
Abstract
The structure, binding energy, magnetic moments, and electronic structure of Fe m Ir n (2 ≤ m + n ≤ 4) nano clusters are investigated using first principles density functional theory techniques. Fully unconstrained structural relaxations are undertaken by considering all possible non-equivalent cluster structures. The optimized clusters are all compact, indicating a clear tendency to maximize the number of nearest neighbor Fe–Ir pairs. The binding energy shows an increment with cluster size. All the clusters preserve ferromagnetic order after optimization and the average magnetic moment shows a general increment with Fe concentration. An enhancement of the local Fe moments in Ir-rich environment is observed, while that of Ir is minimal. The highest occupied molecular orbital–lowest unoccupied molecular orbital energy gaps show a general reduction with alloying, indicating more metallicity for the doped clusters than the pure ones.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Studies on the structural and magnetic properties of small metal clusters are carried out with great interest, both experimentally (Parks et al. 1988; Liyanage et al. 2003) and theoretically (Chen et al. 1991; Gong and Zheng 1995; Ma et al. 2007). These studies are important as clusters play an important bridge between the atom and bulk, thus in understanding the transition from small to big. In small clusters, most of the atoms reside at the surface and hence the structures can be very different from the fragments of bulk phases, and different structures may have quite different magnetic and electronic properties. Owing to the large surface to volume ratio, clusters are more effective catalysts. Apart from the catalytic efficiency, electron localization caused by the small size of the clusters gives rise to larger magnetic moments. Often non-magnetic metals can exhibit magnetism in the cluster form. The high surface to volume ratio of these clusters gives rise to fewer bonds per metal atom and hence frees up non-bonded valence electrons, leading to enhanced magnetic moments if left unpaired. Therefore, the magnetic moments in these clusters are very much sensitive to the geometry of the cluster and the chemical environment (Rao et al. 1990; Rollmann et al. 2006; Longo et al. 2008). In this scenario, clusters of transition metals attract particular attention as they are known to display enhanced magnetic moments relative to the bulk counterparts, hence are relevant candidates for magnetic storage devices. They find applications in heterogeneous catalysis too. Among transition metals, Fe clusters receive wide attention due to its distinctive magnetic properties. Small FeN clusters are studied both experimentally (Montano et al. 1980; Purdum et al. 1982; Moskovits and Diella 1980; Cox et al. 1985) and theoretically (Chen et al. 1991; Castro and Salahub 1994; Oda et al. 1998; Ballone and Jones 1995; Chen et al. 1991; Yu et al. 2007; Dieguez et al. 2001) using different techniques, and showed compact structure with enhanced magnetic moments of about 3μB/atom. Studies on small Fe clusters carried out by Chen et al. (1991) have shown a ferromagnetic ground state with enhanced magnetic moments and smaller inter-atomic distances compared to the bulk phase. Apart from the clusters of a single element, combinations of transition metals are also studied due to the interesting properties that can arise due to alloying (Munoz-Navia et al. 2009). The binary metal clusters have been shown to exhibit projected magnetic moments that are larger or smaller than the corresponding free metal atoms, depending on the composition of the hetero nuclear cluster (Mokkath and Pastor 2012). Our interest in this context is devoted to the study of clusters consisting of ferromagnetic 3d and 5d atoms. Presently, we focus on clusters of Fe and Ir. Most studies on Iridium clusters give emphasis to the catalytic activity and reaction with surfaces (Seets et al. 1997; Sitz and Mullins 2002). Studies have shown that there exists a low translational energy pathway for the dissociation of methane using Ir as the catalyst (Reeves et al. 2001). Iridium clusters of various sizes are being studied both experimentally (Watzky and Finke 1997; Wang and Ehrlich 1997) and theoretically (Feng et al. 1997; Bussai et al. 2005; Pawluk et al. 2005; Stevanovic et al. 2012). Feng et al. (1997) investigated the reactivity of Ir clusters from 4 to 10 atoms. Another study on Ir clusters supported on Ir (111) substrate showed that the shape and size affects the stability and higher stabilities are achieved for larger clusters containing 19 and 39 atoms (Wang and Ehrlich 1997). Bussai et al.’s (2005) analysis on the binding energies of four atom iridium clusters showed a square planar configuration as the most stable structure using a scalar relativistic variant of density functional theory (DFT) method. Another DFT study of Ir clusters with sizes ranging n = 2–64 (Pawluk et al. 2005) showed that the magnetic moments and binding energies showed oscillatory behavior with increase in cluster size. On the other hand, an increase in binding energy with cluster size is also reported in DFT studies (Stevanovic et al. 2012). A DFT study involving Fe clusters supported on Ir (111) layers (Bornemann et al. 2012) have shown that atomic magnetic moments are induced in the Ir substrate due to the presence of Fe atoms. Experimentally, Ir4 clusters showing a tetrahedral geometry are synthesized on substrates (Deutsch et al. 1997). However, there are very few investigations on how the energetic, stability and magnetic properties vary when Ir clusters are alloyed with another transition metal. In this paper, we present first principles DFT investigations of free standing Fe m Ir n (2 ≤ m + n ≤ 4) clusters. The aim is to investigate how the structure, stability, and magnetic properties of the clusters will modify when a ferromagnetic metal (Fe) is bonded with a metal that is non-magnetic (Ir) in the bulk phase. Also, Ir is an important catalyst material, and thus investigating the properties of alloyed clusters containing Ir will be interesting. Complete geometry optimization of the clusters by including all non equivalent combinations and chemical orders will help us to generate the most stable cluster geometries. The magnetic and electronic properties of the optimized geometries are calculated. The computational methodology is presented in the next section followed by the results and discussions. The results are compared with other theoretical and experimental data wherever possible. Finally, our conclusions are presented.
Methodology
First principles DFT calculations are carried out for Fe–Ir clusters using the plane wave DFT program PWSCF (ESPRESSO Version 5) (Baroni et al. 2012). The Rappe–Rabe–Kaxiras–Joannopoulos (RRKJ) ultra soft pseudo potential with non-linear core correction (Rappe et al. 1990) is used for Fe and Ir with the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) correction (Perdew et al. 1996) formalism employed for the exchange-correlation functional. Both of these pseudo potentials are obtained from the PWSCF pseudo potential reference and the atomic electronic configuration of Fe and Ir atoms used in the pseudo potential are 3d74s1 and 5d76s2, respectively. The main difficulty in the theoretical study of binary metal clusters is the identification of the low energy configurations accurately as there exist diverse geometrical conformations and distributions of the different atoms in the cluster. Various ordered and disordered structures may exist with different degrees of intermixing and segregation. Hence, all possible initial geometries and topologies with all possible distributions of the Fe and Ir atoms should be taken into account. These structures are then need to be relaxed in fully unconstrained manner until the equilibrium configuration is reached. In the present study concerning Fe m Ir n (2 ≤ m + n ≤ 4), we have considered all possible topologies and chemical orders of the Fe and Ir atoms within the cluster including the pure Fe m and Ir n limits. We have generated all possible geometries as described by Mokkath and Pastor (2012). Even though different initial geometries are considered, after the unconstrained relaxations the energetically stable ones are considered for further analysis and presented in the paper. The computational parameters such as the size of the super cell, the kinetic energy cut off, the cut off for the electron density and the force convergence threshold are checked for convergence. The clusters were placed in a simple cubic super cell of size 20 × 20 × 20 Å3, assuring that the interaction between the clusters and their replicas in neighboring cells are negligible. This also enables us to consider only the Gamma (Γ) point (k = 0) when integrating over the Brillouin zone. The real space grid for the numerical calculations involving the electron density is described by an energy cut off of 250 Ry. All the clusters are fully relaxed until the inter atomic forces are smaller than 10−3 eV/Å. The stability of each cluster is assessed based on the binding energy per atom. The binding energy per atom is calculated as,
where \( E({\text{Fe}}) \) and \( E({\text{Ir}}) \) are the total energy correspond to the m Fe and n Ir isolated atoms. \( E({\text{Fe}}_{m} {\text{Ir}}_{n} ) \) is the total energy corresponding to the cluster. The binding energy of the cluster (in eV/atom) is defined as the difference in total energy between the interacting atom system and the free atom system. Hence clusters with more binding energy are more stable and vice versa. To aid the convergence, Marzari–Vanderbilt smearing method (Marzari et al. 1999) is used, with a Gaussian broadening of 0.001 Ry. The average as well as the local magnetic moments is calculated by spin polarized total energy calculations. Localized electron population analysis is carried out to calculate the local charges and local magnetic moments on the atoms. It is to be noted that in this study, only the atomic spin magnetic moments are computed, i.e., spin orbit interactions are not taken into account, due to the limitation of computational resources. The inclusion of spin-orbit coupling (SOC) is needed for the calculations involving magnetic anisotropic energy (MAE), as MAE originates from the coupling between the spin and orbital degrees of freedom. SOC is also known to affect the highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) gaps in 5d clusters as has been shown by Sebetci (2008). However, the theme of the present study is to calculate the relative stability and magnetic moment of the clusters and calculation of orbital moments and MAE may be the subject of a future study. It is worth to mention here the work of Hobbs et al. (2000) who had studied small Fe and Cr clusters in a non-collinear frame work as implemented within the PAW formalism, and obtained a collinear ground state for the Fe trimers. In this study, we have considered the GGA and have not taken approximations beyond, including parameters such as Hubbard Hamiltonian. It is a known fact that for larger clusters, role of quantum fluctuations beyond the mean field and appearance of non-collinear spin configurations should be considered for accurate results (Ojeda et al. 1999). However, for the smaller clusters considered in the present study, GGA is considered to deliver accurate results as has been shown by previous studies (Rollmann et al. 2006; Longo et al. 2008). Ab initio total energy calculations by Rollmann et al. (2006) has shown that relaxation in small Fe clusters leads to stabilization of collinear magnetic structures. In another study by Longo et al. (2008), also the non-collinear magnetic order is not found to exist in Fe–Mn clusters. In view of the above results, a non-collinear magnetic structure is not expected for the Fe–Ir clusters studied in the present work and the calculations are done in the collinear frame work. The energy difference between the HOMO and the LUMO are calculated and this energy gap is used to understand the stability of the clusters. The HOMO–LUMO energy gap E g (in units of eV) is a characteristic quantity of the electronic structure of the cluster and is a measure of the stability of the clusters to undergo activated chemical reactions with small molecules. The magnitude of HOMO–LUMO energy gap may vary with both the size and composition of the cluster and the manner in which these orbitals getting filled determines the properties of the clusters.
Results and discussion
In this section, the investigations on the ground state structure, stability, and magnetic properties of Fe m Ir n clusters are described. The main focus is to understand the effects of size and chemical composition on the stability and magnetism. The similarity and differences in properties between clusters of various sizes are enumerated. The geometry of the clusters after optimization and their properties are summarized in Tables 1, 2, and 3. Before proceeding with the cluster calculations, we have calculated the lattice parameter and cohesive energy of bulk Fe and Ir to test the accuracy of computational scheme. The calculated (measured) lattice constant and cohesive energy of bulk and Fe and Ir are 2.87 Å (2.86 Å), 4.41 (4.28 eV), 3.85 Å (3.84 Å), and 6.90 eV (6.94 eV) (Kittel 2008), respectively. The good agreement with experimental values shows that the computational method is appropriate to investigate the Fe–Ir clusters.
We have started our calculations with the Fe dimer, and then replaced one Fe atom by Ir to understand the change in properties with alloying. The results are summarized in Table 1. It is seen that the binding energy increases from Fe–Fe to Ir–Ir, indicating that the bonding in Fe2 is the weakest and the bonding resulting from the Fe–Ir pair is stronger than the Fe–Fe bonds. The binding energy obtained for Fe2, 1.05 eV (using GGA in the present study) turns out to be larger than the experimental value, (0.65 eV/atom) (Moskovits and Diella 1980) but is in agreement with that reported in other theoretical studies (Gong and Zheng 1995; Castro and Salahub 1994; Oda et al. 1998). This over estimation of binding energy compared to experimental results may be related to the choice of exchange and correlation functional employed. The bond length analysis also indicates the same trend as that of binding energy. The Fe–Fe bond length, 2.00 Å, is in consistent with the experimental values 1.87 Å (Montano and Shenoy 1980) and 2.02 Å (Purdum et al. 1982) obtained in different studies and theoretical results of Gong and Zheng (1995), which has shown a bond length of 2.09 Å. However, the calculated bond length is considerably smaller than the nearest neighbor distance, 2.48 Å in bulk bcc Fe (Kittel 2008). On close analysis, it can be seen that the bond length follows the trend of the atomic radii (atomic radii of Fe being 1.72 Å, whereas that of Ir is 1.87 Å (Stevanovic et al. 2012), such that dIr–Ir > dFe–Ir > dFe–Fe. The trend of the Fe2 and Fe–Ir to be more compact compared to the Ir2 indicate a more metallic character of the 3d bonding and a tendency of a more covalent and directional bonding in the case of 5d ones. The HOMO–LUMO energy difference of the dimers are large, indicating a lower reactivity, however, the alloying seems to reduce the energy gap. The average magnetic moment varies from 3.38 to 2.51 μB as one goes from Fe2 to Ir2, indicating nearly full polarization of all the d electrons. The magnetic moment per atom for Fe2, 3.38 μB is in agreement with other DFT results (Chen et al. 1991; Oda et al. 1998; Ballone and Jones 1995), and is in keeping with the value obtained in experiments (3.3 ± 0.5) (Cox et al. 1985). For the pure Fe and Ir dimers, the local and average magnetic moments are equal and it indicates that the spin density,
is quite localized around the atoms. The difference between the average and local magnetic moments give a measure of the spin density \( m(\vec{r}) \), which is spilled off to the immediate environment of the atoms. The local magnetic moment of Fe and Ir in the mixed Fe–Ir dimer is quite remarkable, particularly if one compares them with the pure dimers. The Fe local moment in Fe–Ir, 3.53 μB is 0.15 μB larger than Fe2 and even larger than the Fe atom, while the Ir moment is reduced by a similar amount (0.14 μB) as can be seen from Table 1. It can also be seen from the Table that the electronic charge of Fe decreases where as that of Ir increases. The reduction in Ir magnetic moment is attributed to the transfer of d electrons from Fe to Ir, which enhances the number of d holes and allows the Fe atom to develop a significantly larger spin moment. This increase in Fe magnetic moment occurs at the expense of Ir atom and this observation is qualitatively in agreement with the larger Pauling electro negativity of the Ir atom (χFe = 1.83, χIr = 2.20) (Pauling 1932).
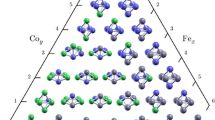
We have considered both linear and triangular geometry for the trimers, however, after optimization, the most stable geometry turned out to be an equilateral triangle and this is true for all compositions. The linear geometries relaxed into triangular geometries, favoring a higher coordination number and the results of the stable configurations for each composition are presented in Table 2. The energy gain in going from the most stable configuration to the least stable one is calculated and is shown in the Table. The Fe2Ir cluster shows maximum relative stability for the triangular configuration and with increase in Ir content, the energy gain decreases and the lowest energy difference is shown by the pure Ir cluster. This indicates that the relative stability of Fe-rich clusters is more in the triangular configuration. One observes that the inter atomic distances follow the trends in the atomic radii as in the case of dimers. For Fe3, the lowest energy structure is an equilateral triangle with bond length 2.19 Å, in good agreement with previous DFT studies, which have reported bond lengths of 2.15, 2.37, 2.04, 2.10 and 2.11 Å (Chen et al. 1991; Gong and Zheng 1995; Castro and Salahub 1994; Oda et al. 1998; Ballone and Jones 1995) respectively. Compared to Fe2, the bond length shows a tendency to increase, indicating that the system always favors the highest dimension for longer bond length. The Fe-rich trimers tend to be more compact indicating more metallic character of bonding as in the case of dimers. The binding energy shows a monotonous dependence on composition similar to the case of dimers, indicating that the Ir–Ir bonds are the strongest in the trimers too. However, the trend of increase in binding energy with increase in number of Ir atoms indicates that stability increases with increase in number of Fe–Ir bonds. It is to be noted that FeIr2 is more stable than Fe2Ir because Ir–Ir bonds are in general stronger than those between Fe atoms. The HOMO–LUMO energy gap shows a monotonous decrease with Ir composition, indicating more chemical activity. The average magnetic moment of Fe3, 3.45μB/atom, is in agreement with previous DFT studies (Gong and Zheng 1995; Ma et al. 2007; Castro and Salahub 1994; Oda et al. 1998; Ballone and Jones 1995) and close to the experimental results (Cox et al. 1985). The ground state of Ir3 is equilateral with local as well as average magnetic moment of 2.22μB/atom. This indicates that the spin polarization is dominated by electrons occupying localized states and the spill off contributions is not important in the case of Ir3, unlike the other trimers. It is to be noted that the local magnetic moments in the trimers always show a ferromagnetic coupling. The average and local magnetic moments of Ir3 clusters, 2.22μB/atom are smaller than that obtained for the pure Ir dimer. For mixed compositions, the presence of Fe–Ir bonds enhances the local Fe magnetic moments beyond 3μB/atom. The substitution of one Ir atom in Fe3 to yield Fe2Ir increases the local Fe magnetic moment to 3.56μB/atom even though the average magnetic moment shows a monotonous decrease with Ir addition. Similar to the dimers, this is mainly due to the charge transfer from Fe to Ir. It can be seen that, quantitatively, local Fe and Ir magnetic moments are similar to the values calculated in dimers. The local magnetic moments of Ir atoms in the mixed trimers are slightly enhanced compared to pure Ir3 cluster due to the proximity to Fe atoms. However, this increase is not significant as the magnetic moments are already quite large in the pure Ir trimers.
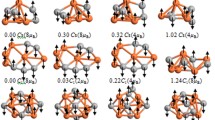
The tetramers are also studied in detail as they are the smallest clusters with a three dimensional geometric structure and the results are summarized in Table 3. We have inspected different possible isomers of the Fe, Ir, and Fe–Ir tetramers, such as tetrahedral, rhombus, square planar, and a linear chain structure, each of them having different coordination number. In the tetrahedral structure, the coordination number of all atoms is 3, while in the rhombus, two atoms have coordination number 3 and two atoms have 2. In the square planar structure, all atoms have coordination number 2. Despite considering all non equivalent cluster geometries for each composition, after complete geometry optimization, the most stable ones turned out to be the tetrahedrons. In this structure, the tetrahedral topology ensures the freedom of permuting the Fe and Ir atoms within the cluster without altering the chemical order. The energy difference (ΔE) between the most stable and least stable structures is enumerated in the Table. We have seen that, among the various configurations considered in the present study, the linear geometry turned out to be the lowest in energy, like in the case of trimers and the energy gain is of the order of ~2 eV. The most stable isomer for the Fe4 cluster is a regular tetrahedron with average magnetic moment of 3.55μB, and is comparable to previous studies (Gong and Zheng 1995), which showed the stable structure to be a regular (Castro and Salahub 1994; Chen et al. 1991) or distorted tetrahedron (Oda et al. 1998; Ballone and Jones 1995). The obtained bond length of Fe4, 2.34 Å is in keeping with other DFT results (Chen et al. 1991; Gong and Zheng 1995). The increasing trend of bond length from Fe2 to Fe3 to Fe4 indicates that the clusters always prefer the highest dimension for longer bond length and maximum pairs of nearest neighbor bonds. Previous studies on of Ir4 clusters show contradictions regarding the ground state structure. The predicted stable geometries in theoretical studies include tetrahedral (Feng et al. 1997), a butterfly structure (Bussai et al. 2005), rhombus (Pawluk et al. 2005), and square (Stevanovic et al. 2012) whereas experiments has showed tetrahedral as the most stable structure having a bond length of 2.71 Å (Deutsch et al. 1997). We have seen that the tetrahedral structure is the most stable structure in agreement with this experimental result and theoretical study which obtained tetrahedron having bond length of 2.79 Å (Feng et al. 1997). For the alloyed tetramers also, the tetrahedron structure came out to be the most stable of all the configurations studied. The binding energy exhibits a non-monotonous dependence on composition, unlike the situation in dimers and trimers. This shows that with the increase in number of Ir atoms, the Fe–Ir bond strength increases. Considering the change in binding energy from Fe4 to Ir4, the relative stability of the clusters can be correlated to the number of homogeneous and heterogeneous bonds. It can be understood by looking at the most stable composition, FeIr3, which has three Fe–Ir and Ir–Ir bonds. Coming to Fe2Ir2, replacing one Ir by one Fe implies replacing two Ir–Ir bonds by a relatively stronger Fe–Ir bond and a weaker Fe–Fe bond. In this way, the binding energy is not altered significantly. A visible decrease in binding energy is obtained only for larger Fe concentration. We have noticed that the average bond length increases from 2.0 Å for the Fe dimer to 2.34 Å for the Fe tetramer, indicating a tendency to approach the nearest neighbor distance in the bulk bcc Fe with the increase in cluster size. The calculated bond length (2.44 Å) and the magnetic moment/atom (2.13μB) of Ir4, is comparable to that obtained by Stevanovic et al. (2012) (2.31 Å and 2.00μB, respectively), however, they have obtained octahedral geometry as the stable structure. Yet another study showed the square planar structure with large binding energy (Pawluk et al. 2005) while we have observed that the square planar structure upon complete geometry optimization transforms into a tetrahedral geometry. These variations in stability can be attributed to the difference in approximations involved in the choice of pseudo potential and exchange correlation functional. The HOMO–LUMO energy gap shows a reduction with alloying, thus favoring more metallicity in the alloyed clusters. Turning to the magnetic properties, it is seen that a ferromagnetic order is observed in all the clusters and the average magnetic moments are larger than that obtained in dimers and trimers. This increase in magnetic moment can be related to the increase in bond length with cluster size. We have noticed that with the increase in compactness, magnetic moment decreases. This is due to the fact that, in compact clusters, the number of nearest neighbors is larger than more open structures, and to have large magnetism, more energy is required. Hence the compact structures tend to possess small magnetic moments. The average as well as local magnetic moment of pure Fe tetramer, is enhanced (3.55μB/atom) compared to Fe2 and Fe3 in agreement with the value reported by Gong and Zheng (1995) for the square isomer and slightly larger than the value reported by Yu et al. (2007) and Dieguez et al. (2001). The Ir4 clusters show average and local magnetic moment of 2.13μB/atom, in agreement with experimental values (2.04μB/atom) reported for the tetrahedral Ir4 cluster (Stevanovic et al. 2012). Pawluk et al. (2005) also reported 2μB/atom for the square isomer, however, they have obtained zero magnetic moment for the tetrahedral structure. For the tetramers also, the presence of Fe in the neighborhood enhances the Ir moments and the average magnetic moment increases with Fe concentration. It is worth to mention here that, the ferromagnetic ordering shown by the small clusters may change, with the increase in dimension. Even though we have seen an increase in magnetic moment with the cluster size, while going from the dimers to tetramers, to obtain a critical cluster size with regard to the magnetic moment needs further calculations considering larger clusters with more number of atoms and which may be attempted in a future study. However, this study is relevant as knowledge of the magnetic behavior of these small clusters can be useful while addressing clusters of larger dimension.
Conclusions
In summary, investigations on the structure, binding energy, magnetic moments and electronic structure of Fe m Ir n (2 ≤ m + n ≤ 4) clusters are performed using DFT techniques. All the clusters are fully relaxed to their ground structures and the optimized geometries are in general compact, with a strong tendency to intermix to maximize the number of heterogeneous Fe–Ir bonds. Even though the bond length increases with the increase in size for the pure Fe and Ir clusters, it is far from the bulk nearest neighbor distance of Fe (2.48 Å) and Ir (2.71 Å) (Kittel 2008). The magnetic order in the optimized clusters is found to be ferromagnetic with the average magnetic moment showing an overall increment with increase in cluster size. The trends in magnetism are controlled to a large extent by the Fe content in the alloyed clusters. An enhancement of the local Fe moments in an Ir-rich environment is observed. On the other hand, the Ir moments are already large in the pure Ir clusters and hence does not show significant enhancement with Fe doping. The metallicity trends of the clusters are reflected in the variation of the HOMO–LUMO energy gap. A reduction in HOMO–LUMO gap is observed with alloying of the clusters, and which indicates that metallicity of the clusters increases with doping.
References
Ballone P, Jones RO (1995) Structure and spin in small iron clusters. Chem Phys Lett 233:63
Baroni S, Corso AD, De Gironcoli S, Gianozzi P (2012) PWSCF http://www.quantum-espresso.org/
Bornemann S, Sipr O, Mankovsky S, Polesya S, Staunton JB, Wurth W, Ebert H, Minar J (2012) Trends in the magnetic properties of Fe, Co., and Ni clusters and monolayers on Ir(111), Pt(111), and Au(111). Phys Rev B 86:104436
Bussai C, Kruger S, Vayssilov G, Rosch N (2005) The cluster Ir4 and its interaction with a hydrogen impurity. A density functional study. Phys Chem Chem Phys 13:2656
Castro M, Salahub DR (1994) Density-functional calculations for small iron clusters: Fe n , Fen+, and Fen− for n ≤ 5. Phys Rev B 49:11842
Chen JL, Wang CS, Jackson KA, Pederson MR (1991) Theory of magnetic and structural ordering in iron clusters. Phys Rev B 44:6558
Cox DM, Trevor DJ, Whetten RL, Rohlfing EA, Kaldor A (1985) Magnetic behavior of free-iron and iron oxide clusters. Phys Rev B 32:7290
Deutsch SE, Mestl G, Knozinger H, Gates BC (1997) MgO-supported tetrairidium clusters: evidence of the metal-support interface structure from X-ray absorption spectroscopy. J Phys Chem B 101:1374
Dieguez O, Alemany MMG, Rey C, Ordejon P, Gallego LJ (2001) Density-functional calculations of the structures, binding energies, and magnetic moments of Fe clusters with 2 to 17 atoms. Phys Rev B 63:205407
Feng JN, Hunang XR, Li Z-S (1997) A theoretical study on the clusters Ir n with n = 4, 6, 8, 10. Chem Phys Lett 276:334
Gong XG, Zheng QQ (1995) Local spin-density electronic structures and magnetic properties of small iron clusters. J Phys Condens Matt 7:2421
Hobbs D, Kresse G, Hafner J (2000) Fully unconstrained noncollinear magnetism within the projector augmented-wave method. Phys Rev B 62:11556
Kittel C (2008) Introduction to solid state physics. Wiley, New York
Liyanage R, Griffin JB, Armentrout PB (2003) Thermodynamics of ammonia activation by iron cluster cations: guided ion beam studies of the reactions of Fen+(n = 2–10,14) with ND3. J Chem Phys 119:8979
Longo RC, Alemany MMG, Vega A, Ferrer J, Gallego LJ (2008) Engineering the magnetic structure of Fe clusters by Mn alloying. Nanotechnology 19:245701
Ma QM, Xie Z, Wang J, Liu Y, Li YC (2007) Structures, binding energies and magnetic moments of small iron clusters: a study based on all-electron DFT. Solid State Commun. 142:114
Marzari N, Vanderbilt D, De Vita A, Payne MC (1999) Thermal contraction and disordering of the Al(110) surface. Phys Rev Lett 82:3296
Mokkath JH, Pastor GM (2012) First-principles study of structural, magnetic, and electronic properties of small Fe–Rh alloy clusters. Phys Rev B 85:054407
Montano PA, Shenoy GK (1980) EXAFS study of iron monomers and dimers isolated in solid argon. Solid State Commun 35:53
Moskovits M, Diella DP (1980) Di-iron and nickel iron. J Chem Phys 73:4917
Munoz-Navia M, Dorantes-Davila J, Zitoun D, Amiens C, Jaouen N (2009) Tailoring the magnetic anisotropy in CoRh nanoalloys. App Phys Lett 95:233107
Oda T, Pasquarello A, Car R (1998) Fully unconstrained approach to noncollinear magnetism: application to Small Fe Clusters. Phys Rev Lett 80:3622
Ojeda MA, Davila JD, Pastor GM (1999) Noncollinear cluster magnetism in the framework of the Hubbard model. Phys Rev B 60:6121
Parks EK, Weiller BH, Bechthold PS, Hoffman WF, Nieman GC, Pobo LG, Riley SJ (1988) Chemical probes of metal cluster structure: reactions of iron clusters with hydrogen, ammonia, and water. J Chem Phys 88:1622
Pauling L (1932) The nature of the chemical bond. The energy of single bonds and the relative electro negativity of atoms. J Am Chem Soc 54:3570
Pawluk T, Hirata Y, Wang L (2005) Studies of Iridium nanoparticles using density functional theory calculations. J Phys Chem B 109:20817
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865
Purdum H, Montano PA, Shenoy GK, Morrison T (1982) Extended-X-ray-absorption-fine-structure study of small Fe molecules isolated in solid neon. Phys Rev B 25:4412
Rao BK, Khanna SN, Jena P (1990) Clusters-A new phase of matter. Phase Transit 24:35
Rappe AM, Rabe KM, Kaxiras E, Joanopoulos JD (1990) Optimized pseudopotentials. Phys Rev B 41:1227
Reeves CT, Seets DC, Mullins CB (2001) Low translational energy mechanisms in the dissociative chemisorption of methane on iridium and platinum surfaces. J Mol Catal A: Chem 167:207
Rollmann G, Entel P, Sahoo S (2006) Competing structural and magnetic effects in small iron clusters. Comp Mater Sci 35:275
Sebetci A (2008) Does spin–orbit coupling effect favor planar structures for small platinum clusters. Phys Chem Chem Phys 11:921–925
Seets DC, Wheeler MC, Mullins CB (1997) Mechanism of the dissociative chemisorption of methane over Ir(110): trapping-mediated or direct? Chem Phys Lett 266:431
Sitz GO, Mullins CB (2002) Molecular dynamics simulations of the influence of surface temperature on the trapping of methane on iridium single-crystalline surfaces. J Phys Chem B 106:8349
Stevanovic V, Sljivancanjn Z, Baldereschi A (2012) Role of adsorbed H, C, O, and CO on the atomic structure of free and MgO(100)-supported Ir4 clusters: an ab Initio Study. J Phys Chem C 114:15653
Wang SC, Ehrlich G (1997) Equilibrium shapes and energetics of iridium clusters on Ir (111). Surf Sci 391:89
Watzky MA, Finke RG (1997) Nanocluster size-control and “Magic Number” investigations. experimental tests of the “Living-metal polymer” concept and of mechanism-based size-control predictions leading to the syntheses of iridium(0) nanoclusters centering about four sequential magic numbers. Chem Mater 9:3083
Yu S, Chen S, Zhang W, Yu L, Yin Y (2007) Theoretical study of electronic structures and magnetic properties in iron clusters (n ≤ 8). Chem Phys Lett 446:217
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Aravindh, S.A. First principles studies of Fe m Ir n (2 ≤ m + n ≤ 4) nano clusters. Appl Nanosci 4, 593–600 (2014). https://doi.org/10.1007/s13204-013-0232-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13204-013-0232-y