonds) significantly strengthen the ligand–protein interaction (Chen et al. 2016). The hydrogen bonds involved in the molecular interactions between the best five peptides (AVP1056, AVP1059, HIP755, AVP1801, and AVP1225) and the omicron-RBD are depicted in Table 1.
Investigation of physicochemical and allergenicity parameters of peptides
The cationic (+2 to +9) charge of peptides significantly impacts their affinity for binding to the anionic target site (Fry 2018). AMPs’ cationic nature causes electrostatic attraction to the negatively charged phospholipids of microbial membranes, and their hydrophobicity facilitates integration into the microbial cell membrane (Cederlund et al. 2011). Furthermore, the solubility of peptides and proteins is proportional to their pI. Peptide aggregation will not allow it to interact with the viral receptors. A pI between 11 and 13 at physiological pH prevents self-aggregation, which is required for a peptide to interact well with the cellular receptor (Giangaspero et al. 2001). Therefore we determine the net charge and the theoretical pI of all the 177 peptides using the PepCalc server.
The Th2 response is triggered when our body is exposed to allergenic stimuli, causing B cells to produce IgE, which binds with eosinophil receptor FceRI to promote eosinophil activation. Inflammation and tissue damage are associated with activated eosinophils (Brock 1995) (Dimitrov et al. 2013; Huby et al. 2000). Therefore, we evaluated the 177 antiviral peptides’ allergenicity properties using the AllerTop server. The AllerTop server contains a training set of 2427 recognized allergens from various species and 2427 non-allergens to evaluate the antigenic property of the query peptides (Dimitrov et al. 2013). The above investigations demonstrated that 18 antiviral peptides out of 177 have a net charge range between +2 and +9, a pI range between 11 and 13, and were non-allergenic (Table 2). This indicates that these 18 peptides may be suitable for peptide-based therapeutics.
Description of the interactions of the five lead peptides with the omicron RBD
AVP1056
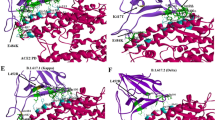
Costin et al. (2010) designed a peptide AVP1056 (DN57opt), which interacts with the Dengue Virus E protein and inhibits viral infection in culture. The peptide AVP1056 inhibits virus binding to cells through binding directly to the E protein (Düzgüneş et al. 2021). In our molecular docking study, AVP1056 is ranked first (docking score of -305.87) and interacts with ARG107, PHE104, SER108, TYR63, SER60, and SER110 residues of the modeled RBD of omicron (Fig. 2). Notably, the peptide can strongly intervene in the viral infection by binding with four crucial amino acids including ARG104, Tyr63, SER60, and SER110 (Table 1) of the omicron RBD involved in the ACE2 binding process.

Representation of molecular interaction diagram of AVP1056 in the binding site of RBD
AVP1059
The AVP1059 (DNBLK1) is a light-chain dynein binding protein with an arginine tail (8R) at the N-terminus that was designed to compete with p54 for cytoplasmic dynein interaction during ASFV infection developed by Bruno et al. (2013) to combat the African swine fever virus (ASFV) infection (Alonso et al. 2001; Hernáez et al. 2010). In our search, we found that AVP1059 has a high affinity (docking score of -277.71) for the omicron RBD, which can neutralize the viral attachment by binding with the amino acid residues such as ARG112, GLU79, LEU66, and ARG80 of the omicron RBD (Fig. 3).

Illustration of molecular interaction diagram of AVP1059 in the binding site of RBD
AVP1801
AVP1801 is a glycoprotein gH (gHH3) peptide from the Marek's disease virus (MDV) of the Herpesviridae family. AVP1801 exhibited potent antiviral activity for MDV in plaque formation assays in vitro and lesion formation assays in vivo (Wang et al. 2011). In the HDOCK and PTACHDOCK molecular docking studies, the AVP1801 has shown a better docking score than the control. Moreover, the AVP1801 forms six H-bonds at SER108, TYR109, ARG112, TYR67, and ASN31 residues of RBD SARS-CoV-2 omicron (Table 1, Fig. 4). We predict that the peptide can potentially neutralize the viral infection by binding with the three amino acids, including ARG112, TYR67, and ASN31 (Fig. 4), which are crucial for ACE2 binding.

Illustration of molecular interaction diagram of AVP1801 in the binding site of RBD
AVP1225
AVP1225 (GBVA3) is a peptide derived from the GB Virus A (GBV-A), belonging to the Flaviviridae family. AVP1225 was shown to inhibit HCV infection by targeting its entry into hepatocytes in human hepatoma-derived HuH-7 cell lines (Huh7.5.1) (Liu et al. 2013). In our study, AVP1225 showed a binding score of -260.42 with the RBD of the spike protein of omicron. AVP1225 forms six H-bonds with TYR115, ARG107, THR84, GLU85, and SER108 residues of RBD of SARS-CoV-2 omicron (Fig. 5). Notably, it potentially interacts with two amino acids (TYR115 and ARG107) involved in the ACE2 binding process.

Illustration of molecular interaction diagram of AVP1225 in the binding site of RBD
HIP755
HIV755 (D94) peptide has been tested for its ability to inhibit Human Immunodeficiency Virus (HIV) entry (Buckheit et al. 1995). In our study, HIP755 showed a stronger affinity for the binding site of omicron RBD (docking score of -271.13). Notably, this peptide does not interact with the crucial amino acids of RBD involved in the ACE2 binding (Fig. 6); hence may interfere with the viral attachment with ACE2 by blocking the interacting site of omicron RBD.

Illustration of molecular interaction diagram of HIP755 in the binding site of RBD
Molecular dynamics simulation
The top five docked complexes were selected for the MD simulation to study the conformational stability of the complexes through the analysis of RMSD, RMSF, Rg, SASA, and hydrogen bond parameters. After completing the simulation of five protein-peptide complexes, we analyzed the trajectory of each simulated complex. The average RMSD of C–α atoms for all the five complexes AVP1056-RBD, AVP1059-RBD, AVP1255-RBD, AVP1801-RBD, and HIP755-RBD were estimated to be ~0.314 nm, ~0.317 nm, ~0.297 nm, ~0.261 nm, and ~0.366 nm, respectively (Fig. 7A). It was found that the average fluctuations detected in the backbone of all the complexes are below 3 Å or 0.3 nm, which suggests that the binding of the antiviral peptides at the RBD does not alter the protein's backbone (Banavath et al. 2014; Pandey et al. 2017). Hence, RMSD analysis revealed that the binding of the five peptides with the omicron RBD is stable and consistent throughout the 100 ns of the simulation period. The dynamic nature of each amino acid residue of the RBD protein was analyzed with the help of RMSF parameters. Figure 7B illustrates the RMSF plot for each complex with respect to the average time position. From the graph, it is clear that there are no noticeable fluctuations in the ligand-binding residues of the complexes. Although for AVP1059, there was a slight fluctuation up to 1 nm seen in the initial five residues, after which it was stable with no significant fluctuations. The average RMSF values for AVP1056, AVP1059, AVP1225, AVP1801, and HIP755 docked complexes were found to be ~0.160, ~0.193, ~0.158, ~0.159, and ~0.201 nm, respectively. Notably, key interface interacting residues of RBD with the hACE2 receptor show a minimal fluctuation of less than 0.5 nm (Fig. 7B), indicating the favorable binding of antiviral peptides with RBD (Pundir et al. 2021; Naik et al. 2020). The radius of gyration or Rg parameter was analyzed to study the compactness of the protein-peptide complexes in a dynamic environment. Figure 7C shows the relation between Rg values of all the five complexes with respect to the 100 ns time range. It is seen that the trajectories of all the five complexes remain constantly compacted, specifying the high binding compatibility of each peptide within the active site of the RBD. The average Rg for AVP1056 and AVP1059 is ~1.69 nm, while the average Rg values for AVP1225, AVP1801, and HIP755 were found to be ~1.64, ~1.67, and ~1.66 nm, respectively which fall within the acceptable range of ~1.64 ± 0.2 nm (Lobanov et al. 2008). Furthermore, the SASA estimates the free energy of nonpolar solvation of each atom in the molecule throughout the simulation (Kumari and Subbarao 2021). Figure 7D shows the SASA plot of the complexes in relation to simulation time, indicating stable surface accessibility of the protein complexes throughout the simulation time. The calculated average values of SASA for AVP1056, AVP1059, AVP1225, AVP1801, and HIP755 docked complexes were 87.65, 87.31, 84.73, 85.01, and 89.51 nm2, respectively. The consistent solvent accessibility of these complexes throughout the simulation period indicates the least conformational changes and hence stable binding between peptides and protein (Marsh Joseph and Teichmann Sarah 2011; Naik et al. 2021).

Graphical representation of A Root Mean Square Deviation (RMSD), B Root Mean Square Fluctuation (RMSF), C Radius of Gyration, and D Solvent Accessible Surface Area (SASA) through the molecular dynamics simulation of AVP1056-RBD (black), AVP1059-RBD (red), AVP1225-RBD (green), AVP1801-RBD (blue), and HIP755-RBD (yellow) docked complexes
Figure 8 plots the no of hydrogen bonds formed between the protein-peptide complexes with respect to the simulation time (100 ns). The average no of H-bonds formed by AVP1056, AVP1059, AVP1225, AVP1801, and HIP755 docked complexes were ~7, ~3, ~8.4, ~1.8, and ~5.2, respectively. The hydrogen bond interactions turn out to be one of the major forces for inhibition (Panda et al. 2021). The number of hydrogen bond formations between the protein and peptides is directly proportional to the stability of the complex (Al-Karmalawy et al. 2021). H-bond indicates the formation of strong interactions during the simulation period. This simulation study reveals that the potential antiviral peptides can strongly bind with the RBD of omicron without creating any significant conformational changes, thus providing evidence for stable complex formation between peptides-RBD of omicron (Pundir et al. 2021).

Hydrogen bonds graphs of protein–ligand interaction of five top-ranked complexes A AVP1056-RBD (black), B AVP1059-RBD (red), C AVP1225-RBD (green), D AVP1801-RBD (blue), and E HIP755-RBD (yellow) showing the presence of H-bond throughout the time scale of 100 ns MD simulations
Discussion
Peptides and peptide-based inhibitors as therapeutics are an appealing alternative to small molecules since peptides exhibit numerous advantages such as high specificity and affinity, low associated toxicity, low immunogenicity, and comparatively short development time (Lau et al. 2018; Lee et al. 2019; Jitendra et al. 2011; Loganathan et al. 2020; Lu et al. 2020). That resulted in the rise of FDA approval of peptide-based therapeutics in the last decade (Sharma et al. 2020). Hence, peptide-based therapeutics have gained more interest, and many peptides of various origins are now being studied to combat viral infection (Nag et al. 2021). Peptides of natural origin exist as part of the innate immune system like protegrin, defensins, and LL-37 (Karthik et al. 2014; Vilas Boas et al. 2019; Wilson et al. 2013; Wang et al. 2020a), whereas some of the peptides, including endogenous peptide-based inhibitors of VIRIP (Eggink et al. 2019; Münch et al. 2007) and CXCR4 (Buske et al. 2015), are released from precursor proteins by proteolytic cleavage (Buske et al. 2015). Additionally, peptides can be designed based on virus-associated proteins’ function and structure like myrcludex B targeted against hepatitis-B (Schulze et al. 2010; Blank et al. 2016) and T20 targeted against HIV-1 (Eggink et al. 2019).
The continuous evolution of SARS-CoV-2 results in the emergence of a new variant of concerns, and omicron is one of them characterized by 30 mutations, 15 of which occur in the RBD of the spike protein (Singhal 2022). The protein–protein interaction between the RBD of S protein of SRAS-CoV2 and omicron with hACE2 is the sole cause of the COVID-19 pandemic and has been considered a promising target for therapeutic interventions. Peptides have been shown to efficaciously disrupt protein–protein interactions (PPIs) as they have a larger surface area bound explicitly to the interface-binding region (Kaspar and Reichert 2013). Unlike small-molecule drugs, single mutations on target sites do not render peptide drugs non-functional (Sorolla et al. 2020).
Targeting the RBD−ACE2 interaction with linear peptides has been considered a strategy for virus neutralization supported by previous in-silico and experimental studies (Byrnes et al. 2020; Shi et al. 2020; Ju et al. 2020; Yuan et al. 2020). This study aims to identify the peptide inhibitors targeting omicron RBD from two experimentally validated AVPdbs such as AVPdb and HIPdb. For that, we opted molecular docking method to screen the peptides against omicron RBD. The molecular docking approach allows positioning peptide/ligands into a receptor structure in various orientations, conformations, and positions, resulting in a complex with a reduced binding free energy (Meng et al. 2011; Singh et al. 2021b) that can be ranked and grouped using the scoring function of the software.
Molecular docking analysis using the HDOCK program revealed that 177 peptides have a better binding affinity for the omicron RBD than the control peptide. As the next step in this study, we examined physiochemical features such as the net charge, the pI, and allergenicity to identify peptides suitable for therapeutics. The study demonstrated that 18 peptides fulfilled the required physicochemical parameters (Table 2) for peptide-based therapeutics. The molecular interaction analysis of the best five antiviral peptides (AVP1056, AVP1059, AVP1225, AVP1801, and HIP755) reveals that all the peptides have relevant interactions with omicron RBD (Figs. 2, 3, 4, 5, 6). Furthermore, the MD simulation, RMSF, Rg, and SASA analysis of these five peptides with omicron RBD revealed that the peptide-protein complexes are stable and compact in dynamic conditions (Fig. 7A–D). As these peptides are targeted for the viral surface protein and need not penetrate the host cell, they may not interact with the proteins within the cell, thereby having less chance of cross-reactivity. However, as these peptides have antiviral activity, they may target other viral infections if the host has co-infection. Computer-aided peptide-based drug development targeting SARS-COV-2 RBD has been shown to be successful in SARS-COV-2. For instance, Valiente et al. (2021) computationally designed two D-peptides with an affinity toward SARS-COV-2 RBD. They have tested these peptides experimentally and showed successful viral neutralization. Another such study identifies the potential of Nsp1 inhibitors to promote the complete cessation of host protein translation by aminoarylbenzosuberene molecules (Singh et al. 2021a). Such studies showed that in-silico lead candidates have potential efficacy, and experimental results align with in-silico results for many cases (Singh et al. 2022). However, our molecular docking study lacks experimental validation. We propose that further in vitro and in vivo studies may reveal the therapeutic potential of our lead peptides to block the SARS-CoV omicron virus infection.
Conclusion
Even after 3 years of SARS-CoV-2, this pandemic continues to cause overwhelming death and infection worldwide and demands effective therapeutics against present SARS-CoV-2 VOCs. This study has implemented a molecular docking approach to investigate antiviral peptides’ inhibitory potential as effective leads for developing new therapeutics against SARS-CoV-2 omicron. To initiate our investigation, we screened peptide libraries AVPdb and HIPdb against the RBD of SARS CoV-2 omicron. The assessment of the physicochemical properties of the resultant antiviral peptide molecules revealed the drug-likeness properties of 18 peptide molecules among 177 peptides. Further molecular dynamic simulation study revealed that the top five antiviral peptides (AVP1059, AVP1056, AVP1225, AVP1801, and HIP755) form a stable and rigid peptide-protein complex with omicron RBD. Thus, these findings may be valuable for developing efficient antiviral therapeutics to combat SARS-CoV-2 omicron at the first site of infection.
References
Adewole K, Ishola A, Olaoye I (2022) In silico profiling of histone deacetylase inhibitory activity of compounds isolated from Cajanus cajan. Beni-Suef University J Basic Appl Sci 11(1):1–22
Al-Karmalawy AA, Dahab MA, Metwaly AM, Elhady SS, Elkaeed EB, Eissa IH, Darwish KM (2021) Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front Chem 9:6612–6630. https://doi.org/10.3389/fchem.2021.661230
Alonso C, Miskin J, Hernáez B, Fernandez-Zapatero P, Soto L, Cantó C, Rodríguez-Crespo I, Dixon L, Escribano JM (2001) African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light-chain dynein. J Virol 75(20):9819–9827. https://doi.org/10.1128/jvi.75.20.9819-9827.2001
Baig MS, Alagumuthu M, Rajpoot S, Saqib U (2020) Identification of a potential peptide inhibitor of SARS-CoV-2 targeting its entry into the host cells. Drugs R&d 20(3):161–169
Banavath HN, Sharma OP, Kumar MS, Baskaran R (2014) Identification of novel tyrosine kinase inhibitors for drug resistant T315I mutant BCR-ABL: a virtual screening and molecular dynamics simulations study. Sci Rep 4:6948. https://doi.org/10.1038/srep06948
Blank A, Markert C, Hohmann N, Carls A, Mikus G, Lehr T, Alexandrov A, Haag M, Schwab M, Urban S (2016) First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J Hepatol 65(3):483–489
Brauer F, Schmidt K, Zahn RC, Richter C, Radeke HH, Schmitz JE, von Laer D, Egerer L (2013) A rationally engineered anti-HIV peptide fusion inhibitor with greatly reduced immunogenicity. Antimicrob Agents Chemother 57(2):679–688
Brock JH (1995) Immunobiology: the immune system in health and disease, CA Janeway Jr, P. Travers (eds.), Garland Publishers (1994),£ 18.95 (xvii+ 575 pages) ISBN 0 865 42811 5. Elsevier Current Trends
Bruno BJ, Miller GD, Lim CS (2013) Basics and recent advances in peptide and protein drug delivery. Ther Deliv 4(11):1443–1467. https://doi.org/10.4155/tde.13.104
Buckheit RW Jr, Kinjerski TL, Fliakas-Boltz V, Russell JD, Stup TL, Pallansch LA, Brouwer WG, Dao DC, Harrison WA, Schultz RJ et al (1995) Structure-activity and cross-resistance evaluations of a series of human immunodeficiency virus type-1-specific compounds related to oxathiin carboxanilide. Antimicrob Agents Chemother 39(12):2718–2727. https://doi.org/10.1128/aac.39.12.2718
Buske C, Kirchhoff F, Münch J (2015) EPI-X4, a novel endogenous antagonist of CXCR4. Oncotarget 6(34):35137
Byrnes JR, Zhou XX, Lui I, Elledge SK, Glasgow JE, Lim SA, Loudermilk RP, Chiu CY, Wang TT, Wilson MR, Leung KK, Wells JA (2020) Competitive SARS-CoV-2 Serology Reveals Most Antibodies Targeting the Spike Receptor-Binding Domain Compete for ACE2 Binding. mSphere 5 (5). doi:https://doi.org/10.1128/mSphere.00802-20
Cederlund A, Gudmundsson GH, Agerberth B (2011) Antimicrobial peptides important in innate immunity. FEBS J 278(20):3942–3951
Chen D, Oezguen N, Urvil P, Ferguson C, Dann SM, Savidge TC (2016) Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci Adv 2(3):e1501240
Chen J, Wei G-W (2022) Omicron BA.2 (B.1.1.529.2): high potential to becoming the next dominating variant. J Phys Chem Lett 13:3840–3849
Costin JM, Jenwitheesuk E, Lok S-M, Hunsperger E, Conrads KA, Fontaine KA, Rees CR, Rossmann MG, Isern S, Samudrala R (2010) Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl Trop Dis 4(6):e721
Cowan R, Grosdidier G Visualization tools for monitoring and evaluation of distributed computing systems. In: Proc. of the International Conference on Computing in High Energy and Nuclear Physics, Padova, Italy, 2000.
Das S, Samanta S, Banerjee J, Pal A, Giri B, Kar SS, Dash SK (2022) Is Omicron the end of pandemic or start of a new innings? Travel Med Infect Dis 48:102332. https://doi.org/10.1016/j.tmaid.2022.102332
Dimitrov I, Flower DR, Doytchinova I (2013) AllerTOP–a server for in silico prediction of allergens. BMC Bioinformatics 14(6):S4. https://doi.org/10.1186/1471-2105-14-s6-s4
Dimitrov I, Naneva L, Doytchinova I, Bangov I (2014) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30(6):846–851
Duhovny D, Nussinov R, Wolfson HJ Efficient Unbound Docking of Rigid Molecules. In: Guigó R, Gusfield D (eds) Algorithms in Bioinformatics, Berlin, Heidelberg, 2002// 2002. Springer Berlin Heidelberg, pp 185–200
Düzgüneş N, Fernandez-Fuentes N, Konopka K (2021) Inhibition of viral membrane fusion by peptides and approaches to peptide design. Pathogens 10(12):1599
Dyer O (2021) Covid-19: South Africa’s surge in cases deepens alarm over omicron variant. BMJ 375:n3013. https://doi.org/10.1136/bmj.n3013
Eggink D, Bontjer I, de Taeye SW, Langedijk JP, Berkhout B, Sanders RW (2019) HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry. J Biol Chem 294(15):5736–5746
Fry DE (2018) Antimicrobial peptides. Surg Infect 19(8):804–811. https://doi.org/10.1089/sur.2018.194
Giangaspero A, Sandri L, Tossi A (2001) Amphipathic α helical antimicrobial peptides. A systematic study of the effects of structural and physical properties on biological activity. Eur J Biochem 268(21):5589–5600
Gurtovenko AA, Vattulainen I (2005) Pore formation coupled to ion transport through lipid membranes as induced by transmembrane ionic charge imbalance: atomistic molecular dynamics study. J Am Chem Soc 127(50):17570–17571
Han Y, Král P (2020) Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano 14(4):5143–5147
Hernáez B, Tarragó T, Giralt E, Escribano JM, Alonso C (2010) Small peptide inhibitors disrupt a high-affinity interaction between cytoplasmic dynein and a viral cargo protein. J Virol 84(20):10792–10801. https://doi.org/10.1128/JVI.01168-10
Huang J, MacKerell AD Jr (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34(25):2135–2145. https://doi.org/10.1002/jcc.23354
Huby RD, Dearman RJ, Kimber I (2000) Why are some proteins allergens? Toxicol Sci 55(2):235–246
Jitendra P, Bansal S, Banik A (2011) Noninvasive routes of proteins and peptides drug delivery. Indian J Pharm Sci 73(4):367–375
Jones G, Willett P (1995) Docking small-molecule ligands into active sites. Curr Opin Biotechnol 6(6):652–656
Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, Yu J, Shan S, Zhou B, Song S, Tang X, Yu J, Lan J, Yuan J, Wang H, Zhao J, Zhang S, Wang Y, Shi X, Liu L, Zhao J, Wang X, Zhang Z, Zhang L (2020) Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584(7819):115–119. https://doi.org/10.1038/s41586-020-2380-z
Karthik L, Kumar G, Keswani T, Bhattacharyya A, Chandar SS, Bhaskara Rao K (2014) Protease inhibitors from marine actinobacteria as a potential source for antimalarial compound. PLoS ONE 9(3):e90972
Kaspar AA, Reichert JM (2013) Future directions for peptide therapeutics development. Drug Discovery Today 18(17–18):807–817
Kumar A, Ramanathan K (2015) Analyzing resistance pattern of non-small cell lung cancer to crizotinib using molecular dynamic approaches. Indian J Biochem Biophys 52:23–28
Kumari M, Subbarao N (2021) Identification of novel multitarget antitubercular inhibitors against mycobacterial peptidoglycan biosynthetic Mur enzymes by structure-based virtual screening. J Biomol Struct Dyn 7:1–12. https://doi.org/10.1080/07391102.2021.1908913
Kumar S, Thambiraja TS, Karuppanan K, Subramaniam G (2022) Omicron and Delta variant of SARS-CoV-2: a comparative computational study of spike protein. J Med Virol 94(4):1641–1649
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581(7807):215–220. https://doi.org/10.1038/s41586-020-2180-5
Lau JL, Dunn MKJB, chemistry m (2018) Therapeutic peptides: Historical perspectives, current development trends, and future directions Bioorg Med Chem 26(10):2700-2707
Lee AC-L, Harris JL, Khanna KK, Ji-Hong H (2019) A comprehensive review on current advances in peptide drug development and design Int J Mol Sci 20(10):2383
Li Q, Zhang M, Liang Z, Zhang L, Wu X, Yang C, An Y, Tong J, Liu S, Li T, Cui Q, Nie J, Wu J, Huang W, Wang Y (2022) Antigenicity comparison of SARS-CoV-2 Omicron sublineages with other variants contained multiple mutations in RBD. MedComm 3(2):e130. https://doi.org/10.1002/mco2.130
Liu X, Huang Y, Cheng M, Pan L, Si Y, Li G, Niu Y, Zhao L, Zhao J, Li X, Chen Y, Yang W (2013) Screening and rational design of hepatitis C virus entry inhibitory peptides derived from GB virus A NS5A. J Virol 87(3):1649–1657. https://doi.org/10.1128/JVI.02201-12
Liu Z, Liu Y, Zeng G, Shao B, Chen M, Li Z, Jiang Y, Liu Y, Zhang Y, Zhong H (2018) Application of molecular docking for the degradation of organic pollutants in the environmental remediation: A review. Chemosphere 203:139–150
Lobanov MY, Bogatyreva NS, Galzitskaya OV (2008) Radius of gyration as an indicator of protein structure compactness. Mol Biol 42(4):623–628. https://doi.org/10.1134/S0026893308040195
Loganathan SK, Schleicher K, Malik A, Quevedo R, Langille E, Teng K, Oh RH, Rathod B, Tsai R, Samavarchi-Tehrani PJS (2020) Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 367(6483):1264–1269
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu NJTl, (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395(10224):565–574
Mark P, Nilsson L (2001) Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A 105(43):9954–9960
Marsh Joseph A, Teichmann Sarah A (2011) Relative solvent accessible surface area predicts protein conformational changes upon binding. Structure 19(6):859–867. https://doi.org/10.1016/j.str.2011.03.010
Meng X-Y, Zhang H-X, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7(2):146–157. https://doi.org/10.2174/157340911795677602
Moradi S, Khani S, Ansari M, Shahlaei M (2019) Atomistic details on the mechanism of organophosphates resistance in insects: insights from homology modeling, docking and molecular dynamic simulation. J Mol Liq 276:59–66
Münch J, Ständker L, Adermann K, Schulz A, Schindler M, Chinnadurai R, Pöhlmann S, Chaipan C, Biet T, Peters T (2007) Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 129(2):263–275
Nag A, Paul S, Banerjee R, Kundu R (2021) In silico study of some selective phytochemicals against a hypothetical SARS-CoV-2 spike RBD using molecular docking tools. Comput Biol Med 137:104818. https://doi.org/10.1016/j.compbiomed.2021.104818
Naik B, Gupta N, Ojha R, Singh S, Prajapati VK, Prusty D (2020) High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. Int J Biol Macromol 160:1–17. https://doi.org/10.1016/j.ijbiomac.2020.05.184
Naik B, Mattaparthi VSK, Gupta N, Ojha R, Das P, Singh S, Prajapati VK, Prusty D (2021) Chemical system biology approach to identify multi-targeting FDA inhibitors for treating COVID-19 and associated health complications. J Biomol Struct Dyn:1–25. doi:https://doi.org/10.1080/07391102.2021.1931451
Panda SK, Sen Gupta PS, Biswal S, Ray AK, Rana MK (2021) ACE-2-Derived biomimetic peptides for the inhibition of spike protein of SARS-CoV-2. J Proteome Res 20(2):1296–1303. https://doi.org/10.1021/acs.jproteome.0c00686
Pandey RK, Narula A, Naskar M, Srivastava S, Verma P, Malik R, Shah P, Prajapati VK (2017) Exploring dual inhibitory role of febrifugine analogues against Plasmodium utilizing structure-based virtual screening and molecular dynamic simulation. J Biomol Struct Dyn 35(4):791–804. https://doi.org/10.1080/07391102.2016.1161560
Patel B, Singh V, Patel D (2019) Structural bioinformatics. In: Essentials of Bioinformatics, Vol I. Springer, pp 169–199
Pundir H, Joshi T, Joshi T, Sharma P, Mathpal S, Chandra S, Tamta S (2021) Using Chou’s 5-steps rule to study pharmacophore-based virtual screening of SARS-CoV-2 Mpro inhibitors. Mol Diversity 25(3):1731–1744. https://doi.org/10.1007/s11030-020-10148-5
Qureshi A, Thakur N, Kumar M (2013) HIPdb: a database of experimentally validated HIV inhibiting peptides. PLoS ONE 8(1):e54908
Qureshi A, Thakur N, Tandon H, Kumar M (2014) AVPdb: a database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res 42(D1):D1147–D1153
Schulze A, Schieck A, Ni Y, Mier W, Urban S (2010) Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J Virol 84(4):1989–2000
Schütz D, Ruiz-Blanco YB, Münch J, Kirchhoff F, Sanchez-Garcia E, Müller JA (2020) Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv Drug Deliv Rev 167:47–65
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31(13):3381–3385
Shahlaei M, Zamani P, Farhadian N, Balaei F, Ansari M, Moradi S (2021) Cholesterol-lowering drugs the simvastatin and atorvastatin change the protease activity of pepsin: an experimental and computational study. Int J Biol Macromol 167:1414–1423
Sharifkashani S, Bafrani MA, Khaboushan AS, Pirzadeh M, Kheirandish A, Yavarpour-BaliHessamiSaghazadehRezaei HAAN (2020) Angiotensin-converting enzyme 2 (ACE2) receptor and SARS-CoV-2: potential therapeutic targeting. Eur J Pharmacol 884:173455
Sharma P, Singh H, Bal C, Kumar R (2020) THE NEW DRUGS of 2019 The 48 medicines represent another highly productive year for the pharmaceutical industry, with cancer and rare-disease drugs again dominating the list (vol 98, pg 32. Chem Eng News 98(7):4–4
Shi R, Shan C, Duan X, Chen Z, Liu P, Song J, Song T, Bi X, Han C, Wu L, Gao G, Hu X, Zhang Y, Tong Z, Huang W, Liu WJ, Wu G, Zhang B, Wang L, Qi J, Feng H, Wang F-S, Wang Q, Gao GF, Yuan Z, Yan J (2020) A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584(7819):120–124. https://doi.org/10.1038/s41586-020-2381-y
Singh R, Bhardwaj VK, Das P, Bhattacherjee D, Zyryanov GV, Purohit R (2022) Benchmarking the ability of novel compounds to inhibit SARS-CoV-2 main protease using steered molecular dynamics simulations. Comput Biol Med 146:105572. https://doi.org/10.1016/j.compbiomed.2022.105572
Singh R, Bhardwaj VK, Das P, Purohit R (2021a) A computational approach for rational discovery of inhibitors for non-structural protein 1 of SARS-CoV-2. Comput Biol Med 135:104555. https://doi.org/10.1016/j.compbiomed.2021.104555
Singh R, Bhardwaj VK, Purohit R (2021b) Potential of turmeric-derived compounds against RNA-dependent RNA polymerase of SARS-CoV-2: an in-silico approach. Comput Biol Med 139:104965. https://doi.org/10.1016/j.compbiomed.2021.104965
Singhal T (2022) The emergence of omicron: challenging times are here again! Indian J Pediatr 89(5):490–496. https://doi.org/10.1007/s12098-022-04077-4
Sorolla A, Wang E, Golden E, Duffy C, Henriques ST, Redfern AD, Blancafort PJO (2020) Precision medicine by designer interference peptides: applications in oncology and molecular therapeutics. Oncogene 39(6):1167–1184
Tossi A, Scocchi M, Skerlavaj B, Gennaro R (1994) Identification and characterization of a primary antibacterial domain in CAP18, a lipopolysaccharide binding protein from rabbit leukocytes. FEBS Lett 339(1–2):108–112
Unciti-Broceta JD, Del Castillo T, Soriano M, Magez S, Garcia-Salcedo JA (2013) Novel therapy based on camelid nanobodies. Ther Deliv 4(10):1321–1336
Valiente PA, Wen H, Nim S, Lee J, Kim HJ, Kim J, Perez-Riba A, Paudel YP, Hwang I, Kim K-D, Kim S, Kim PM (2021) Computational design of potent D-peptide inhibitors of SARS-CoV-2. J Med Chem 64(20):14955–14967. https://doi.org/10.1021/acs.jmedchem.1c00655
Variant O SARS-CoV-2 B. 1.1. 529 (Omicron) Variant—United States, December 1–8, 2021.
Vilas Boas LCP, Campos ML, Berlanda RLA, de Carvalho NN, Franco OL (2019) Antiviral peptides as promising therapeutic drugs. Cell Mol Life Sci 76(18):3525–3542
Wang X, Chi X, Wang M (2011) Structural characteristics and antiviral activity of multiple peptides derived from MDV glycoproteins B and H. Virol J 8(1):1–13
Wang H, Dommert F, Holm C (2010) Optimizing working parameters of the smooth particle mesh Ewald algorithm in terms of accuracy and efficiency. J Chem Phys 133(3):034117. https://doi.org/10.1063/1.3446812
Wang H, Li X, Li T, Zhang S, Wang L, Wu X, Liu J (2020ba) The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur J Clin Microbiol Infect Dis 39(9):1629–1635
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen KY, Wang Q, Zhou H, Yan J, Qi J (2020dc) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181(4):894-904.e899. https://doi.org/10.1016/j.cell.2020.03.045
Wang C, Wang S, Li D, Wei D-Q, Zhao J, Wang J (2020a) Human intestinal defensin 5 inhibits SARS-CoV-2 invasion by cloaking ACE2. Gastroenterology 159 (3):1145–1147. e1144
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303
Wilson SS, Wiens ME, Smith JG (2013) Antiviral mechanisms of human defensins. J Mol Biol 425(24):4965–4980
World Health Organization (WHO): Tracking SARS-CoV-2 variants. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q (2020a) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367(6485):1444–1448
Yan Y, Tao H, He J, Huang S-Y (2020b) The HDOCK server for integrated protein–protein docking. Nat Protoc 15(5):1829–1852. https://doi.org/10.1038/s41596-020-0312-x
Yan Y, Wen Z, Wang X, Huang SY (2017a) Addressing recent docking challenges: a hybrid strategy to integrate template-based and free protein-protein docking. Proteins 85(3):497–512. https://doi.org/10.1002/prot.25234
Yan Y, Zhang D, Zhou P, Li B, Huang S-Y (2017b) HDOCK: a web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res 45(W1):W365–W373
Yuan M, Wu NC, Zhu X, Lee CD, So RTY, Lv H, Mok CKP, Wilson IA (2020) A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science (new York, NY) 368(6491):630–633. https://doi.org/10.1126/science.abb7269
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Singh, S., Banavath, H.N., Godara, P. et al. Identification of antiviral peptide inhibitors for receptor binding domain of SARS-CoV-2 omicron and its sub-variants: an in-silico approach. 3 Biotech 12, 198 (2022). https://doi.org/10.1007/s13205-022-03258-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-022-03258-4