
Abstract
A soil bacterium, Bacillus subtilis, isolated from the rhizosphere of Chilli, showed high antagonistic activity against Colletotrichum gloeosporioides OGC1. A clear inhibition zone of 0.5–1 cm was observed in dual plate assay. Microscopic observations showed a clear hyphal lysis and degradation of fungal cell wall. In dual liquid cultures, the B. subtilis strain inhibited the C. gloeosporioides up to 100 % in terms of dry weight. This strain also produced a clear halo region on chitin agar medium plates containing 0.5 % colloidal chitin, indicating that it excretes chitinase. The strain also produced other mycolytic enzymes—glucanase and cellulase, demonstrated by a clear zone of hydrolysis of yeast cell wall glucan (YCW 0.1 % v/v) and carboxymethylcellulose (CMC 0.1 % v/v). In liquid cultures, the strain showed appreciable levels of chitinase, glucanase and cellulase activities and hydrolytic activity with C. gloeosporioides OGC1 mycelia as the substrate. The role of the B. subtilis strain in suppressing the fungal growth in vitro was studied in comparison with a UV mutant of that strain, which lacked both antagonistic and hydrolytic activity. The mycolytic enzyme mediated antagonism of B. subtilis was further demonstrated by heat inactivation (70–100 °C), treatment with trypsin and TCA of the crude enzyme extract which lacked antifungal property also. Treatment of the chilli seeds with Bacillus sp. culture showed 100 % germination index similar to the untreated seeds. The treatment of the seed with co-inoculation of the pathogen with Bacillus sp. culture showed 65 % reduction in disease incidence by the treatment as compared to the seed treated with pathogen alone (77.5 %).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rhizosphere bacteria are excellent agents to control soil-borne plant pathogens. Bacterial species such as Bacillus, Pseudomonas, Serratia and Arthrobacter have been proved in controlling the fungal diseases (Joseph et al. 2007) Earlier reports showed that microorganisms capable of lysing chitin, which is a major constituent of the fungal cell wall, play an important role in biological control of fungal pathogens (Yu et al. 2002; Zhang and Fernando 2004; Abdullah et al. 2008). Fungi such as Trichoderma and bacteria such as Bacillus, Serratia and Alteromonas were reported to have chitinolytic activity (Mabuchi et al. 2000; Someya et al. 2001; Wen et al. 2002; Huang et al. 2005; Viterbo et al. 2001). Non-pathogenic soil Bacillus species offer several advantages over other organisms as they form endospores and hence can tolerate extreme pH, temperature and osmotic conditions. Bacillus species were found to colonize the root surface, increase the plant growth and cause the lysis of fungal mycelia (Turner and Backman 1991; Podile and Prakash 1996; Takayanagi et al. 1991).
Chilli, Capsicum annum L. cultivation has existed for several 100 years as a sustainable form of agriculture in India and in many other countries. It is an annual herbaceous vegetable and spice grown in both tropical and subtropical regions. The crop is grown in almost all states of India, such as Andhra Pradesh, Maharashtra, Karnataka, Gujarat, Tamil Nadu and Orissa. India accounts for 25 % of the world’s total production of chilli (http://agropedia.iitk.ac.in/content/area-and-production-chilli-world-2008-09).
The crop is a significant source of income making India the world’s single largest producer and exporter to the USA, Canada, UK, Saudi Arabia, Singapore, Malaysia, Germany and many more countries across the world. The sustainability of chilli-based agriculture is threatened by a number of factors. Main biotic stresses such as bacterial wilt, anthracnose, viruses and several insect pests have been reported to impair the crop productivity (Isaac 1992).
The genus Colletotrichum and its teleomorph Glomerella contain an extremely diverse number of fungi including both plant pathogens and saprophytes. Plant pathogenic species are important worldwide, causing pre- and post-harvest losses of crops (Bosland and Votava 2003). These fungi cause diseases commonly known as anthracnose of grasses, legumes, vegetables, fruits and ornamentals. The disease can occur on leaves, stems and fruit of host plants (Sutton 1992). Anthracnose disease is a major problem in India and one of the more significant economic constraints to chilli production worldwide, especially in tropical and subtropical regions (Than et al. 2008).
Economic losses caused by the disease are mainly attributed to lower fruit quality and marketability. Although infected fruits are not toxic to humans or animals, severely affected fruits showing blemishes are generally considered unfit for human consumption. This is because the anthracnose causes an unpleasant colour and taste in chilli products. Management of the disease under the prevailing farming systems in India has become a recurrent problem to chilli growers (Thind and Jhooty 1985).
Effective control of anthracnose disease involves the use of one of, or a combination of, the following: resistant cultivars, cultural control and chemical control. The intensive use of fungicides has resulted in the accumulation of toxic compounds potentially hazardous to humans and the environment, and also in the build-up of resistance of the pathogens. In view of this, investigation and the application of biological control agents (BCAs) seems to be one of the promising approaches (Cook 1985). Biocontrol involves the use of naturally occurring non-pathogenic microorganisms that are able to reduce the activity of plant pathogens and thereby suppress diseases. Hence, controlling this pathogen using biocontrol agents will help in enhancing the yield of the crop.
B. subtilis JN032305, isolated from chilli rhizosphere produced appreciable levels of three mycolytic enzymes—chitinase, glucanase and cellulase and showed broad spectrum antagonism against potent bacterial and fungal phytopathogens (Srividya et al. 2012). The production of all three enzymes of this strain have been optimised using Plackett-Burman approach (Ashwini and Srividya 2012a, b). The objective of the present study was to ascertain the concerted role of these three mycolytic enzymes (chitinase, β1,3-glucanase and cellulase) mediated antagonism of B. subtilis against C. gloeosporioides.
Materials and methods
Isolation of rhizospheric Bacillus sp.
Chilli rhizosphere soils were collected from in and around Bangalore and Bacillus sp. were isolated by soil dilution method. The dilutions were heat treated at 80 °C for 20 min to ensure that only heat resistant strains remained. The different isolates obtained were screened for chitinase production based on the halo produced on plates with minimal salts medium amended with chitin (1 % chitin) (Cook 1985). The Bacillus sp., thus, obtained were maintained on Nutrient agar (NA) amended with chitin (1 %).
Morphological and phenotypic characterization of Bacillus sp.
This strain was characterised morphologically and biochemically by following Bergey’s Manual of Systematic Bacteriology (Sneath 1986) and was found to be a Bacillus sp. It was grown and maintained on NA at 30 °C. Polymerase chain reaction (PCR) was performed to amplify a partial 16S rRNA gene of the bacteria, and partial 16S rDNA sequencing was used to assist in the identification of the isolate. Isolation of genomic DNA, PCR amplification and sequencing of PCR product for analysis of 16S rRNA were conducted according to Marchesi et al. (1998). A similarity search for the nucleotide sequence of 16S rRNA of the test isolate was carried out using a blast search at NCBI (Altschul et al. 1990).
Preparation of colloidal chitin
Colloidal Chitin was prepared from crab shell chitin powder (Roberts and Selitrennikoff 1988). A few modifications were made as described: 10 g of chitin powder was added slowly into 100 mL of concentrated HCl under vigorous stirring for 2 h. The mixture was added to 1,000 mL of ice-cold 95 % ethanol with rapid stirring and kept overnight at 25 °C and then stored at −20 °C until use. When needed, the filtrate was collected and washed with 0.1 M phosphate buffer (pH 7) until the colloidal chitin became neutral (pH 7) and used for further applications.
Phytopathogens and chilli seeds
Colletotrichum gloeosporioides (OGC1) and chilli seeds (Arka Shweta variety) were obtained as a kind gift from IIHR, Hessarghatta, Bangalore.
Dual plate assay
The fungal growth inhibition capacity of Bacillus sp. strains was determined (Huang and Hoes 1976). A few modifications were made to suit the need. One 5-mm disc of a pure culture of the pathogen was placed at the centre of a Petri dish containing PDA. The Bacillus sp. was inoculated at two opposing corners. Plates were incubated for 72 h, at 28 °C, and growth diameter of the pathogen was measured and compared to control growth, where the bacterial suspension was replaced by sterile distilled water. Each experiment using a single pathogen isolate was run in triplicate. Results were expressed as the means of the percentage of inhibition of growth of the corresponding pathogen isolate in the presence of any of the strain of Bacillus sp.
Percent inhibition was calculated using the following formula:

Detection of extracellular hydrolytic activity
Plates with minimum salts medium (MSM) containing (1 % w/v) carboxy methyl cellulose (CMC) was prepared. The Bacillus sp. was spot inoculated in the centre of the plate. After an appropriate incubation period at 30 °C for 48 h, the agar medium was flooded with an aqueous solution of Congo red for 15 min. The Congo red solution was then poured off and plates containing CMC were visualised for zones of hydrolysis (Shanmugaiah et al. 2008; Moataza 2006; Teather and Wood 1982) detecting β-1,4 cellulase. Yeast Glucan containing plates (Chen et al. 1995) was used to detect β-1,3 cellulase activity. MSM with (1 % v/v) yeast cell glucan was prepared and spot inoculated with the isolate. Development of a clear zone surrounding the colony indicated enzyme production.
Assay of hydrolytic enzymes
Chitinase enzyme assay
Chitinase activity was measured with colloidal chitin as a substrate. The culture broth was centrifuged and enzyme solution of 1 ml was added to 1.0 ml of substrate solution, which was made by suspending 1 % of colloidal chitin in Phosphate buffer (pH 7.0). The mixture was incubated at 50 °C for 30 min. 1 ml of DNS was added and incubated at 100 °C in boiling water bath. The amount of reducing sugar produced in the supernatant was determined by dinitrosalicylic acid method (DNS) (Miller 1959). One unit of chitinase activity was defined as the amount of enzyme that produced 1 μmol of reducing sugars per min (Annamalai et al. 2008).
Cellulase β-1,3 assay
The specific activity of β-1,3-cellulase was determined by measuring the amount of reducing sugars liberated using dinitrosalicylic acid solution (DNS) (Annamalai et al. 2008). The culture broth was centrifuged and enzyme solution of 1 ml was added to 1.0 ml of substrate solution which contained 1 ml of yeast cell wall extract (YCW 1 % v/v). The mixture was incubated in a water bath at 50 °C for 30 min and the reaction was terminated by adding 1 ml of DNS solution and incubated in boiling water bath for 10–15 min till the development of the colour of the end product. Reducing sugar concentration was determined by optical density at 540 nm (Gadelhak et al. 2005).
Cellulase β-1,4 assay
The specific activity of β-1,4-glucanase was determined by measuring the amount of reducing sugars liberated using dinitrosalicylic acid solution (DNS) (Annamalai et al. 2008). The culture broth was centrifuged and enzyme solution of 1 ml was added to 1.0 ml of substrate solution which contained 1 ml of carboxy methyl cellulose solution (CMC 1 % v/v). The mixture was incubated in a water bath at 50 °C for 30 min and the reaction was terminated by adding 1 ml of DNS solution and incubated in boiling water bath for 10–15 min till the development of the colour of the end product. Reducing sugar concentration was determined by optical density at 540 nm (Moataza 2006).
Induction with fungal mycelium
The Bacillus sp. was grown on Nutrient broth supplemented with dead fungal mycelium (Colletotrichum gloeosporioides OGC1) as inducer for enzymes production at a concentration of 1.0 % and dispensed in Erlenmeyer flasks (250 ml), each flask containing 50 ml of medium. The flasks were autoclaved and inoculated with 1.0 ml of a pre-cultured Bacillus sp. culture. The culture was incubated in a shaker (120 rpm.), at 28 ± 2 °C. Aliquots from the flask were analyzed daily for chitinase and cellulases (β-1,3 and β-1,4) for a period of 5 days (Moataza 2006).
Hydrolytic assay
Preparation of hyphal wall
The pathogenic fungal culture (C. gloeosporioides) was inoculated into 50 ml of PDB broth and incubated at 30 °C for 5 days under shaking conditions. After incubation, the mycelia were collected by filtration. The mycelia were thoroughly washed with autoclaved distilled water and homogenised on ice, with a homogenizer for 5 min. The mycelia suspension was centrifuged at 10,000 rpm for 20 min at 4 °C (Remi: C 24). The pellet was resuspended in sodium phosphate buffer (0.1 M, pH 7.0). This preparation was used as substrate for the hydrolytic assay (Moataza 2006).
Hydrolytic activity of B. subtilis (wild type and mutants) culture filtrate
For assessing the hydrolytic activity, the reaction mixture (1 ml) containing 1 mg/ml of fungal mycelia with 1.0 ml of crude enzyme (from B. subtilis grown on NB + CMC) was incubated at 30 °C for 24 h. The released total reducing sugars (Miller 1959) in control and treated fungal cell wall was estimated using the DNS method.
Dual culture method
The differences in dry weights between the fungal cultures grown with B. subtilis strain or the control culture grown without any bacterium were recorded according to Broekaert et al. 1990. For this, 48 h grown dual cultures were passed through the pre-weighed Whatman No 1 filter paper. It was dried for 24 h at 70 °C and weights were measured (Saleem and Kandasamy 2002).
Microscopy
The fungal culture grown on NA agar plate without any bacterial culture served as control. The damage caused by the bacterium to the fungal mycelium by dual plate assay was studied microscopically. The mycelium along with the agar disc present in the inhibition zone and control mycelium was taken, stained with lactophenol cotton blue and observed under a Nikon Trinocular microscope (Saleem and Kandasamy 2002).
UV mutagenesis
To characterise the antagonistic mechanism by this bacterium, a mutant of this bacterium was developed, which lost its antagonistic activity. UV mutagenesis of B. subtilis was carried out following the procedure of Miller (1992). During UV mutagenesis, the log phase culture was exposed to short wavelength UV light (280 nm, Philips TUV 30 W, G3018, Holland) from a distance of 30 cm for various time intervals (for 0.1 % UV survivors). From the serial dilutions of the mutagenized culture, 0.1 ml was plated on NA plates for isolated mutant colonies. The isolated colonies were further screened for the loss of antifungal activity using dual plate assay. The mutants were also screened for mycolytic enzyme activity and hydrolytic activity with the pathogen mycelium.
Sensitivity of the culture supernatant of wild type B. subtilis to proteolytic enzymes, TCA and heat
The crude supernatant (1 mL) was subjected to treatments for 1 h at 37 °C (for enzymes) or room temperature (for TCA). The proteolytic enzymes (Sigma) were used at a final concentration of 1 mg ml−1 in 10 mmol−1 potassium phosphate buffer, pH 7.0. The crude supernatant in buffer without enzymes as well as the enzyme solutions was exposed to the same conditions. For the heat treatment, the preparations were incubated at 70, 80 and 90 °C and autoclaving for 20 min. Antifungal activities were checked before and after all treatments on a test plate made with C. gloeosporioides as mentioned earlier (Tendulkar et al. 2007).
Seed testing
Germination efficiency and antagonism of the Bacillus sp. against C. gloeosporioides was checked on chilli seeds in vitro. The water agar plates were seeded with the following: Set 1-Seeds control-plain seeds coated with carboxy methyl cellulose (CMC); Set 2- Seeds coated with CMC and C. gloeosporioides spores; Set 3- Seeds coated with CMC and Bacillus sp. culture and Set 4- Seeds coated with CMC and both C. gloeosporioides spores and Bacillus sp. culture. Chilli seeds were surface sterilised successively with sterile distilled water and 0.1 % HgCl2. To remove the residual HgCl2, the seeds were washed with sterile distilled water. The isolate was inoculated into NB medium and incubated for 24 h at 30 °C (Moataza 2006). C. gloeosporioides was inoculated onto PDA plates and incubated at 28 °C for 3–4 days. The above three sets of treated seeds were seeded onto 1 % water agar plates. Plain CMC coated seeds on water agar were used as control. The four sets were monitored regularly for germination and growth. After 1 week, the sets were observed for germination and biocontrol against C. gloeosporioides coated seeds by the isolate.
Reproducibility
All the experiments were done in triplicates and the values represented statistically are in ANOVA form.
Results and discussion
15 chilli rhizosphere soils samples were collected from in and around Bangalore, heat treated and plated on chitin amended plates. Out of 15 samples plated, 9 chitinolytic colonies were isolated based on the clearance zones that they formed.
Dual plate assay
These chitinolytic bacteria were subjected to dual plate assay with nine different chilli fungal pathogens, one particular isolate designated as isolate two showed inhibition of all the nine pathogens and this was chosen for further work. The selected isolate showed broad spectrum antagonism against Alternaria (3) spp. (55 %), C. gloeosporioides (57 %), Phytophthora capsici (55 %), Rhizoctonia solani (42 %), Fusarium solani (42 %), Fusarium oxysporum (40 %) and Verticillium sp. (36 %), the range of percentage inhibition varied from 40 to 62 (Table 1). The isolate was checked for other hydrolytic enzyme production and was found to produce both Cellulase β-1,3 and β-1,4 on the basis of clearance zones produced on yeast glucan plates as well as CMC plates, respectively (Fig. 1).

Plates showing zone of hydrolysis for β1,4-glucanase; chitinase and β 1,3 glucanase by WT B. subtilis
Strain identification
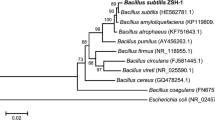
This strain was characterised morphologically and biochemically by following Bergey’s Manual of Systematic Bacteriology and was found to be a Bacillus sp. It was grown and maintained on NA at 30 °C. The isolate upon Gram’s staining was identified as a gram positive, spore forming rod. The isolate was motile and answered positive for catalase, nitrate reduction, Voges Proskauer, starch hydrolysis and growth on 6.5 % NaCl medium and negative for parasporal crystal formation and citrate utilisation, hence, was tentatively identified as Bacillus sp. Further, 16SrDNA sequencing identified the isolate to be B. subtilis and the Gene-Bank accession no. for the nucleotide sequence is JN032305 (Fig. 2) (Ashwini et al. 2012).

Phylogenetic tree constructed with the 16S rDNA sequences
UV mutagenesis
To characterise the antagonistic mechanism by this bacterium, a mutant of this bacterium was developed, which lost its antagonistic activity. Out of 61 putative mutant colonies tested, six showed no antagonistic property against C. gloeosporioides (Fig. 3). These mutant isolates also did not grow over the mycelial mat and were named as M17, M22, M23, M27, M28 and M30.

Bacillus strain mutants M17, M22, M23, M27, M28 and M30 not showing inhibition to Colletotrichum on PDA
Microscopy
The fungal mycelium grown with Bacillus BC2 culture showed damage, swelling and distortions as compared to the control (Fig. 4a, b). The mycelium which grew with Bacillus BCUVM cultures and the control mycelium which was not grown with any bacterial culture did not show these abnormal features (Fig. 4c). This clearly indicates the mycolytic activity of the Bacillus BC2 culture. A similar observation has been made in the antagonism of Arthrobacter sp. to Fusarium sp. (Barrows-Broaddus and Kerr 1981). Similarly, Podile and Prakash (1996) reported the lysis and dissolution of fungal mycelium of Aspergillus niger by B. subtilis AF1 strain.

Light microscopic observations of mycelium inhibited by BC2 strain (1,000×). Mycelium of Colletotrichuma grown on PDA (control), b present in the inhibition zone, when grown with Bacillus strain BC2 on PDA, c grown with mutant strains on PDA medium
Mycolytic enzyme activities
The induction profile of the Bacillus sp. was checked with autoclaved C. gloeosporioides mycelium used as the carbon source in the medium. Data represented in (Fig. 5) showed that the lysis of dead mycelia of C. gloeosporioides was very efficient by the Bacillus sp. Appreciable levels of all the three enzymes were observed in the presence of the autoclaved mycelia—chitinase peaked on day 1 (2.84 U/mL) and gradually decreased (2 U/mL by day 2 and 1.4 U/mL by day 3); β-1,3 cellulase was detected from day 1 (2.8 U/mL) which peaked on day 2 (3.2 U/mL) followed by a gradual decrease (2.7 U/mL by day 3 and 1 U/mL by day 4) and β-1,4 cellulase was also detected from day 1 (6 U/mL) and increased to a maximum of 13.21 U/mL by day 3 followed by a gradual decrease on day 4 (8 U/mL) suggesting the possible role of these enzymes in antibiosis of the mycelia. Moataza 2006 also reported varied levels and types of mycolytic enzymes by different Pseudomonas strains with different pathogens such as P. capsici and R. solani. Further, the mutants were studied for their mycolytic enzyme activities under shake flask conditions. All the mutants showed significant loss of all three mycolytic enzyme activities (Fig. 5). The mutants also exhibited low levels of hydrolytic activity with C. gloeosporioides mycelia as compared to the wild type strain indicating clearly the mycolytic enzyme mediated antagonism of this strain (Fig. 6).

Comparison of mycolytic enzyme activities of the mutants and the wild strain BC2

Comparison of hydrolytic activities of the mutants and the wild strain BC2. Activities of all three enzymes on day 1 by WT B. subtilis is represented
Dual culture method
To test the antifungal activity of the Bacillus strain BC2, dual liquid culture method was employed. The differences in dry weights between the fungal cultures grown with BC2 strain or the mutant strains or the control culture grown without any bacterium were recorded according to Broekaert et al. (1990). There was almost 100 % reduction in dry weight of the culture grown with BC2 strain when compared to the control. There was very little reduction in dry weight of the culture when grown with the mutant strains. This clearly shows that the reduction in dry weight of the fungus when grown with the BC2 strain is due to the antifungal activity of this bacterium. The mutant strains which had lost the antifungal activity could not reduce the dry weight of the fungus.
Sensitivity of the culture supernatant of B. subtilis to proteolytic enzymes, TCA and heat
The sensitivity of the WT crude culture filtrate of B. subtilis BC2 was tested with TCA, heat and proteolytic enzymes. The results revealed that the activity was not preserved. Further, when such treated extracts were subjected to antagonism assay, the extract had lost its antagonistic property which supported the mycolytic enzyme mediated antagonism of the fungal pathogen.
Seed bacterization
Treatment of the chilli seeds (Arka variety, obtained as a kind gift from IIHR, Bangalore) with Bacillus sp. culture showed 100 % germination index similar to the untreated seeds (Table 2). The treatment of the seed with co-inoculation of the pathogen with Bacillus sp. culture showed 65 % reduction in disease incidence by the treatment as compared to the seed treated with pathogen alone (77.5 %). Kamil et al. (2007) reported that the seed coat treatment of sunflower seeds with B. licheniformis induced high reduction in percentage of infection of R. solani damping off (from 60 to 25 %) as compared with the pathogen alone. Our observations also comply with these reports.
Statistical analysis
The analysis of variance (ANOVA) has been performed for all three mycolytic enzymes and hydrolytic activity by the wild type and mutants of B. subtilis. P value was found to be very low at both P = 0.05, which indicated that there is significant difference in mycolytic enzyme and hydrolytic activity between the strains (Table 3).
In a similar study, Saleem and Kandasamy (2002) showed the role of the Bacillus strain BC121 in suppressing the fungal growth in vitro when studied in comparison with a mutant of that strain, which lacks both antagonistic activity and chitinolytic activity. Another study by Balasubramanian et al. (2010) reported that on testing the biocontrol efficacy of the mutants and wild strain against phytopathogens such as Fusarium oxysporum, Bipolaris oryzae, Rhizoctonia solani and Alternaria sp. by dual culture assay on PDA medium, the UV H11 mutant and adapted mutant showed increased biocontrol activity when compared to wild strain. Balasubramanian et al. (2010) further reported that the antagonism of these two mutants with F. oxysporum, R. solani, B. oryzae and Alternaria sp. were varied and could be related with lytic enzyme production with fast growing ability. However, Lorito et al. (1993) reported chitinolytic enzymes contributing to the ability of Trichoderma sp to act as biocontrol agents. Graeme Cook and Faull (1991) reported that high antibiotic production by two T. harzianum mutant strains, BC10 and BC63, increased inhibition of hyphal growth of R. solani and P. ultimum; while Papavizas et al. (1982) have shown UV-induced benomyl resistant mutant to suppress the saprophytic activity of R. solani more effectively than the wild strain (Papavizas et al. 1982).
Conclusion
The selection of effective antagonistic organisms is the first and foremost step in biological control. On the basis of these studies, it is concluded that the Bacillus BC2 isolate is showing antagonistic property probably through the enzyme mediated lytic mechanism, which has been proved to be an effective mechanism in controlling the fungal pathogens (Chet et al. 1990). The in vitro seed bacterization studies also have revealed the success of biocontrol of the pathogen. These observations and further studies will help in developing the Bacillus BC2 isolates as a potential biological control agent against C. gloeosporioides.
References
Abdullah TM, Ali YN, Suleman P (2008) Biological control of Sclerotinia sclerotiorum (Lib.) de Bary with Trichoderma harzianum and Bacillus amyloliquefaciens. Crop Prot 27:1354–1359
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Annamalai N, Giji S, Arumugam M, Balasubramanian T (2008) Purification and characterization of chitinase from Micrococcus sp.AG84 isolated from marine environment. Afr J Microbiol Res 4(24):2822–2827
Ashwini N, Srividya S (2012a) Optimization of chitinase produced by a biocontrol strain of B. subtilis using Plackett-Burman design. Eur J Exp Biol 2(4):861–865
Ashwini N, Srividya S (2012b) Optimization of fungal cell wall degrading glucanases produced by a biocontrol strain of B. subtilis using Plackett-Burman design. In: Proceeding Intl Conf Biol Active Mol, Excel India Publisher New Delhi, pp 430–433
Ashwini N, Samantha S, Deepak B, Srividya S (2012) Enhancement of mycolytic activity of an antagonistic Bacillus subtilis through ethyl methane sulfonate (EMS) mutagenesis. Turk J Biol. doi:10.3906/biy-1209-20
Balasubramanian N, Thamil Priya V, Gomathinayagam S, Shanmugaiah V, Jashnie J, Lalithakumari D (2010) Effect of chitin adapted and ultra violet induced mutant of trichoderma harzianum enhancing biocontrol and chitinase activity. Aust J Basic Appl Sci 4(10):4701–4709
Barrows-Broaddus J, Kerr TJ (1981) Inhibition of Fusarium moniliforme var.subglutinans the causal agent of pine pitch canker, by the soil bacterium Arthrobacter sp. Can J Microbiol 27:20–27
Bosland PW, Votava EJ (2003) Peppers: vegetable and spice capsicums. CAB International, Oxford, p 233
Broekaert WF, Franky RG, Terras Bruno PA, Cammue J (1990) An automated quantitative assay for fungal growth inhibition. FEMS Microbiol Lett 69:55–60
Chen MH, Shen Bobin S, Kahn PC, Lipke PN (1995) Structure of Saccharomyces cerevisiae a-agglutinin. J Biol Chem 270:26168–26177
Chet I, Ordentlich A, Shapira Oppenheim A (1990) Mechanism of biocontrol of soil borne plant pathogens by rhizobacteria. Plant Soil 129:85–92
Cook RJ (1985) Biological control of plant pathogens: theory to application. Phytopathology 75:25–29
Gadelhak G, Khaled A, El-Tarabily KA, Fatma K, Al-Kaabi FK (2005) Insect control using chitinolytic soil actinomycetes as biocontrol agents. Int J Agri Biol 7(4):627–633
Graeme Cook KA, Faull JL (1991) Effect of ultraviolet induced mutants of T. harzianum with altered antibiotic production of selected pathogens in vitro. Can J Microbiol 37:659–664
Huang HC, Hoes JA (1976) Penetration and infection of Sclerotinia sclerotiorum by Coniothyrium minitans. Can J Bot 54:406–410
Huang CJ, Wang TK, Chung SC, Chen CY (2005) Identification of an antifungal chitinase from a potential biocontrol agent, Bacillus cereus. J Biochem Mol Biol Sci 38:82–88
Isaac S (1992) Fungal plant interaction. Chapman and Hall Press, London, p 115
Joseph B, Patra RR, Lawrence R (2007) Characterization of plant growth promoting Rhizobacteria associated with chickpea (Cicer arietinum L). Intern J Plant Prod 1(Suppl 2):141–152
Kamil Z, Rizk M, Saleh M, Moustafa S (2007) Isolation and Identification of Rhizosphere Soil Chitinolytic bacteria and their Potential in Antifungal Biocontrol. Global J Mol Sci 2:57–66
Lorito M, Harman GE, Hayes CK, Broadway RM, Tronsmo A, Woo SL, Di Pietro A (1993) Chitinolytic enzymes produced by Trichoderma harzianum: Antifungal activity of purified endochinitinase and chitobiosidase. Phytopathology 83:302–307
Mabuchi N, Hashizume I, Araki Y (2000) Characterization of chitinases exerted by Bacillus cereus CH. Can J Microbiol 46:370–375
Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, Hiom SJ, Wade WJ (1998) Design and evaluation of useful bacterium-specific pcr primers that amplify genes coding for bacterial 16 s rrna. Appl Environ Microbiol 64(2):795–799
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Miller H (1992) A short course in bacterial genetics. A laboratory manual and handbook for E.coli and related bacteria. Cold Spring Harbor Laboratory Press, New York
Moataza MS (2006) Destruction of Rhizoctonia solani and Phytophthora capsici causing tomato root-rot by Pseudomonas fluorescences lytic enzymes. Res J Agri Biol Sci 2(6):274–281
Papavizas GC, Lewis JA, Moity Abd- Ei (1982) Evaluation of new biotypes of Trichoderma harzianum for tolerance of Benomyl and enhanced biocontrol capabilities. Phytopathology 72(1):127–132
Podile AR, Prakash AP (1996) Lysis and biological control of A. niger by Bacillus subtilis AF1. Can J Microbiol 42:533–538
Roberts WK, Selitrennikoff CP (1988) Plant and bacterial chitinases differ in antifungal activity. J Gen Microbiol 134:169–176
Saleem B, Kandasamy U (2002) Antagonism of Bacillus species (strain BC121) towards Curvularia lunata. Curr Sci 82(12):1457–1463
Shanmugaiah V, Mathivanan N, Balasubramanian N, Manoharan PT (2008) Optimization of Cultural conditions for production of Chitinase by Bacillus laterosporous MMI2270 isolated from rice rhizosphere soil. Afr J of Biotechnol 7(15):2562–2568
Sneath PHA (1986) In Bergey’s manual of systematic bacteriology. In: Sneath PHA et al (eds) Williams and Wilkins, vol 2. Baltimore, pp 1104–1207
Someya N, Nakajima M, Hirayae K, Hibi T, Akutsu K (2001) Synergistic antifungal activity of chitinolytic enzymes and prodigiosin produced by the biocontrol bacterium Serratia marcescens strain B2 against the gray mold pathogen, Botrytis cinerea. J Gen Plant Pathol 67:312–317
Srividya S, Sasirekha B, Ashwini N (2012) Multifarious antagonistic potentials of rhizosphere associated bacterial isolates against soil borne diseases of Tomato. Asian J Plant Sci Res 2(2):180–186
Sutton BC (1992) The genus Glomerella and its anamorph Colletotrichum, In: Bailey JA, Jeger MJ (eds) Colletotrichum—biology, pathology, and control, CAB International, Wallinngford, pp 1–26
Takayanagi T, Ajisaka K, Takiguchi Y, Shimahara K (1991) Isolation and characterization of thermostable chitinases from Bacillus licheniformis X-7u. Biochem Biophys Acta 12:404–410
Teather RM, Wood PJ (1982) Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl Environ Microbiol 43:777–780
Tendulkar SR, Saikumari YK, Patel V, Raghotama S, Munshi TK, Balaram P, Chattoo BB (2007) Isolation, purification and characterization of an antifungal molecule produced by Bacillus licheniformis BC98, and its effect on phytopathogen Magnaporthe grisea. J Appl Microbiol 103:2331–2339
Than PP, Jeewon R, Hyde KD, Pongsupasamit S, Mongkolporn O, Taylor PWJ (2008) Characterization and pathogenicity of Colletotrichum species associated with anthracnose on chilli (Capsicum spp.) in Thailand. Plant Pathol 57:562–572
Thind TS, Jhooty JS (1985) Relative prevalence of fungal disease of chilli fruit in Punjab. Ind J Mycol Plant Path 15:305–307
Turner JT, Backman PA (1991) Factors relating to peanut yield increases after seed treatment with Bacillus subtilis. Plant Dis 75:347–353
Viterbo A, Haran S, Friesem D, Ramot O, Chet I (2001) Antifungal activity of a novel endochitinase gene (chit36) from Trichoderma harzianum Rifai TM. FEMS Microbiol Lett 200:169–174
Wen C, Tseng MCS, Cheng CY, Li YK (2002) Purification, characterization and cloning of a chitinases from Bacillus sp. NCTU2. Biotechnol Appl Biochem 35:213–219
Yu GY, Sinclair GL, Hartman GL, Bertagnolli BL (2002) Production of iturin A by Bacillus amyloliquefaciens suppressing Rhizoctonia solani. Soil Biol Biochem 34:955–963
Zhang Y, Fernando WGD (2004) Zwittermicin A detection in Bacillus spp. controlling Sclerotinia sclerotiorum on canola. Phytopathology 94:S116
Acknowledgments
The authors thank the management of Jain University for providing the necessary facilities for carrying out this work.
Conflict of interest
The authors declare that they have no conflict of interest in the publication.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ashwini, N., Srividya, S. Potentiality of Bacillus subtilis as biocontrol agent for management of anthracnose disease of chilli caused by Colletotrichum gloeosporioides OGC1. 3 Biotech 4, 127–136 (2014). https://doi.org/10.1007/s13205-013-0134-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13205-013-0134-4