
Abstract
Human death receptors (TNFR1, FAS, DR3, DR4, DR5, DR6 and TNFBR), primarily from tumor necrosis receptor super family, play an essential role in the process of the extrinsic pathway of apoptosis. We performed conserved domain, amino acid residues analysis in which cysteine residues were found to be highly conserved for all the family members. Sixteen (16) highly conserved residues were observed in TNFR1, DR3 and TNFBR; and in case of Fas, only seven (7) residues are highly conserved. From molecular phylogenetics, we found that DR5 and DR4, TNFR1 and DR3 and TNFR1 and DR3 had the same point of origin. Alternatively, Fas was found to be distant from the rest of the death receptors. A network map was developed to explain these proteins are not only interlinked among themselves, but also interlinked with several other proteins. We have also observed from this system that scores of all the nodes ranges from 0.996 to 0.999.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The families of death receptor members belong to the tumor necrosis factor (TNF) or nerve growth factor receptor super family. Death receptors are known to initiate the process of the extrinsic pathway of apoptosis, and ligand-bound death receptors triggered the extrinsic pathway (Thorburn et al. 2004; Fesik 2000; Krueger et al. 2001) This death receptors family contains a sequence of 2–5 cysteine-rich extracellular repeats. These receptors also include an intracellular death domain (DD). This DD is required for transduction of the apoptotic signal. TNFR1, Fas, DR3, DR4, DR5, DR6, TNFBR (tumor necrosis factor beta receptor) are among the well-known human death receptors (Schulze-Osthoff et al. 1998; Thorburn et al. 2004; Krueger et al. 2001). The death receptors identify their ligand, based on structural uniqueness, which forms DISC (Death Inducing Signaling Complex) (Harper et al. 2003). Usually, functionality or transmission of the death signal is by the result of binding of the specific ligand to the death receptor which is then followed by the attachment of adaptor protein molecules. This results in activation of pro-caspases to mediate various signaling pathways, depending on the chosen adaptor protein (Sandra et al. 2005). These receptors have other non-apoptotic functions as well, for example, inflammatory responses, cell proliferation, cell immune responses, receptor internalization (Los et al. 2001; Algeciras-Schimnich et al. 2002). FasL binds to CD95 receptor, while TNF alpha and lymphotoxin forms the ligand receptor complex with TNFR1. TRAIL2 (Apo2L) receptor 1 and 2 forms close association with both the death receptors—DR4 and DR5, whereas DR3 only show its specificity by binding to Apo3L (Hongxia et al. 2009).
Evolutionary history can be studied through molecular phylogenetics and it can be explored further by molecular approach through amino acid sequencing in human (Kumar and Hedges 1998). It is a well recognized method for conservation genetics. This method plays a remarkable role in understanding the applied evolution; because genetic patterns can lead to an evolutionary process (Latta 2008; Chakraborty et al. 2012). Conservation especially evolutionary conservation of a protein sequence is directly linked with the conserved regions of protein sequence particularly conserved amino acids which has structural and functional significance (Chakraborty et al. 2011; Ashkenazy et al. 2010). The presence of the conserved domain not only tells about the functional aspect of the respective protein, but also enables to get an idea of its evolution (Branden and Tooze 1999). It has been well acknowledged that cognate ligand binding and the intracellular N-terminal domain, extracellular C-terminal region are correlated with the conserved amino acid residues (Tanaka et al. 2006). However, very few works on conserved domains of the human death receptors have been reported on the structural aspect (Marchler-Bauer et al. 2005). Networking of member proteins in a family or related to biological pathway of a disease is very much important to understand the drug target discovery (Chakraborty et al. 2010). However, no data are available on the network between human death receptors family.
In this study, we provide information about the conserved domain, amino acid residues and also relate the sequence similarity with the evolutionary divergence of the different death receptors. We also performed molecular phylogenetics to understand the relationship between the family members of death receptors family. We also performed multiple sequences alignment (MSA) to understand sequence similarity and we developed a network to understand associations among the members by using computational biology.
Materials and methods
Data collection
We have collected human death receptors gene and protein sequence data as available in the public repository of the National Centre for Biotechnology Information (NCBI) database (Wheeler et al. 2007; Sayers et al. 2011). The protein sequences were collected with the corresponding accession number from the database and analyzed further. The protein sequences collected were in FASTA format for our use.
Multiple sequences alignment
The protein sequences were analyzed using the well-known multiple alignment tool ClustalW (Chenna et al. 2003) to observe the similarity between the sequences. The graphical output of the tool is visualized using JalView of ClustalW; we have also tried to study using another multiple sequence alignment tool, MUSCLE, to locate the conserved pattern across the sequences of MUSCLE (Edgar 2004). Using the multiple sequence alignment technique, we have observed the similarity in the sequences and their respective alignment scores have been elucidated. In this analysis, seven sequences have been used and TNFR1, Fas, DR3, DR4, DR5, DR6, TNFBR sequences has been represented as Seq1, Seq2, Seq3, Seq4, Seq5, Seq6, Seq7, respectively. We have used notation Seq (x:y) meaning alignment score between sequence x, and sequence y.
Phylogenetic tree and computational analysis
For extensive study of human receptors, we have used POWER (Phylogenetic Web Repeater), a tool based on the concept of ancestral relationship using the genetic distance (Lin et al. 2005). This tool performs multiple sequence analysis and tree building based on ClustalW, PHYLIP (PhyloDraw) (Choi et al. 2000), BLAST and PSI-BLAST (Altschul et al. 1997). A phylogenetic tree (phylogram) is developed to show the distances between protein sequences of human death receptors. We have also developed another phylogenetic tree, i.e., cladogram (ignoring branch length). This cladogram has been used for algorithm analysis based on Aldous as well as Bereg and Wang algorithms (Aldous 1996; Sandvik 2009).
Conservation pattern of structures and calculation of highly conserved amino acids in human death receptors’ family members
ConSurf Server (Ashkenazy et al. 2010; Glaser et al. 2003) enabled us to calculate the conservation pattern in the structure of human death receptors family (TNFR) members. The conservation scores which have been calculated by ConSurf Server not only discuss the extent of conservation, but also reveal the evolutionary rate. It represents the output in colored format where each conserved position in the chain is represented by different color. It performs the task of sequence to structure relationship by performing multiple sequence analysis, creating phylogenetic tree based on NJ method. It further guides about the rate of evolution of each amino acid residue in the target sequence using either Bayesian method or Maximum likelihood approach.
Protein–protein network design between the human death receptors
Using STRING (http://string-db.org/), a database of known and predicted protein interactions, we have developed a landscape networking between the human death receptor family members. This web-based database dedicated to protein–protein interactions includes direct (physical) and indirect (functional) associations among the members (Jensen et al. 2009).
Results
Collected data
Human death receptor proteins and their genes were compiled using the services provided by the NCBI data bank. Human death receptor genes, their protein IDs, locus, accession number, version, GI have been documented (Table 1).
Multiple sequence alignment (MSA)
Multiple sequence alignment was generated to analyze the similarities and differences among the death receptors. The output shows that the sequences share certain conserved regions. These regions were found to be starting from 75–115, 149–151, 190–200, 299–313, 437–451, 493–509, 522–528, 537–549 and 554–566. Certain positions like 181, 233 and 254 were highly conserved. Using multiple sequence alignments, scores have been generated (Fig. 1). The sequence alignment shows highest similarity score of 51 in both the sequences 4 and 5. These results not only indicate the sequence similarity between DR4 and DR5, but also show the excellent sequence match. Lowest similarity score of 9 was observed between the sequences 1 and 7 which illustrate the huge difference between the TNFR1 and TNFBR sequence.

MSA scores of protein sequences of different human death receptors. a MSA score between two sequences (the information such as Seq (x:y) meaning MSA score between sequence x, and sequence y). b Scatter distribution of MSA score, and c MSA score connected by smoothed line without marker
Phylogenetic tree and computational analysis
The constructed phylogenetic tree is shown in Fig. 2. Phylogenetic tree represents that DR5 and DR4 have same point of origin while on the other hand TNFR1 and DR3 share similar point of origin. The result also depicts that DR6 and TNFBR have same point of origin. The result shows three subgroups according to their common point of origin. Furthermore, we have depicted cladogram (Fig. 3a, b) (“without any distance”) from our phylogram (Fig. 2). From the phylogram, we developed cladogram (Fig. 3a). For the generation of the algorithm, we have depicted a binary tree figure (Fig. 3b) from the cladogram. Here, we assume that the figure is a binary tree and this tree is a level 4 binary tree. The leaf nodes containing DR6 and TNFBR are located at level 2; TNFR1, DR3, Fas at level 3; and DR4, DR5 at level 4, respectively.

Phylogenetic relationship of the different human death receptors. a Using POWER, Phylogenetic Web Repeater, the phylogenetic tree has been constructed

Development of phylogenetic tree. a Generated cladogram for tree algorithm analysis. b Generated binary tree equivalent to cladogram
Conservation pattern and calculation of highly conserved amino acids in human death receptors family
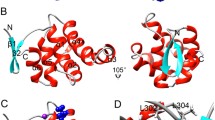
The structural data comprising the conserved amino acid residues in the human death receptors is represented in the Fig. 4. Here, we have shown conservation patterns in 3D structure and backbone structures with the help of highly conserved residues of death receptors. Figure 5 represents the graphical representation of highly conserved cysteine residues which are common among the death receptors. It describes the position of highly conserved cysteine residues of the amino acid which are common among the different receptors. In this study, the structural data of DR6 has not been predicted by ConSurf Server. Therefore, it could not be included in this paper. We have tried to represent the highly conserved amino acid residues present in each receptor in a separate table (Table 2). We have again recorded the number of highly conserved amino acid residues in death receptors which is represented in Fig. 6. It has been noted from the figure that the highest number of highly conserved residues was shared by TNFR1, DR3 and TNFBR which is 16. On the other hand, the lowest count of 7 was observed in case of Fas for highly conserved residues.

Conservation prototype and backbone structures analysis of the proteins of death receptors. a A common conservation prototype with highly conserved amino acids in 3D structure of the death receptors. Amino acid conservation scores have been categorized into nine levels and the color of residue indicates that conservation prototype of the death receptors. b Backbone structures of the of the death receptors where we have indicated highly conserved amino acids

List of amino acid residues which are highly conserved among the death receptors

Number of conserved amino acid residues among the death receptors
Protein–protein network design between the human death receptors
Protein–protein networking was generated between the human death receptors’ family members (Fig. 7). It clearly shows that these proteins are not only interlinked among themselves, but also interlinked with several other proteins. We have also observed from this network that scores of all the nodes ranges from 0.996 to 0.999. Therefore, each node of this network is strongly interconnected.

Protein–protein network between the proteins of death receptors. We have used STRING software (http://string-db.org/) for the generation of the network where we provided input as protein of death receptors. It shows a networking layer is not only related between them (protein cascades of the node), but also related to the several other proteins in other signaling pathways
Discussion
Apoptosis or programmed cell death is carried out along with several pathways which play a significant role in numerous physiological processes, particularly in development processes. A number of diseases are associated with either excess or inadequate apoptosis, such as AIDS, cancer, and autoimmunity (Krammer 2000; Vaux and Korsmeyer 1999). The two main apoptotic pathways were identified that activate caspases for programmed cell death (Thorburn 2004). One is “intrinsic pathway”, a stress-derived pathway, that involves mitochondrial proteins such as cytochrome c (Wang 2001; Huang and Strasser 2000; Cory and Adams 2002) and the other apoptotic pathway is the “extrinsic pathway” that is commenced by stimulus of ‘death receptors’ in the plasma membrane (Hengartner 2000; Sayers 2011). In the second pathway, ligand-bound death receptors, for example, TNF, Fas or TRAIL receptors initiate the process. This pathway was thought to be much easier and well comprehended (Ashkenazi and Dixit 1998). In this case, apoptosis can be started through the stimulation of death receptors which incorporate Fas, TNFRα, DR3, DR4, and DR5 by their respective ligands. Till date, seven known death receptors—Fas, TNFR, DR3, DR4, DR5, DR6, TNFBR—are known to have an intracellular globular protein interaction domain also named as death domain (DD). Ligand binding to the death receptors is perhaps in the form of pre-associated receptor complex (Siegel et al. 2000; Chinnaiyan et al. 1995). The complex activated death receptors hire an adaptor protein entitled Fas-Associated Death Domain (FADD) (Siegel et al. 2000). In this case, we have studied death receptors and their conserved domain, residues as well as evolutionary relations. However, there may be a correlation between conserved domains and Fas-Associated Death Domain (FADD) for death receptors. Chan et al. 2000 established evolutionary relationship between death receptors. Their phylogenetic analysis indicates that the member of the death receptor family represent an ancient divergence. Actually, death domains consisting of 80- 100-residue extended motifs can be seen in cytoplasmic proteins. These proteins belonging to the TNF-receptor super family are trans-membrane proteins and are known by some other names, this has been recorded in (Table 3). Sometime death domains provide as employing modules through their capacity to heterodimerize the DD of distinct proteins, including adaptor proteins such as Fas-associated protein with Death Domain (FADD), TNF Receptor-Associated protein with Death Domain (TRADD) and Receptor Interacting Protein (RIP) (Bridgham et al. 2003). It has been reported that death receptors are characterized by the presence of intracellular death domain (Ryan and Aksentijevich 2009) and this death domain consists of cysteine-rich residues. According to Itoh et al. 1991, members of the TNF-R1 family include 1–5 extracellular cysteine-rich domains. From our study, it is very clear that CYS76 could be considered as one of the highly conserved amino acid which is found to be common among TNFR1, TNFBR, DR3 and DR5, whereas CYS137, CYS129 and CYS139 are only present in TNFR1, DR3 and DR4. CYS76 is the only residue common among death receptors TNFR1, DR3 and DR5. Moreover, we have found that all those residues which are conserved in TNFR1 are also conserved in DR3. Fas seem to be the only death receptor which had none of the conserved residues common to the rest of the human death receptors. Our phylogenetic analysis as well as highly conserved amino acid analysis supports the view of cysteine-rich residues. We have noted that CYS129, CYS137 and CYS139 are shared by the TNFR1, DR3 and DR4. The result of phylogenetic tree as well as alignment scores represents that DR5 and DR4 form one subgroup, while TNFR1 and DR3 forms another subgroup and the third subgroup comprises DR6 and TNFBR. On the other hand, Fas is found to be distant with the rest of the caspase receptors. Phylogenetic analysis validates the point given by the ConSurf Server where the tree is apparently showing the right pathway of diversion as well as evolution because all the amino acid like CYS70, 73, 88, 76, 96, 98, 114, 117, 120, 129, 137, 139, 156 are shown common to both the TNRF1 and DR3. In order to study the conservation pattern in the structure of receptor proteins, we have used ConSurf server. This software enables to explore the 3D structure from the protein sequence data. This server uses the sequence data provided by PDB (Berman et al. 2000) file and further allows the user to go for stepwise calculation of evolutionary conserved residues from closely related homologous amino acid sequences using PSI-BLAST. The rate of evolution is calculated using either distance-based method or character-based method. It also determines the conservation score of a particular amino acid at a particular position. ConSurf server uses either the Maximum likelihood approach or the Empirical Bayesian method (Mayrose et al. 2004) to study the rate of evolution at each position. This tool is quite user-friendly and enables to visualize and analyze the receptor protein structure using its feature First Glance in JMOL.
Conclusions
Several fascinating queries about the conserved domains and evolutionary relationship between these receptor proteins need comprehensive understanding. It has been found that all the conserved domains indicate either structural or functional relevance in terms of evolutionary change. So, we performed an in silico study using sequence and structure analysis from the various tools of bioinformatics. Even though the cell death signaling pathways have been studied for the past few years, there is not much data available specifically on the human death receptors, their conserved domains and even with respect to their structures. We know about the pathways and also know a number of the proteins that may be involved in the reaction. But, we have to understand more about the evolutionary relationship as well as structural and functional relationship between these family members. To address this, in silico analysis was carried out to understand the conserved domain, residues, evolutionary relation and landscape networking of death receptors. This work is a preliminary effort to know the structural and functional relationship. In this analysis, we applied a pioneering and quick method to apprehend the structural, functional and phylogenetic association among the death receptors family. However, we have to go long way to understand the structural and functional relationship between the death receptors and further study is required in this area. Current study may provide great help to future researchers to progress on more findings between the structural and functional relationship of the death receptors.
Abbreviations
- TNFR1:
-
Tumor necrosis factor receptor 1
- DR:
-
Death receptor
- TNFBR:
-
Tumor necrosis factor beta receptor
- DD:
-
Death domain
References
Aldous D (1996) Probability distributions on cladograms. In Random discrete structures (Minneapolis, MN, 1993). IMA Vol Math Appl 76:1–18 Springer
Algeciras-Schimnich A, Barnhart BC, Peter ME (2002) Apoptosis-independent functions of killer caspases. Curr Opin Cell Biol 14:721–726
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281:1305–1308
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
Boldin MP, Mett IL, Wallach D (1995) A protein related to a proteasomal subunit binds to the intracellular domain of the p55 TNF receptor upstream to its ‘death domain. FEBS Lett 367:39–44
Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, Hopf C, Huhse B, Mangano R, Michon AM, Schirle M, Schlegl J, Schwab M, Stein MA, Bauer A, Casari G, Drewes G, Gavin AC, Jackson DB, Joberty G, Neubauer G, Rick J, Kuster B, Superti-Furga G (2004) A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol 6:97–105
Branden C, Tooze J (1999) Introduction to protein structure, 2nd edn. Garland Publication, New York
Bridgham JT, Wilder JA, Hollocher H, Johnson AL (2003) All in the family: evolutionary and functional relationships among death receptors. Cell Death Differ 10:19–25
Carpentier I, Coornaert B, Beyaert R (2008) Smurf2 is a TRAF2 binding protein that triggers TNF-R2 ubiquitination and TNF-R2-induced JNK activation. Biochem Biophys Res Commun 374:752–757
Chakraborty C, Roy SS, Hsu CH, Wen ZH, Lin CS (2010) Network building of proteins in a biochemical pathway: a computational biology related model for target discovery and drug-design. Curr Bioinform 5(4):290–295
Chakraborty C, Agoramoorthy G, Hsu MJ (2011) Exploring the evolutionary relationship of insulin receptor substrate family using computational biology. PLoS One 6(2):e16580
Chakraborty C, Roy SS, Hsu MJ, Agoramoorthy G (2012) Can computational biology improve the phylogenetic analysis of insulin? Comput Methods Programs 108:860–872
Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ (2000) A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288:2351–2354
Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L (1997) Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 7:821–830
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the clustal series of programs. Nucleic Acids Res 31:3497–3500
Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81:505–512
Choi JH, Jung HY, Kim HS, Cho HG (2000) PhyloDraw: a phylogenetic tree drawing system. Bioinformatics 16:1056–1058
Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2(9):647–656
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Fesik SW (2000) Insights into programmed cell death through structural biology. Cell 103(2):273–282
Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19:163–164
Harper N, Hughes M, MacFarlane M, Cohen GM (2003) Fas- associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem 278:25534–25541
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776
Hostager BS, Bishop GA (2002) Role of TNF receptor-associated factor 2 in the activation of IgM secretion by CD40 and CD120b. J Immunol 168:3318–3322
Hsu H, Shu HB, Pan MG, Goeddel DV (1996a) TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84:299–308
Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV (1996b) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4:387–396
Huang DS, Strasser A (2000) BH3-Only proteins-essential initiators of apoptotic cell death. Cell 103:839–842
Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW (1996) NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 384:638–641
Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O’Connell M, Kelley RF, Ashkenazi A, de Vos AM (1999) Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell 4:563–571
Imtiyaz HZ, Zhou X, Zhang H, Chen D, Hu T, Zhang J (2009) The death domain of FADD is essential for embryogenesis, lymphocyte development, and proliferation. J Biol Chem 284(15):9917–9926
Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66:233–243
Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C (2009) STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37:D412–D416
Jung YS, Kim KS, Kim KD, Lim JS, Kim JW, Kim E (2001) Apoptosis-linked gene 2 binds to the death domain of Fas and dissociates from Fas during Fas-mediated apoptosis in Jurkat cells. Biochem Biophys Res Commun 288:420–426
Kitson J, Raven T, Jiang YP, Goeddel DV, Giles KM, Pun KT, Grinham CJ, Brown R, Farrow SN (1996) A death-domain-containing receptor that mediates apoptosis. Nature 384:372–375
Krammer PH (2000) CD95's deadly mission in the immune system. Nature 407:789–795
Krueger A, Baumann S, Krammer PH, Kirchhoff S (2001) FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol (24):8247–54
Kumar S, Hedges B (1998) A molecular timescale for vertebrate evolution. Nature 392:917–920
Latta RG (2008) Conservation genetics as applied evolution: from genetic pattern to evolutionary process. Evol Appl 1:84–94
Lin CY, Lin FK, Lin CH, Lai LW, Hsu HJ, Chen SH, Hsiung CA (2005) Nucleic Acids Res 33:553–556
Los M, Stroh C, Janicke RU, Engels IH, Schulze-Osthoff K (2001) Caspases: more than just killers? Trends Immunol 22:31–34
Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Song JS, Simonyan V, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH (2005) CDD: a conserved domain database for protein classification. Nucleic Acids Res 33:D192–D196
Mayrose I, Graur D, Ben-Tal N, Pupko T (2004) Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol Biol Evol 21:1781
Miyazaki T, Reed JC (2001) A GTP-binding adapter protein couples TRAIL receptors to apoptosis-inducing proteins. Nat Immunol 2:493–500
Munroe ME, Bishop GA (2004) Role of tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) in distinct and overlapping CD40 and TNF receptor 2/CD120b-mediated B lymphocyte activation. J Biol Chem 279:53222–53231
Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457:981–989
Pan G, Bauer JH, Haridas V, Wang S, Liu D, Yu G, Vincenz C, Aggarwal BB, Ni J, Dixit VM (1998) Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett 431(3):351–356
Pype S, Declercq W, Ibrahimi A, Michiels C, Van Rietschoten JG, Dewulf N, de Boer M, Vandenabeele P, Huylebroeck D, Remacle JE (2000) TTRAP, a novel protein that associates with CD40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-kappa B activation. J Biol Chem 275(24):18586–18593
Ryan JG, Aksentijevich I (2009) Tumor necrosis factor receptor-associated periodic syndrome: toward a molecular understanding of the systemic autoinflammatory diseases. Arthritis Rheum 60(1):8–11
Sandra F, Degli Esposti M, Ndebele K, Gona P, Knight D, Rosenquist M, Khosravi-Far R (2005) Tumor necrosis factor-related apoptosis-inducing ligand alters mitochondrial membrane lipids. Cancer Res 65(18):8286–8297
Sandvik H (2009) Anthropocentricisms in cladograms. Biol Philos 24(4):425–440
Sayers TJ (2011) Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother 60(8):1173–1180
Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J (2011) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 39:D38–D51
Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, Holler N, Tschopp J (1997) TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 7:831–836
Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME (1998) Apoptosis signaling by death receptors. Eur J Biochem 254:439–459
Shu HB, Halpin DR, Goeddel DV (1997) Casper is a FADD- and caspase-related inducer of apoptosis. Immunity 6:751–763
Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, Tsien RY, Lenardo MJ (2000) Science 288(5475):2354–2357
Tanaka N, Tani Y, Tada T, Lee YF, Kanaori K, Kunugi S (2006) The roles of conserved amino acids on substrate binding and conformational integrity of ClpB N-terminal domain. Biochemistry 45:8556–8561
Thomas LR, Stillman DJ, Thorburn A (2002) Regulation of Fas-associated death domain interactions by the death effector domain identified by a modified reverse two-hybrid screen. J Biol Chem 277:34343–34348
Thorburn A (2004) Death receptor-induced cell killing. Cell Signal 16(2):139–144
Vaux DL, Korsmeyer SJ (1999) Cell death in development. Cell 96:245–254
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15(22):2922–2933
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Geer LY, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Ostell J, Miller V, Pruitt KD, Schuler GD, Sequeira E, Sherry, ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L and Yaschenko E (2007) Nucleic Acids Res 35:D5–12
Acknowledgments
The authors take this opportunity to thank the management of Vellore Institute of Technology and Galgotias University for providing the facilities and encouragement to carry out this work. The authors have declared that no conflict of interest exists.
Author information
Authors and Affiliations
Corresponding authors
Additional information
J. Tomar and C. Chakraborty contributed equally.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Tomar, J., Chakraborty, C., Doss, C.G.P. et al. Understanding the conservation patterns and molecular phylogenetics of human death receptors family through computational biology. 3 Biotech 4, 177–187 (2014). https://doi.org/10.1007/s13205-013-0141-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13205-013-0141-5