
Abstract
The discovery of transition metal-catalysed polymerisation of olefins in the 1950s by Ziegler and Natta was of huge importance. Over the last 30 years, the interest in homogenous polymerisation has not only grown but has changed focus from primarily studying the metallocene complexes of Group 4 to widespread exploration of post-metallocene systems. This is a consequence of extensive patenting of the metallocene catalyst systems, as well as for general interest in the improvement of polyolefin catalysis. The plastics industry produces more than 107 tonnes of polyethylene, polypropylene and polystyrene each year worldwide, and the study of well-defined homogenous catalysts is invaluable in uncovering the mechanism of polymerisation and the fundamental properties of the active species. This short Tutorial Review gives an overview of homogenous transition metal Ziegler–Natta catalysis in general as well as an overview of a selection of Group 4 post-metallocene catalysts. An overview of the synthesis and reactions of alkyl cations relevant to olefin polymerisation is also given. Finally, this review summarises recent advances in bimetallic catalysts for olefin polymerisation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction to Ziegler–Natta catalysis
Ziegler and Natta were awarded the Nobel Prize in Chemistry in 1963 “for their discoveries in the field of the chemistry and technology of high polymers”. Ziegler and Natta [1–3] discovered that heterogeneous polymerisation of ethylene and propylene could be achieved by mixtures of transition metal and trialkylaluminium compounds; such systems included TiCl4/AlEt3. To this day, the majority of industrially produced polyolefins are produced in this way. These systems have been widely developed over the last 50 years and act as efficient and selective catalysts for many types of olefin polymerisation. The main problem with these catalysts is the lack of definition in the active site. Without a well-defined active site, rational catalyst design and characterisation is difficult, and the polymer produced by such a system has a broad molecular weight distribution. This is where the study of homogenous catalysis finds its benefits. The molecules which now act as the active sites for catalysis can be completely defined and characterised by methods such as NMR spectroscopy or X-ray crystallography with relative ease leading to logical catalyst design. This short tutorial review, focussing on Group 4 metals and metallocene and half-sandwich types of compounds in particular, is not intended by any means to provide an exhaustive account of the literature. The reader is directed to several review articles for a more comprehensive overview of homogenous Ziegler–Natta catalysis [4–14].
Metallocene catalysts
Sir Geoffrey Wilkinson [15], who won a Nobel prize in 1973 together with Ernst Otto Fischer “for their pioneering work, performed independently, on the chemistry of the organometallic, so called sandwich compounds”, reported the syntheses of Cp2TiBr2 and Cp2ZrBr2 in 1953. In 1957, Natta and Breslow [16, 17] both reported that the bent metallocene Cp2TiCl2 in conjunction with Et2AlCl or AlEt3 could polymerise ethylene but not propylene. This system was not ideal, however, as the polymerisation was slow. Unexpectedly in 1980, Sinn and Kaminsky [18–20] discovered that addition of a small amount of water to metallocene/alkylaluminum systems vastly increased the system’s polymerisation activity. This was due to the formation of methyl aluminoxane (MAO) which is formed from the partial hydrolysis of trimethylaluminium (TMA) and can be prepared prior to polymerisation or in situ. MAO can be used in general to activate a variety of Group 4 metallocenes towards ethylene or α-olefin polymerisation [21, 22].
Metallocene systems activated with MAO are also capable of polymerising propylene. One such system is the zirconocene system Cp2ZrCl2/MAO. An extra consideration when polymerising propylene, due to the presence of an extra methyl group per monomer, is tacticity. This is defined by the relative chiral arrangement of adjacent pendant methyl groups. Examples of the principal different stereochemistry possible in polypropene can be seen in Fig. 1.

The main types of tacticity in polypropylene
Constrained geometry catalysts (CGCs) and ansa-metallocenes
Following the discovery of metallocene catalysts, the 1980s saw a considerable amount of research into catalysts that could control the stereochemistry and thus microstructure of the resulting polyolefin. Variation of metallocene symmetry (see Fig. 2 for examples) was discovered to be the main factor in determining the tacticity of the final polymer [10].

Ansa-metallocene catalysts which can control the tacticity of polypropylene
The ligand sets investigated were metallocene based, but the two supporting ligands were linked by an ansa-bridging moiety. This connection prevents rotation of the ligands helping to well define the steric environment of the active site. Kaminsky and Brintzinger [23] developed the C 2 symmetric zirconocene catalyst 1 which produced highly isotactic polypropylene; the sterics defined in the active site control the insertion of each monomer to give this microstructure. Ewen et al. [24] later found that C s symmetric complex 2 produced highly syndiotactic polypropylene. Although it is not an ansa compound, in 1995, Coates and Waymouth [25] described a sterically crowded metallocene compound 3 which acted in a similar fashion; although this was not constrained by a physical bridge, the bulky groups led to restricted rotation. In the absence of a physical link, the top and bottom ligands could rotate intermittently, and thus, the catalyst fluctuates between chiral and achiral producing a block polymer containing isotactic and atactic sections.
Limited thermal stability and the production of low molecular weight polymers under commercially relevant conditions by the metallocene ansa-catalysts resulted in the development of a new catalyst type, the constrained geometry catalysts (CGCs) [10, 26]. Steven et al. [26, 27] define a CGC as “a complex in which a π-bonded moiety is linked to one of the other ligands on the same metal centre in such a way that the angle measured between the centroid of the π-system and the additional ligand is smaller than the comparable unbridged complex”. This important class of catalyst was developed by Dow [26–30] and Exxon [31–34] following the discoveries of silane-bridged cyclopentadienyl-amido scandium and titanium catalysts by Bercaw and Okuda [35, 36], respectively. The general form of a CGC is illustrated in Fig. 3.

General form of a constrained geometry catalyst (CGC)
CGC catalysts have outstanding performance in ethylene polymerisation and in copolymerisations with higher α-olefins. The reasons for superior performance in these areas compared with metallocenes are a more open coordination sphere, a small Cpcent-M-N angle and a reduced propensity to undergo chain transfer [26]. The amido group acts as a maximum three-electron donor, cf. cyclopentadienyl (five-electron donor), which yields a highly electrophilic metal centre and thus results in its high productivity as a catalyst. CGCs can be used in high-temperature polymerisation processes because of their high thermal stability and ability to produce higher molecular weight polymers than their metallocene counterparts [37, 38].
Post-metallocene catalysts and the isolobal analogy
Due in part to extensive patenting of the more traditional Group 4 metallocene systems and CGCs, new post-metallocene systems were of great research interest during the 1990s and onwards [9, 11]. Research efforts directed at new types of catalysts also allowed for a chance to improve and control resultant polymer properties and to explore new monomer combinations. Post-metallocene systems aim to replace one or more of the cyclopentadienyl ligands on the metal centre with a different moiety. A strategy that has been successfully employed in new ligand design is the isolobal analogy developed by Hoffman [39] for which he was awarded a Nobel prize alongside Fukui “for their theories, developed independently, concerning the course of chemical reactions”. When two electronically defined systems are isolobal with each other, the symmetry, shape and energies of the frontier orbitals are similar. This principle, when combined with the correct steric properties, can be powerful in ligand design for new catalysts.
Ligands relevant to this project, which have been employed because they fulfil the criteria put forward by the isolobal analogy, consist of an anionic heteroatom binding to the metal centre in a κ1-mode. If one considers the cyclopentadienide ligand and compares its electronic properties to the general form of a κ1-bound anionic donor, for example, an imido ligand, the isolobal analogy can be seen in operation (see Fig. 4). The frontier orbitals of each ligand comprise a single σ-donor and two π-donor orbitals which can interact with the metal centre.

Occupied frontier orbitals of cyclopentadienide and NR2− ligands
Previous work in the Mountford group has utilised this analogy in the synthesis of titanium imido compounds such as Ti(NtBu)(Me3[9]aneN3)Me2 (4, Me3[9]aneN3 = 1,4,7-trimethyl triazacyclononane) or related compounds [13, 40–43]. In this compound, the [NR]2− ligand is isolobal with Cp−, but since it is formally dianionic, the other supporting ligand, for example, a 1,4,7-triazacyclononane, must be neutral to keep the charges balanced in the compound. DFT calculations show that that the frontier molecular orbitals of dicationic imido species [Ti(NMe)(H3[9]aneN3)]2+ (H3[9]aneN3 = 1,4,7-triazacyclononane) are broadly similar to that of [Cp2Ti]2+ in line with the isolobal analogy [44]. For in-depth computational studies on [Cp2M]n+, the reader is directed to a detailed review article [45]. 
Non-CGC half-sandwich catalysts
Group 4 complexes of the form (η5-C5R5)M(L)X2 (M = Group 4 metal, L = supporting ligand, X = halide or alkyl), which are supported by one cyclopentadienyl and one κ1-bound formally monoanionic ligand, have been well studied [9, 11, 46]. The monoanionic ligands act as an isolobal analogue of cyclopentadienide and also possess the same charge; thus, they can be directly substituted without the need to change the other supporting ligand. Prominent examples comprise amide (5) [47–49]-, ketimide (6) [50–54]-, guanidinate (7) [55]-, iminoimidazolidide (8) [56–61]-, phosphinimide (9) [62–70]- and aryloxide (10) [71–76]-supported half-sandwich compounds (Fig. 5). The κ1-bound N-donor ligands will be discussed further below.

Half-sandwich post-metallocene precatalysts
The first half-sandwich ketimide complex of the form CpRM(NCR2)Cl2 (CpR = C5R′5, R′ = H or alkyl, R = alkyl) was reported in 1986 by Leigh et al. [77], namely CpTi{NC(nBu)tBu}Cl2. Since then, a large number of compounds of this type have been reported, and the area has been reviewed [54]. In CpTi{NC(nBu)tBu}Cl2, the coordination of the ketimide to the metal is approximately linear [Ti–N–C, 171.3(4)°] indicating that the nitrogen atom is effectively sp hybridised and suggests the possibility of Ti–N multiple bonding. Another important system where the ketimide-supporting ligand is –NCtBu2 was reported by Dias, Nomura and Zhang [50, 52, 53, 78]. However it was over a decade between the first reported synthesis of the half-sandwich ketimide complexes and their recognition as polyolefin catalysts.
Another important and comprehensively studied ligand is phosphinimide (9, –NPR3, R = alkyl or aryl). Stephan et al. [63] have investigated CpTi(NPR3)X2 (R = alkyl or aryl, X = halide or alkyl) complexes since the 1990s regarding their general reactivity and polymerisation ability. Phosphinimide ligands have been shown to bind in a linear fashion to the metal centre with a short M–N bond [69, 70]. These attributes are evidence of a σ+2π-bonding mode between ligand and metal just like Cp. The steric influence of the phosphinimides have also been shown to be comparable with Cp, and it is argued that the steric influence of the ligand is the most important factor in determining the polymerisation activity in these particular systems [65, 67].
The steric influence of the ligand is also thought to be important in aryloxide-supported half-sandwich systems (10), but in others it appears to be the electronic effects that are considered key to catalyst productivity. Hessen [57], Tamm [59, 60] and Nomura [58] have all investigated iminoimidazolidide ligands (cyclic guanidinates, 8, Fig. 5), and it has been suggested that an increase in π-bonding between the ligand and titanium can lead to an increase in catalytic activity [57–60]. The significant π-bonding in these complexes originates from formal lone pair donation of the amino nitrogens towards the πCN system, which means the ligand can be considered up to a five-electron donor via a σ+2π interaction (Fig. 6). This electronic arrangement is supported experimentally by short Ti–N bond lengths and linear Ti–N–C angles. The activity of complex CpTi{NC(N(Xyl)CH2)2}Bn2 is higher than ketimide-supported CpTi(NCtBu2)Bn2 (1600 and 1128 kg mol−1 h−1 bar−1, respectively) due to the addition of amino nitrogen donors [57].

Metal–ligand bonding interactions for guanidinate-supported complexes (7)

[Tp]− ligand shown alongside donor orbitals of σ- and π-symmetries for a fac-L3 moiety
Other Group 4 post-metallocene catalysts
Many post-metallocene catalysts have also been developed, which do not contain a cyclopentadienyl-supporting ligand [9, 11]. In some instances, they have contained ligands that still aim to imitate Cp, for example, boratobenzene [79–81]- and phosphacyclopentadienyl [82]-supported catalysts 11 and 12. 
Boratobenzenes are formed by the addition of a boron atom into a cyclopentadienide ring. Since boron contributes only an empty p-orbital to the π system, the aromaticity is not disturbed. When activated with MAO, the catalysts give reasonably high activities (105 kg mol−1 h−1 bar−1) [79]. When changing the NiPr2 substituent on boron (11) to less sterically demanding substituents Ph or OEt, there is an increased rate of β-hydrogen elimination resulting in more α-olefin production [79–81]. Phosphacyclopentadienes are obtained by the exchange of a single carbon in a cyclopentadiene ring by a P atom; this maintains the same ring size as that of Cp, as well as the aromaticity. Catalysts supported by these ligands can give productivities similar to their Cp analogues. Tropidinyl ligands have been utilised by Bergman et al. [83] as a Cp mimic in complexes such as 13 despite having separate σ and π systems. An alternative ligand that can be used in place of Cp is the monoanionic tris(pyrazol)borate (Tp) [84–86]. Although this is a face-capping ligand, it is isolobal with Cp−; the relevant σ- and π-orbitals are shown alongside the ligand (14 –) in Fig. 7.
These ligands have been used in the Mountford group as Cp surrogates in production of Group 4 polymerisation catalysts that are supported by Tp and κ1-amidinate ligands [87, 88]. Other Tp-supported Group 4 polyolefin catalysts have also been investigated by other groups [89–92]. In addition, Group 4 non-cyclopentadienyl catalysts supported by κ2 N,N′-amidinate ligands have also been reported [93–96].
Metallocene and post-metallocene alkyl cations
It is generally acknowledged that the active species in Group 4-based olefin polymerisation catalysis is of the form [LnMR]+ (L = supporting ligands, M = Group 4 metal, R = alkyl group). The nature of the active species is of great importance in the fundamental understanding of the mechanism of olefin polymerisation. Once one understands the mechanisms, deactivation reactions and structure–activity relationships, then, in principle, better catalysts can be designed. This section will present a concise introduction into the area of cationic Group 4 metal alkyl complexes and their reactivities. This subject area has been extensively reviewed [4, 8, 97, 98].
Historical background
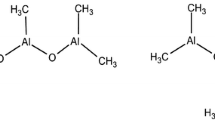
The suggestion of a cationic active species in olefin catalysis occurred not long after initial reports of the polymerisation activity of TiCl4/AlEt3 by Ziegler in 1953. Newburg, Long and Breslow [99, 100] suggested that the active agent was a highly polarised bimetallic species (15) containing Ti and Al bridged by chlorine (Scheme 1).

Insertion of ethylene into a highly polarised heterobimetallic species (15)
Conductivity and electrodialysis studies by Shilov et al. [101, 102] suggested that the catalytically active species in polymerisations is actually cationic. Direct evidence for the involvement of a cationic active species was provided by electrochemical synthesis of [Cp2TiMe]+ in CH2Cl2 carried out by Dyachovskii [103]: polymerisation only occurred only in the cathode chamber. The idea of an ionic active species in a process which was carried out in non-polar solvents was not well accepted at the time. Curiosity was revitalised in 1985 when Eisch et al. [104] provided chemical evidence for the existence of a cationic active species by isolating [Cp2Ti{Me3SiCC(Ph)Me}][AlCl4] (16); this was synthesised by the insertion of the alkyne PhCCSiMe3 into the then postulated [Cp2TiMe]+ (Scheme 2). This type of reaction models the insertion of ethylene into the Ti–C bond.

The first example of an isolable insertion product (16) formed by insertion into the Ti–C bond of a titanium methyl cation
In 1986, both Bochmann and Jordan made important contributions in this area. Jordan et al. [105] reported the synthesis of [Cp2ZrMe(THF)][BPh4] (17) which was structurally characterised and was able to polymerise ethylene. This was the first time that Group 4 cations were shown to polymerise ethylene without alkyl aluminium cocatalysts. Bochmann et al. [106, 107] reported titanocene analogues in the same year, [Cp2TiMe(L)][X] (L = NH3, NCR, pyridine, X = BPh4, PF6); however, the presence of the stronger Lewis bases meant that these complexes showed no activity towards ethylene polymerisation.
Preparation of Group 4 alkyl cations
The possibility of performing polymerisation in the absence of MAO combined with the potential to undertake mechanistic investigations has led to a large amount of research into the different methods of preparing monoalkyl cations. There are a number of general synthetic routes to Group 4 alkyl cations which have been reviewed and will be discussed below [97].
Protonolysis
Ammonium salts have been used to generate alkyl cations via protonolysis of M–R bonds; an example of this is the generation of [Cp2TiMe(NH3)][PF6] from Cp2TiMe2 and [NH4][PF6] reported by Bochmann et al. [106]. Other ammonium salts have been used to this effect such as [PhNMe2H]+ and [nBu3NH]+. Treatment of Cp2ZrMe2 with [PhNMe2H][BF20] ([BF20]− = [B(C6F5)4]−) produces the cation [Cp2ZrMe]+ with a [BF20]− counterion [97, 108]. Using protonolysis to generate the alkyl cation leads to the generation of a Lewis base side product which can bind to the cation. The Lewis base must be removed from the cation to allow binding of the olefin for polymerisation, and therefore the presence of Lewis bases reduces catalytic activity. As an alternative to ammonium salts, phosphonium salts can be used in the same vein [109]. If one employs bulky phosphonium ions such as [Cy3PH]+ and [Mes3PH]+, the resulting phosphine does not coordinate to the generated cation which then leads to a productive catalytic system [109].
Lewis acids
Tris(pentafluorophenyl)borane (“BF15”) is a strong Lewis acid and as such can be used for the activation of Group 4 alkyl-based olefin polymerisation precatalysts. This was employed by Marks et al. [110] in 1991 to synthesise [{1,2-(CH3)2C5H3}2ZrMe][MeBF15] (18) which was the first example of an isolable and crystallographically characterised cation-like “Lewis base-free” species. Activating in this fashion leads to no side products that could interfere with the cation; on the other hand, the anion itself ([MeBF15]−) is known to interact with the cation. If there is an interaction, the complex is best considered as a zwitterionic molecule containing a methyl bridge between the metal and boron. The extent of interaction between the anion and the cation can be determined by the separation of the meta and para 19F NMR chemical shifts [111]. This type of interaction is important to consider since it can obstruct close approach of an olefin monomer to the metal centre during polymerisation, and therefore generally, the presence of this anion leads to an inhibitory effect on the polymerisation.
Alkyl abstraction
The most commonly employed agent used to abstract methyl groups is the trityl cation, [Ph3C]+. Abstraction of the alkyl group (R) from the precatalyst yields the formation of Ph3CR alongside the corresponding cation [97]. This method of activation is extremely widespread, and Eq. 1 shows activation of rac-ethylenebis(indenyl)dimethylzirconium by way of example. If the trityl cation is coupled with an anion such as tetrakis(pentafluorophenyl)borate ([BF20]−), the product is a cation accompanied by a non-interacting anion, which acts as highly active polymerisation catalyst.

Other methods
Early methods of producing cations involved oxidative reagents such as [Cp2Fe]+ (Eq. 2) or Ag+ [112]. Activation of dimethyl zirconocene was carried out by Jordan et al. [113] in 1986 with Ag[BPh4] yielding ethane and silver metal as side products, which occurred via oxidative cleavage of the Zr–Me bond. The titanium(III) species Cp*2TiMe has also been shown to be oxidised by AgBPh4 in THF to yield the monoalkyl titanocenium(IV); this process occurs via a one-electron oxidation [114].

Another method employed to generate cations is displacement of chloride by strong neutral donors, e.g. PMe3 in complexes such as Cp2TiMe(Cl) and Ind2TiMe(Cl) as shown in Eq. 3.

AlMe3 and its effects on alkyl cations and polymerisation
The alkyl aluminium compound Al2Me6 (from now on referred to as AlMe3) is present in polymerisation reactions that use MAO as a co-catalyst. Although this can help the process by scavenging oxygen and water from the reaction, it can also cause complications. For example, AlMe3 can cause variation in catalytic productivity and also changes in the properties of the polymer [115–117]. Activation of zirconocene complexes with AlMe3 can lead to a small amount of activity but are significantly less productive than their MAO-activated analogues [118]. The presence of AlMe3 leads to the formation of heterobimetallic species of the type [LnM(μ-Me2)AlMe2]+ (M = Group 4 metal, L = supporting ligands), which themselves are not catalytically active, and result in chain transfer and can also act to deactivate the active species; they are also considered to be catalytic resting states [8, 119, 120]. Alkyl aluminium compounds can also have a negative effect on the polymerisation performance of Group 4 catalysts by acting as reducing agents; titanium(III) compounds are not electrophilic enough to achieve high polymerisation activities. The more the AlMe3 is present in solution the more the polymerisation inactive heterobimetallic species will be present. Scheme 3 illustrates the relevant processes.

Illustration of propagation, catalyst resting state and chain transfer to aluminium in MAO-activated ethylene polymerisation catalysis. L n M represents a generic metal–ligand fragment; P represents a growing polymeryl chain
The first adduct of AlMe3 with a Group 4 alkyl cation was reported in 1994 by Bochmann et al. ([Cp2Zr(μ-Me2)AlMe2]+, 19, Fig. 8). However, this was not structurally characterised [121]. In 2006, Mountford et al. [122] reported the first cationic transition metal compound containing a M(μ-Me2)AlMe2 moiety characterised by X-ray crystallography (20, Fig. 8).

To reduce the effect of AlMe3 on the polymerisation process, scavengers can be added; one such scavenger is 2,6-di-tert-butyl-4-methylphenol which is more commonly referred to as butylated hydroxytoluene (BHT-H). This phenol reacts with AlMe3 to produce (BHT)2AlMe [123] which does not interfere with the active site but can still scavenge unwanted impurities [124, 125].
Anion and solvent interactions
Interaction of the anion with the cation is important to consider as it can have a substantial effect on the polymerisation process. These types of interaction occur because of a low number of valence electrons at the metal centre. If a significant interaction exists between the anion and cation, and the active site is blocked, the anion must be removed from the active site before polymerisation can take place [126] which has been shown in various metallocene and post-metallocene systems to involve quite substantial energies [127–129]. It has already been discussed above that [MeBF15]− forms a strong interaction with the cationic metal centre to form a zwitterionic molecule. Equally if the anion reacts with the cation to give a different non-active species, then this too has to be circumvented by the use of a different anion. It is also worth noting that anions that are usually considered non-coordinating such as [BF4]− and [PF6]− have been shown to react with [Cp2MR]+, e.g. reaction of [Cp2ZrMe(CH3CN)]+ with [PF6]− produces Cp2ZrMeF via fluoride abstraction [113]. Even anions such a [BPh4]− have been shown to interact with the cationic centres. Treatment of ZrBn4 (Bn = CH2Ph) with dimethylanilinium tetraphenylborate leads to the formation of [ZrBn3(η6-C6H5)BPh3)] (21) in which the cationic centre is stabilised by the anion. This type of interaction has also been demonstrated with {C2H4(Ind)2}ZrMe2 [130]. Hlatky and Turner [108] reported that [BPh4]− can even react with cationic species such as [Cp*2ZrMe]+ to form 22 which is formed as a result of C–H activation. 
In a search of non-interacting and non-coordinating anions perfluorinated species, the most popular of which is [BF20]−, were investigated [130–132]. Indeed in some cases, it has been shown that a molecule of solvent would rather bind to the vacant site of the cation than [BF20]−; this is a testament to the anion’s non-coordinating nature [133–135]. Despite its weakly coordinating nature, there have been reports of reactivity with [BF20]−, for example, by McConville and Stephan [64, 136] yielding 23 and 24, respectively. Compound 23 was formed by the extraction of C6F5
− from [BF20]− by [(tBu3PN)3Ti]+ followed by phosphinimide ligand abstraction and B–C bond cleavage. 
Work by Brintzinger and Geyer in 1999 showed that, using Pulsed Gradient Spin Echo (PGSE) NMR spectroscopy, the compound [Cp2ZrMe][BF20] exists as a dimer in C6D6—in this case, an ion quadruple. Thus, the weakly coordinated anion [BF20]− can interact with cations sufficiently to form higher aggregates [137]. Marks et al. [138] reported using similar PGSE NMR measurements in C6D6 that [Cp*ThMe][BF20] exists as only monomeric tight ion pairs. PGSE reveals that PPh3, THF, benzene and toluene adducts of various zirconocene type systems can exist as ion quadruples or even hextuples in solution at higher concentrations. It is worth noting that the previously mentioned PGSE NMR experiments were carried out at concentrations above that which is usually used in single-site catalysis, and the existence of these aggregates at lower, more relevant, concentrations is more unlikely. Bochmann has carried out extensive research into a series of more complex fluorinated anions based around BF15 which are of the form: [X(BAr3)2]− [X = CN, NH2, N(CN)2; Ar = C6F5, 2-C6F4(C6F5)]. The idea was to create very weakly coordinating anions by delocalising the negative charge over multiple atoms. Accordingly in the case of [CN(BF15)2]−, the contribution of the anion to the activation energy of propene polymerisation with zirconocene catalysts was actually 1.1 kJ mol−1 less than \(\left[ {{\text{BF}}_{ 20} } \right]^{ - }\). These anions can have an effect on both the activity and stereoselectivity during polymerisations [8, 139–143].
The solvents generally used in this chemistry are polar due to the ionic nature of the compounds and have been shown to form adducts with Group 4 cations in order to stabilise the active species. One such example that has been crystallographically characterised is [Cp2ZrBn(C6H5Cl)][BF20] (25) [144]. Other examples of halocarbon solvents bound to transition metals are known [144–147]. It is possible that the solvent-stabilised cations are resting states in the catalytic cycle and that the solvent needs to be removed from the metal centre before binding and subsequent polymerisation of the olefin can occur [4, 126, 148]. The cations in [Cp*ZrMe2][MeBF15] and [{Me2Si(tBuN)(C5Me4)}ZrMe][BF20] are even stabilised by a molecule of hydrocarbon solvent (toluene) in solution and the former also in the solid state [133–135]. 
Non-classical interactions
Agostic interactions have been shown to be important in stabilising highly electrophilic cations [149, 150]. These interactions have also been proposed to play a key role in the polymerisation process. The μ-Me dication [Cp*2Ti2{NC(NiPr2)ArF2}2Me2][BF20]2 (26-[BF 20 ] 2 , Fig. 9) has been characterised by X-ray crystallography and displays two agostic interactions for each methyl group, respectively [151]. [Cp2TiMe][MeBF15] also exhibits α-agostic interactions which were not crystallographically characterised but determined using IR spectroscopy; neutral compounds such as CpTiMe3 and CpTiMeCl2 have also been shown to display α-agostic interactions using the same method [150].

Structurally characterised compound containing α-agostic interactions [151]
Computational studies by Clot and Eisenstein [152] have predicted that the α-agostic interaction is not so much a result of electron donation from the C–H σ-bond into the electron deficient metal centre, although this does occur, but is in fact better viewed as a second order Jahn–Teller distortion. Agostic interactions are identified by a short M∙∙∙HC distance; these occur not primarily because the C–H bond is acting as a base, but because the tilt of the CH3 group allows the σCH3 orbital to pick up an additional stabilising interaction from a different lower energy d-orbital. The extent to which this tilting occurs is a balance of the overlap and energetics involved. This is illustrated in Fig. 10. Tilting of the CH3 group also allows a more efficient σ(C–H) interaction to occur with the metal centre.

Frontier orbitals involved in the formation of an α-agostic interaction
Jordan et al. [153] have reported the β–C–H agostically stabilised species [Cp′2Zr(PMe3)C2H5]+ (Cp′ = C5H4Me) which contains a structurally characterised β–C–H agostic interaction. Green et al. [148] have used pendant phenyl groups to study agostic interactions in Group 4 alkyl cations. Another somewhat more prominent example of a non-classical interaction is that of the so called ipso-interaction which involves the η2-coodination of a benzyl group. An example of such an interaction was reported by Horton et al. [111] in an alkylzirconium complex supported by a tridentate diamide ligand (27). The Ti∙∙∙C
ipso
interaction manifests itself in the 13C NMR spectrum showing a large 1
J
CH for the ZrCH2 group (142 Hz) and an unusually upfield ipso carbon chemical shift (137.5 ppm). 
Agostic interactions are also possible between Si and Me bonds and the metal centre. Compound 16 synthesised by Eisch et al. [104] displays a β-Si–C agostic interaction with the metal centre which is reflected in the upfield 29Si NMR resonance (δ = −54.9 ppm). Bochmann and Macchioni [154] also suggested their systems contained Si–Me agostic interactions which manifested itself as restricted rotation in the NMR spectra of compound [{rac-Me2Si(1-Ind)2}M{CH2SiMe3}]+ (M = Zr or Hf). Likewise Marks et al. [128] also suspected the presence of an agostic interaction in the complex [(C5H3Me2)2Zr{CH2(SiMe3)}]+ which became evident in the low-temperature NMR spectra. Work by Mountford et al. [44, 155] in 2005 showed that treatment of Ti(NtBu)(Me3[9]aneN3)(CH2SiMe3)2 (Me3[9]aneN3 = 1,4,7-trimethyltriazacyclo-nonane) with TBF20 ([Ph3C][B(C6F5)4]) gave the species [Ti(NtBu)(Me3[9]aneN3)(CH2SiMe3)][BF20] in which the metal centre is stabilised by a β-Si–C agostic interaction as characterised by a high field 29Si NMR resonance (−17.5 ppm).
Reactions of alkyl cations
So far this tutorial review has centred on the formation and solution behaviour of Group 4 alkyl cations. This section gives a brief introduction into the types of reactivity that have been observed with these species. Reactivity of base-free cations is not only interesting from a fundamental point of view but can also help reveal mechanistic aspects of the polymerisation process [156].
Insertion chemistry of alkyl cations
The study of the insertion of unsaturated substrates into the metal alkyl bond is applicable to the understanding of the chain growth processes present in Ziegler–Natta polymerisation. As mentioned previously, in 1985 Eisch et al. showed that insertion of Me3SiCCPh into the Ti–Me bond resulted in the formation of alkenyl complex 16. Shortly after this discovery Bochmann et al. [106, 107] showed that base-stabilised cation [Cp2TiMe(NCMe)]+ would react with CO to yield complex 28 and with tBuNC to yield complex 29. 
Treatment of [Cp2TiMe(NCMe)]+ with nitriles PhCN and tBuCN produced a Lewis base adduct. However, with indenyl-supporting ligands and in the presence of excess nitrile, both PhCN and tBuCN undergo insertion into the Ti–Me bond within 1 h; this increase in reactivity is explained by the increased donor ability of indenyl vs. Cp. Insertion of MeCN and PhCN into the Ti–Me bond of [Cp2TiMe][BPh4] can occur but a longer reaction time is observed. For example, acetonitrile takes 2 weeks to react with the cation [106, 107, 157]. In 2006, Mountford et al. [44] showed that imido complex [Ti(NtBu)(Me3[9]aneN3)(NCMe2)(NCMe)]+, produced initially from the insertion of acetonitrile into the Ti–Me bond of [Ti(NtBu)(Me3[9]aneN3)Me]+, would rearrange to form a β-diketiminato complex (30) over 1 month. Insertion chemistry equivalent to that described above was exhibited by the analogous base-stabilised zirconocene cation 17 + by Jordan et al. [98, 113, 158]. This reactivity is illustrated in Scheme 4.

Reactions of 17 + with unsaturated substrates. [BPh4]− omitted
Donor ligands (e.g. ethylene or nitrile) bond to [Cp2TiMe]+ by interacting with the metal’s d σ orbital [159]. In the case of nitriles, this complex is stable and isolable, whereas with ethylene, this is not the case (with electron-poor metals). The metal–nitrile complexes consequently experience a more significant activation barrier to migratory insertion than the unstable metal–ethylene complex. This crucially results in a much faster insertion into the metal alkyl bond for ethylene than nitriles and thus facile polymerisation of the ethylene. These nitrile reactions, therefore, help show that it is important to have an unstable metal–alkene intermediate in order for polymerisation of unsaturated species to occur. Post-metallocene species have also had their insertion chemistry studied. Stephan et al. [68, 160] have shown their ubiquitous phosphinimide-supported titanium and zirconium complexes will undergo insertion chemistry with carbodiimides and alkynes to give κ2-amidinate and alkenyl complexes, respectively; an example of a carbodiimide insertion to form 31 is shown in Eq. 4.

The insertion of alkenes into M–C bonds of Group 4 cations has also been investigated. In 1994, Pellacchia et al. reported the synthesis of [Zr(Bn)2(CH2CHMeCH2Ph)][PhCH2B(C6F5)3)] by the insertion of propene into the Zr–C bond of [ZrBn3{(η6-PhCH2)B(C6F5)3}]. Compound 32 was also synthesised by the insertion of propene into the Zr–C bond of [Cp*ZrBn2]+ and was isolated and characterised by NMR spectroscopy and shown to contain a back-biting Ph–Zr interaction which acts to stabilise the cationic metal centre; this compound was later characterised by X-ray crystallography. The Ph–Zr interaction results in a stability in the complex towards both β-hydrogen elimination and further propene insertion. Other higher olefin insertion products could also be isolated and characterised [161]. 
Horton et al. reported the products of alkene insertion into the Zr–CH2Ph bond of diamide-supported complex [{Me2Si(NCMe3)2}ZrBn][BF20]. Reaction with 1-hexene produced the isolable product 33; the insertions of ethylene and propylene could be observed on the NMR tube scale but the products could not be isolated. Compound 33 was also found to be stabilised by a Ph–Zr interaction as evident in the 13C NMR. An interesting point to note is that the alkenes only reacted with the monomeric catalyst, and the dimeric catalyst persisted in solution [162]. Baird and Budzelaar [163–165] have also published papers in the past few years regarding the formation of titanium alkyl species resulting from alkene insertion into the Ti–Me bond.
Insertion of 3,3-dimethyl-1-butene (DMB) into the Ti–Me bond of the solvent-stabilised monomethyl cation [Cp2TiMe(CD2Cl2)]+ results in the formation of complex 34 (Fig. 11), which can be characterised by NMR spectroscopy at 195–205 K. Compound 34 is stabilised by an α-agostic interaction which is reflected in the 7-ppm separation of the α-CH2 protons and in the geminal coupling constant. Upon standing, 34 appears to undergo β-hydrogen elimination to give [Cp2TiH]+ which then inserts excess DMB to yield 35 which exists as a β-agostic-stabilised species in fast exchange with its non-agostic analogue [165]. A similar β-agostic-stabilised species is also formed from the reaction of [Cp2TiMe(CD2Cl2)]+ with 2,4-dimethyl-1-pentene (DMP). In this reaction, Cp2TiMe2 is treated with [Ph3C][BF20] (TBF20) in the first step resulting in a mixture of species which are assigned as [Cp2TiMe(CD2Cl2)][BF20], [Cp2TiMe][BF20] and [(Cp2TiMe)2(μ-Me)][BF20]. On addition of alkene, DMP only [Cp2TiMe(CD2Cl2)][BF20] reacted; [Cp2TiMe][BF20] did not react with DMP or did it re-equilibrate to form [Cp2TiMe(CD2Cl2)][BF20]. In the publication, it does note that a reviewer is not compelled by the existence of both [Cp2TiMe(CD2Cl2)][BF20] and [Cp2TiMe][BF20] and suggested the existence of a dimeric species [164].

Cations 34 and 35 stabilised by agostic interactions
Lewis acid–base chemistry
Alkyl cations are well known to form adducts with Lewis bases as previously mentioned, and the early examples of alkyl cations were isolated as adducts as previously discussed. Examples of Lewis bases used in this chemistry include RCN, PR3, THF, pyridine, NR3. Stronger donors such as PR3 will replace moderately labile donors such as MeCN, but THF, being a weaker donor, will not [106]. The number of Lewis base donors that can bond to the metal centre is dependent on the radius of the metal. For example, titanium can form 16 valence electron complexes with a single Lewis base, whereas zirconium can fit another ligand if small donor ligands are used and thus form 18 valence electron cations, e.g. [Cp2ZrMe(PMe3)2]+ [98, 166].
Bimetallic olefin polymerisation catalysts
Since the early 1990s, multimetallic catalysts have been explored in olefin polymerisation. Inspired by the role proximate metal centres play in increasing localised reagent concentration within enzymes, multimetallic species have been introduced to enhance several chemical transformations such as the Diels–Alder [167] and Stecker [168] reactions [169, 170]. In olefin polymerisation, a wide range of bimetallic catalysts have been employed to try and find any cooperative effect that would arise from the complimentary interaction of the polymeric chain between the adjacent metal centres. It took until the turn of the century for any beneficial properties to be observed, most notably, a higher molecular weight and superior chain branching. A particularly successful and readily studied family of bimetallic catalysts are the covalently tethered constrained geometry catalysts: for instance, the ansa-amido-monocyclopentadienyl Ti and Zr catalysts introduced by Li and Marks [171–173] (Fig. 12). This area has been comprehensively reviewed recently, and the reader is referred to this article for further details [169].

Covalently tethered Group 4 bimetallic constrained geometry catalysts (M = Ti or Zr) [170]
A study showed that 36 (Fig. 12) increases product polymer molecular weight by 130 times compared to its mononuclear analogue 37, and that there are 13 and three times more ethyl branches per polymer chain than their mononuclear analogue for catalysts 36 and 38, respectively. A stabilising agostic interaction between the propagating polymeric chain and the proximate metal centre results in a higher barrier to termination, thus generating a polymer with a higher molecular weight [171]. A computational study was carried out by Fragala and co-workers on catalyst 38 where n-octyl was used to represent a propagating chain and n-propyl was used to represent an oligomeric chain. The insertion and chain transfer transition states were analysed, and similar energetic values were seen for ethylene insertion in both cases. A consistent destabilisation (2 kcal mol−1) was seen for the β-hydrogen chain transfer of catalyst 38 with an n-octyl chain, attributable to steric repulsions resulting from the constraints from the stabilising agostic interaction (Fig. 13). Molecular weight is proportional to the ratio of the rates of propagation to termination of the reaction, and therefore, the reduced termination corresponds to an increase in molecular weight [174].

Reaction pathway describing the propagation and termination routes in ethylene homopolymerisation. Zr 1 and Zr 2 represent catalysts 37 and 38, respectively [174]
The increase in branching is proposed to arise from the recapture of the vinylic end of the polymer at the neighbouring metal centre after chain transfer. This process aids an intramolecular re-insertion into the growing polymeric chain. The computational study, introduced above, also refined the geometrical orientation of an oligomeric π-bonded vinyl terminated oligoethylene chain; a non-negligible (2 kcal mol−1) stabilisation energy is observed with the second metal site, a pre-requisite for ethyl branching [174].
A secondary interaction is also seen when alternative monomers are activated. Ethylene/hexene and ethylene/styrene copolymerisations using the titanium analogue of 38 show a higher α-olefin incorporation. In the case of the styrene homopolymerisation, the binuclear catalysts show a significantly higher yield and activity than their mononuclear analogue. In the mononuclear counterpart, the styrene 2,1 insertion product results in the deactivation of the metal centre. However, when using the bimetallic species, 1,2 and 2,1 insertion compete equally. This suggests an unusual insertion regiochemistry, which the authors suggest is a result of a favourable interaction with the proximate metal centre as is depicted in Fig. 14 [175, 176].

Proposed competing regiochemistry forms in the homopolymerisation of styrene [175]
Another explored capacity of the bimetallic catalysts is their use in copolymerisation of severely encumbered isoalkenes without ring opening or rearrangements. Methylenecyclopentane, methylenecyclohexane and 2-methyl-2-butene are all incorporated into the polyethylene backbone under mild conditions with catalysts 38 and 36. The presence of bulky cycloalkane rings along the polymeric chain frustrates the tendency to coil, altering the physical properties of the product polymer [173].
Although so far only one family of Group 4 bimetallic catalysts have been described, a wide range of covalently tethered alternatives have been investigated with varying successes. Phenylene [177–179]-, polymethylene [180–182]-, and silyl [183, 184]-bridging moieties have all been shown to have some cooperative effects resulting in increased molecular weight or improved comonomer incorporation. Configurationally restrained ligands give an alternative to their flexible counterparts by maintaining proximate metal centres. This was effectively shown by Noh and co-workers [185] who introduced a doubly bridged ligand (39, Fig. 15), which produces a higher activity, molecular weight and tacticity in styrene polymerisation, and later by Marks [186] with the use of a napthoxydiiminato ligand (40, Fig. 15).

In 2002, Marks and co-workers demonstrated that using both a titanium and a zirconium mononuclear ansa-monocyclopentadienyl-amido catalyst in situ results in a bimodal GPC (gel permeation chromatography) plot, which corresponds to the two separate active sites. When the bis(borate) cocatalyst [CPh3]2[(C6F5)3B(1,4-C6F4)B(C6F5)3] is employed, only a single site is seen, synonymous with the predominance of a single homogeneous catalytic species [187]. Other heterobimetallic species, using both early and late transition metals, have been synthesised in order to increase the efficiency of “tandem catalysis”, where the polymer properties produced by different metal centres are incorporated into the same polymer product [188–191].
Concluding remarks
This tutorial review has provided an introduction to homogenous Group 4 olefin polymerisation catalysts, starting from the early days of metallocene chemistry, to the so-called post-metallocene systems. It has shown how electron-deficient cations can be generated and trapped, and highlighted a number of key roles of these complexes in catalytic systems. Finally, an introduction to bimetallic catalysts was provided, which suggests one of the way forward strategies to be adopted in the future for developing new catalysts with improved performance.
References
Ziegler K, Holzkamp E, Breil H, Martin H (1955) The Mulheim normal pressure polyethylene process. Angew Chem Int Ed 67:541–547
Wilke G (2003) Fifty years of Ziegler catalysts: consequences and development of an invention. Angew Chem Int Ed 42:5000–5008
Natta G (1956) Stereospecific catalyses and isotactic polymers. Angew Chem Int Ed 68:393–403
Bochmann M (1996) Cationic group 4 metallocene complexes and their role in polymerisation catalysis: the chemistry of well defined Ziegler catalysts. J Chem Soc, Dalton Trans 3:255–270
Kaminsky W (1998) Highly active metallocene catalysts for olefin polymerization. J Chem Soc, Dalton Trans 9:1413–1418
Coates GW (2002) Polymerization catalysis at the millennium: frontiers in stereoselective, metal-catalyzed polymerization. J Chem Soc, Dalton Trans 4:467–475
Brintzinger HH, Fischer D, Mülhaupt R, Rieger B, Waymouth RM (1995) Stereospecific olefin polymerization with chiral metallocene catalysts. Angew Chem Int Ed Engl 34:1143–1170
Bochmann M (2004) Kinetic and mechanistic aspects of metallocene polymerisation catalysts. J Organomet Chem 689:3982–3998
Britovsek GJP, Gibson VC, Wass DF (1999) The search for new-generation olefin polymerization catalysts: life beyond metallocenes. Angew Chem Int Ed 38:428–447
McKnight AL, Waymouth RM (1998) Group 4 ansa-cyclopentadienyl-amido catalysts for olefin polymerization. Chem Rev 98:2587–2598
Gibson VC, Spitzmesser SK (2003) Advances in non-metallocene olefin polymerization catalysis. Chem Rev 103:283–315
Mitani M, Saito J, Ishii S-I, Nakayama Y, Makio H, Matsukawa N, Matsui S, Mohri J-I, Furuyama R, Terao H, Bando H, Tanaka H, Fujita T (2004) FI catalysts: new olefin polymerization catalysts for the creation of value-added polymers. Chem Rec 4:137–158
Bolton PD, Mountford P (2005) Transition metal imido compounds as Ziegler–Natta olefin polymerisation catalysts. Adv Synth Catal 347:355–366
Baier MC, Zuideveld M, Mecking S (2014) Post-metallocenes in the industrial production of polyolefins. Angew Chem Int Ed 53:2–25
Wilkinson G, Pauson PL, Birmingham JM, Cotton FA (1953) Bis-cyclopentadienyl derivatives of some transition elements. J Am Chem Soc 75:1011–1012
Breslow DS, Newburg NR (1957) Bis-(cyclopentadienyl)-titanium dichloride—alkylaluminum complexls as catalysts for the polymerization of ethylene. J Am Chem Soc 79:5072–5073
Natta G, Pino P, Mazzanti G, Giannini U (1957) A crystallizable organometallic complex containing titanium and aluminum. J Am Chem Soc 79:2975–2976
Reichert KH, Meyer KR (1973) Zur kinetik der niederdruckpolymerisation von äthylen mit löslichen Ziegler-katalysatoren. Makromol Chem 169:163–176
Long WP, Breslow DS (1975) Der einfluß von wasser auf die katalytische aktivität von bis(π-cyclopentadienyl)titandichlorid-dimethylaluminiumchlorid zur polymerisation von äthylen. Justus Liebigs Annalen der Chemie 1975:463–469
Andresen A, Cordes H-G, Herwig J, Kaminsky W, Merck A, Mottweiler R, Pein J, Sinn H, Vollmer H-J (1976) Halogen-free soluble ziegler catalysts for the polymerization of ethylene. Control of molecular weight by choice of temperature. Angew Chem Int Ed Engl 15:630–632
Sinn H, Kaminsky W, Stone FGA, Robert W (1980) Ziegler–Natta catalysis. Adv Organomet Chem, Academic Press, vol 18, pp 99–149
Sinn H, Kaminsky W, Vollmer H-J, Woldt R (1980) “Living polymers” on polymerization with extremely productive Ziegler catalysts. Angew Chem Int Ed Engl 19:390–392
Kaminsky W, Külper K, Brintzinger HH, Wild F (1985) Polymerization of propene and butene with a chiral zirconocene and methylalumoxane as cocatalyst. Angew Chem Int Ed Engl 24:507–508
Ewen JA, Jones RL, Razavi A, Ferrara JD (1988) Syndiospecific propylene polymerizations with Group IVB metallocenes. J Am Chem Soc 110:6255–6256
Coates GW, Waymouth RM (1995) Oscillating stereocontrol—a strategy for the synthesis of thermoplastic elastomeric polypropylene. Science 267:217–219
Braunschweig H, Breitling FM (2006) Constrained geometry complexes—synthesis and applications. Coord Chem Rev 250:2691–2720
Stevens JC, Timmers FJ, Wilson DR, Schmidt GF, Nickias PN, Rosen RK, Knight GW, Lai SY (1991) Constrained geometry addition polymerization catalysts, processes for their preparation, precursors therefore, methods of use, and novel polymers formed therewith. Dow Chemical Co., EP416815A2
Stevens JC, Neithamer DR (1991) Cyclopentaedienyl metal complex compounds, process for preparation and their use as polymerization catalysts. Dow Chemical Co., EP418044A2
Rosen RK, Nickias PN, Devore DD, Timmers FJ (1993) Addition polymerization catalysts comprising reduced oxidation state metal complexes. Dow Chemical Co., WO9319104A1
Rosen RK, Nickias PN, Devore DD, Stevens JC, Timmers FJ (1994) Addition polymerization process using stabilized reduced metal catalysts. Dow Chemical Co., US5374696A
Canich JAM (1991) Olefin polymerization catalysts. Exxon Chemical Patents Inc., EP420436A1
Canich JAM (1991) Process for producing crystalline poly(alpha-olefins) with a monocyclopentadienyl transition metal catalyst system. Exxon Chemical Patents Inc., US5026798A
Canich JAM (1992) Monocyclopentadienyl transition metal compounds as olefin polymerization catalysts. Exxon Chemical Patents Inc., US5096867A
Canich JAM, Turner HW, Hlatky GG (1997) Monocyclopentadienyl metal compounds for ethylene/alpha-olefin copolymerization catalysts. Exxon Chemical Patents Inc., US5621126A
Shapiro PJ, Bunel E, Schaefer WP, Bercaw JE (1990) Scandium complex [{η5-C5Me4)Me2Si(η1-NCMe3)}(PMe3)ScH]2: a unique example of a single-component alpha-olefin polymerization catalyst. Organometallics 9:867–869
Okuda J (1990) Functionalized cyclopentadienyl ligands. IV. Synthesis and complexation of linked cyclopentadienyl-amido ligands. Chem Ber 123:1649–1651
Sinnema P-J, van der Veen L, Spek AL, Veldman N, Teuben JH (1997) Titanium dichloro, bis(carbyl), aryne, and alkylidene complexes stabilized by linked cyclopentadienyl-amido auxiliary ligands. Organometallics 16:4245–4247
Millward DB, Cole AP, Waymouth RM (2000) Catalytic carboalumination of olefins with cyclopentadienylamidotitanium dichloride complexes. Organometallics 19:1870–1878
Hoffmann R (1982) Building bridges between inorganic and organic-chemistry (Nobel lecture). Angew Chem Int Ed 21:711–724
Wilson PJ, Blake AJ, Mountford P, Schröder M (1998) Syntheses and structures of a new class of aza-and thio-ether macrocyclic d0 imido complexes. Chem Commun 9:1007–1008
Bigmore HR, Zuideveld M, Kowalczyk RM, Cowley AR, Kranenbrug M, McInnes EJL, Mountford P (2006) Synthesis, structures, and olefin polymerization capability of vanadium(4+) imido compounds with fac-N3 donor ligands. Inorg Chem 45:6411–6423
Adams N, Arts HJ, Bolton PD, Cowell D, Dubberley SR, Friederichs N, Grant CM, Kranenburg M, Sealey AJ, Wang B, Wilson PJ, Zuideveld M, Blake AJ, Schroeder M, Mountford P (2006) Imido titanium ethylene polymerization catalysts containing triazacyclic ligands. Organometallics 25:3888–3903
Bigmore HR, Dubberley SR, Kranenburg M, Lawrence SC, Sealey AJ, Selby JD, Zuideveld MA, Cowley AR, Mountford P (2006) A remarkable inversion of structure-activity dependence on imido N-substituents with varying co-ligand topology and the synthesis of a new borate-free zwitterionic polymerisation catalyst. Chem Commun 4:436–438
Bolton PD, Clot E, Adams N, Dubberley SR, Cowley AR, Mountford P (2006) Experimental and DFT studies of cationic imido titanium alkyls: agostic interactions and C–H bond and solvent activation reactions of isolobal analogues of Group 4 metallocenium cations. Organometallics 25:2806–2825
Green JC (1998) Bent metallocenes revisited. Chem Soc Rev 27:263–272
Nomura K, Liu J (2011) Half-titanocenes for precise olefin polymerisation: effects of ligand substituents and some mechanistic aspects. Dalton Trans 40:7666–7682
Sinnema PJ, Spaniol TP, Okuda J (2000) Non-bridged amido cyclopentadienyl complexes of titanium: synthesis, characterization, and olefin polymerization catalysis. J Organomet Chem 598:179–181
Nomura K, Fujii K (2002) Synthesis of nonbridged (anilide)(cyclopentadienyl)titanium(iv) complexes of the type Cp’TiCl2[N(2,6-Me2C6H3)(R)] and their use in catalysis for olefin polymerization. Organometallics 21:3042–3049
Nomura K, Fujii K (2003) Effect of cyclopentadienyl and amide fragment in olefin polymerization by nonbridged (amide)(cyclopentadienyl)titanium(IV) complexes of the type Cp’TiCl2[N(R1)R2]. Cp’TiCl2[N(R1)R2]-methylaluminoxane (MAO) catalyst systems. Macromolecules 36:2633–2641
Zhang S, Piers WE, Gao X, Parvez M (2000) The mechanism of methane elimination in B(C6F5)3-initiated monocyclopentadienyl-ketimide titanium and related olefin polymerization catalysts. J Am Chem Soc 122:5499–5509
Henderson KW, Hind A, Kennedy AR, McKeown AE, Mulvey RE (2002) Synthesis of zirconocene amides and ketimides and an investigation into their ethylene polymerization activity. J Organomet Chem 656:63–70
Dias AR, Duarte MT, Fernandes AC, Fernandes S, Marques MM, Martins AM, da Silva JF, Rodrigues SS (2004) Titanium ketimide complexes as alpha-olefin homo- and copolymerisation catalysts. J Organomet Chem 689:203–213
Nomura K, Fujita K, Fujiki M (2004) Olefin polymerization by (cyclopentadienyl)(ketimide)titanium(IV) complexes of the type, Cp’TiCl2(N=CtBu2)-methylaluminoxane (MAO) catalyst systems. J Mol Catal A: Chem 220:133–144
Ferreira MJ, Martins AM (2006) Group IV ketimide complexes: synthesis, reactivity and catalytic applications. Coord Chem Rev 250:118–132
van Doremaele GHJ, Zuideveld MA, Quiroga NVF, Leblanc A (2011) Catalyst component having a guanidinate ligand for the polymerization of olefins. LANXESS Elastomers, WO2011054927A1
Kretschmer WP (2004) Olefin polymerization catalyst component and catalyst system and polymerization process using such a catalyst system. Stichting Dutch Polymer Institute, US20040192541A1
Kretschmer WP, Dijkhuis C, Meetsma A, Hessen B, Teuben JH (2002) A highly efficient titanium-based olefin polymerisation catalyst with a monoanionic iminoimidazolidide π-donor ancillary ligand. Chem Commun 6:608–609
Nomura K, Fukuda H, Katao S, Fujiki M, Kim HJ, Kim D-H, Zhang S (2011) Effect of ligand substituents in olefin polymerisation by half-sandwich titanium complexes containing monoanionic iminoimidazolidide ligands-MAO catalyst systems. Dalton Trans 40:7842–7849
Tamm M, Randoll S, Bannenberg T, Herdtweck E (2004) Titanium complexes with imidazolin-2-iminato ligands. Chem Commun 7:876–877
Tamm M, Randoll S, Herdtweck E, Kleigrewe N, Kehr G, Erker G, Rieger B (2006) Imidazolin-2-iminato titanium complexes: synthesis, structure and use in ethylene polymerization catalysis. Dalton Trans 3:459–467
Nomura K, Fukuda H, Apisuk W, Trambitas AG, Kitiyanan B, Tamm M (2012) Ethylene copolymerization by half-titanocenes containing imidazolin-2-iminato ligands–MAO catalyst systems. J Mol Catal A: Chem 363–364:501–511
Stephan DW, Guérin F, Spence REVH, Koch L, Gao X, Brown SJ, Swabey JW, Wang Q, Xu W, Zoricak P, Harrison DG (1999) Remarkably active non-metallocene ethylene polymerization catalysts. Organometallics 18:2046–2048
Stephan DW, Stewart JC, Guérin F, Spence REVH, Xu W, Harrison DG (1999) Phosphinimides as a steric equivalent to cyclopentadienyl: an approach to ethylene polymerization catalyst design. Organometallics 18:1116–1118
Guerin F, Stewart JC, Beddie C, Stephan DW (2000) Synthesis, structure, and reactivity of the phosphinimide complexes (tBu3PN)nMX4-n (M=Ti, Zr). Organometallics 19:2994–3000
Yue NLS, Stephan DW (2001) Phosphinimide ligands: new bulky ligands for ethylene polymerization catalysts. Organometallics 20:2303–2308
Yue N, Hollink E, Guerin F, Stephan DW (2001) Zirconium phosphinimide complexes: synthesis, structure, and deactivation pathways in ethylene polymerization catalysis. Organometallics 20:4424–4433
Stephan DW, Stewart JC, Guerin F, Courtenay S, Kickham J, Hollink E, Beddie C, Hoskin A, Graham T, Wei P, Spence REVH, Xu W, Koch L, Gao X, Harrison DG (2003) An approach to catalyst design: cyclopentadienyl-titanium phosphinimide complexes in ethylene polymerization. Organometallics 22:1937–1947
Hollink E, Wei P, Stephan DW (2004) The effects of activators on zirconium phosphinimide ethylene polymerization catalysts. Organometallics 23:1562–1569
Dehnicke K, Weller F (1997) Phosphorane iminato complexes of main group elements. Coord Chem Rev 158:103–169
Dehnicke K, Krieger M, Massa W (1999) Phosphoraneiminato complexes of transition metals. Coord Chem Rev 182:19–65
Nomura K, Naga N, Miki M, Yanagi K, Imai A (1998) Synthesis of various nonbridged titanium(IV) cyclopentadienyl-aryloxy complexes of the type CpTi(OAr)X2 and their use in the catalysis of alkene polymerization. Important roles of substituents on both aryloxy and cyclopentadienyl groups. Organometallics 17:2152–2154
Nomura K, Naga N, Miki M, Yanagi K (1998) Olefin polymerization by (cyclopentadienyl)(aryloxy)titanium(IV) complexes-cocatalyst systems. Macromolecules 31:7588–7597
Nomura K, Tsubota M, Fujiki M (2003) Efficient ethylene/norbornene copolymerization by (aryloxo)(indenyl)titanium(IV) complexes-MAO catalyst system. Macromolecules 36:3797–3799
Qian Y, Zhang H, Qian X, Huang J, Shen C (2003) Syndiospecific polymerization of styrene catalyzed in situ by alkoxyl substituted half-sandwich titanocene and BF3·Et2O. J Mol Catal A: Chem 192:25–33
Fenwick AE, Phomphrai K, Thorn MG, Vilardo JS, Trefun CA, Hanna B, Fanwick PE, Rothwell IP (2004) Formation of neutral and cationic methyl derivatives of titanium containing cyclopentadienyl and aryloxide ancillary ligation. Organometallics 23:2146–2156
Wang W, Tanaka T, Tsubota M, Fujiki M, Yamanaka S, Nomura K (2005) Effect of cyclopentadienyl fragment in copolymerization of ethylene with cyclic olefins catalyzed by non-bridged (aryloxy)(cyclopentadienyl)titanium(IV) complexes. Adv Synth Catal 347:433–446
Latham IA, Leigh GJ, Huttner G, Jibril I (1986) The chemistry of dinitrogen residues. Part 1. Diazenido-complexes of titanium, and some substituted analogues. X-Ray crystal structures of [Ti(C5H5)Cl2(NNPh)], [Ti(C5H5)Cl2(NCBunBut)], and [Ti(C5H5)Cl2(NPPh3)]. J Chem Soc, Dalton Trans 377–383
Nomura K, Fujita K, Fujiki M (2004) Effects of cyclopentadienyl fragment in ethylene, 1-hexene, and styrene polymerizations catalyzed by half-titanocenes containing ketimide ligand of the type, Cp’TiCl2(N=CtBu2). Catal Commun 5:413–417
Bazan GC, Rodriguez G, Ashe AJ, Al-Ahmad S, Müller C (1996) Aminoboratabenzene derivatives of zirconium: a new class of olefin polymerization catalysts. J Am Chem Soc 118:2291–2292
Lee RA, Lachicotte RJ, Bazan GC (1998) Zirconium complexes of 9-phenyl-9-borataanthracene. Synthesis, structural characterization, and reactivity. J Am Chem Soc 120:6037–6046
Bazan GC, Rodriguez G, Ashe AJ, Al-Ahmad S, Kampf JW (1997) (Phenylboratabenzene)zirconium complexes: tuning the reactivity of an olefin polymerization catalyst. Organometallics 16:2492–2494
Janiak C, Lange KCH, Versteeg U, Lentz D, Budzelaar PHM (1996) Ethene polymerization activity and coordination gap aperture in non-ansa alkyl-substituted cyclopentadienyl- and phospholyl-zirconium/MAO catalysts. Chem Ber 129:1517–1529
Skoog SJ, Mateo C, Lavoie GG, Hollander FJ, Bergman RG (2000) Synthesis of novel Group 4 complexes bearing the tropidinyl ligand: investigations of dynamic behavior, reactivity, and catalytic olefin polymerization. Organometallics 19:1406–1421
Trofimenko S (1966) Boron-pyrazole chemistry. J Am Chem Soc 88:1842–1844
Trofimenko S (1967) Transition metal poly(1-pyrazolyl)borates containing other ligands. J Am Chem Soc 89:3904–3905
Trofimenko S (1967) Boron-pyrazole chemistry. IV. Carbon- and boron-substituted poly[(1-pyrazolyl) borates]. J Am Chem Soc 89:6288–6294
Bigmore H (2007) DPhil Thesis. University of Oxford
Clark L (2008) Part II Thesis, Final Honours School of Chemistry, University of Oxford
Nakazawa H, Ikai S, Imaoka K, Kai Y, Yano T (1998) Polymerization of olefins with titanium and zirconium complexes containing hydrotris(pyrazolyl)borate or hydrotris(3,5-dimethylpyrazolyl)borate. J Mol Catal A: Chem 132:33–41
Gil MP, Dos SJHZ, Casagrande OL Jr (2001) Copolymerization of ethylene with 1-hexene using sterically hindered tris(pyrazolyl)borate titanium (IV) compounds. Macromol Chem Phys 202:319–324
Gil MP, Casagrande OL (2004) Titanium and zirconium complexes containing sterically hindered hydrotris(pyrazolyl)borate ligands: synthesis, structural characterization, and ethylene polymerization studies. J Organomet Chem 689:286–292
Gil MP, Dos SJHZ, Casagrande OL (2004) Polymerization of ethylene by the tris(pyrazolyl)borate titanium (IV) compound immobilized on MAO-modified silicas. J Mol Catal A: Chem 209:163–169
Volkis V, Averbuj C, Eisen MS (2007) Reactivity of Group 4 benzamidinate complexes towards mono- and bis-substituted silanes and 1,5-hexadiene. J Organomet Chem 692:1940–1950
Elkin T, Aharonovich S, Botoshansky M, Eisen MS (2012) Synthesis and characterization of Group 4 fluorinated bis(amidinates) and their reactivity in the formation of elastomeric polypropylene. Organometallics 31:7404–7414
Elkin T, Kulkarni NV, Tumanskii B, Botoshansky M, Shimon LJW, Eisen MS (2013) Synthesis and structure of Group 4 symmetric amidinate complexes and their reactivity in the polymerization of α-olefins. Organometallics 32:6337–6352
Kulkarni NV, Elkin T, Tumaniskii B, Botoshansky M, Shimon LJW, Eisen MS (2014) Asymmetric bis(formamidinate) Group 4 bomplexes: synthesis, structure and their reactivity in the polymerization of α-olefins. Organometallics 33:3119–3136
Chen EY-X, Marks TJ (2000) Cocatalysts for metal-catalyzed olefin polymerization: activators, activation processes, and structure-activity relationships. Chem Rev 100:1391–1434
Jordan RF, Stone FGA, Robert W (1991) Chemistry of cationic dicyclopentadienyl Group 4 metal-alkyl complexes. Adv Organomet Chem, Academic Press, vol 32, pp 325–387
Long WP, Breslow DS (1960) Polymerization of ethylene with bis-(cyclopentadienyl)-titanium dichloride and diethylaluminum chloride. J Am Chem Soc 82:1953–1957
Breslow DS, Newburg NR (1959) Bis-(cyclopentadienyl)-titanium dichloride-alkylaluminum complexes as soluble catalysts for the polymerization of ethylene. J Am Chem Soc 81:81–86
Zefirova AK, Shilov AE (1961) Kinetics and mechanism of reaction of aluminum alkyls with titanium halides. Dokl Akad Nauk 136:599–602
Dyachkovskii FS, Shilova AK, Shilov AE (1967) Role of free ions in reactions of olefins with soluble complex catalysts. J Polym Sci, Polym Symp No. 16: 2333–2339
Dyachkovskii FS (1975) In: Chien JCW (ed) Coordination polymerisation
Eisch JJ, Piotrowski AM, Brownstein SK, Gabe EJ, Lee FL (1985) Direct observation of the initial insertion of an unsaturated hydrocarbon into the titanium carbon bond of the soluble Ziegler polymerization catalyst, Cp2TiCl2–MeAlCl2. J Am Chem Soc 107:7219–7221
Jordan RF, Bajgur CS, Willett R, Scott B (1986) Ethylene polymerization by a cationic dicyclopentadienylzirconium(IV) alkyl complex. J Am Chem Soc 108:7410–7411
Bochmann M, Wilson LM, Hursthouse MB, Short RL (1987) Cationic alkylbis(cyclopentadienyl)titanium complexes. Synthesis, reactions with carbon monoxide and tert-butyl isocyanide, and the structure of [Cp2Ti[.eta.2-C(Me)NBu-tert](CNBu-tert)]BPh4.MeCN. Organometallics 6:2556–2563
Bochmann M, Wilson LM (1986) Synthesis and insertion reactions of cationic alkylbis(cyclopentadienyl)titanium complexes. J Chem Soc, Chem Commun 21:1610–1611
Hlatky GG, Turner HW, Eckman RR (1989) Ionic, base-free zirconocene catalysts for ethylene polymerization. J Am Chem Soc 111:2728–2729
McCahill JSJ, Welch GC, Stephan DW (2009) Sterically hindered phosphine and phosphonium-based activators and additives for olefin polymerization. Dalton Trans 40:8555–8561
Yang X, Stern CL, Marks TJ (1991) “Cation-like” homogeneous olefin polymerization catalysts based upon zirconocene alkyls and tris(pentafluorophenyl) borane. J Am Chem Soc 113:3623–3625
Horton AD, de With J, van der Linden AJ, van de Weg H (1996) Cationic alkylzirconium complexes based on a tridentate diamide ligand: new alkene polymerisation catalysts. Organometallics 15:2672
Jordan RF, LaPointe RE, Bajgur CS, Echols SF, Willett R (1987) Chemistry of cationic zirconium(IV) benzyl complexes. One-electron oxidation of d0 organometallics. J Am Chem Soc 109:4111–4113
Jordan RF, Dasher WE, Echols SF (1986) Reactive cationic dicyclopentadienyl zirconium(IV) complexes. J Am Chem Soc 108:1718–1719
Bochmann M, Jaggar AJ, Wilson LM, Hursthouse MB, Motevalli M (1989) Synthesis of cationic alkyl bis(cyclopentadienyl)titanium complexes by one-electron oxidation of titanium(III) alkyls. The structure of [Cp2*TiMe(THF)]BPh4 and [Cp2*Ti(OH)(H2O)]BPh4.2THF. Polyhedron 8:1838
Barron AR (1995) New method for the determination of the trialkylaluminum content in alumoxanes. Organometallics 14:3581–3583
Zhu F, Huang Q, Lin S (1999) Syntheses of multi-stereoblock polybutene-1 using novel monocyclopentadienyltitanium and modified methylaluminoxane catalysts. J Polym Sci, Part A: Polym Chem 37:4497–4501
Tritto I, Sacchi MC, Locatelli P, Li SX (1996) Low-temperature 1H and 13C NMR investigation of trimethylaluminium contained in methylaluminoxane cocatalyst for metallocene-based catalysts in olefin polymerization. Macromol Chem Phys 197:1537–1544
Resconi L, Bossi S, Abis L (1990) Study on the role of methylalumoxane in homogeneous olefin polymerization. Macromolecules 23:4489–4491
Britovsek GJP, Cohen SA, Gibson VC, van Meurs M (2004) Iron catalyzed polyethylene chain growth on zinc: a study of the factors delineating chain transfer versus catalyzed chain growth in zinc and related metal alkyl systems. J Am Chem Soc 126:10701–10712
Bryliakov KP, Semikolenova NV, Yudaev DV, Zakharov VA, Brintzinger HH, Ystenes M, Rytter E, Talsi EP (2003) 1H-, 13C-NMR and ethylene polymerization studies of zirconocene/MAO catalysts: effect of the ligand structure on the formation of active intermediates and polymerization kinetics. J Organomet Chem 683:92–102
Bochmann M, Lancaster SJ (1994) Monomer–dimer equilibria in homo- and heterodinuclear cationic alkylzirconium complexes and their role in polymerization catalysis. Angew Chem Int Ed Engl 33:1634–1637
Bolton PD, Clot E, Cowley AR, Mountford P (2006) AlMe3 and ZnMe2 adducts of a titanium imido methyl cation: a combined crystallographic, spectroscopic, and DFT studies. J Am Chem Soc 128:15005–15018
Shreve AP, Mulhaupt R, Fultz W, Calabrese J, Robbins W, Ittel SD (1988) Sterically hindered aryloxide-substituted alkylaluminum compounds. Organometallics 7:409–416
Williams VC, Dai C, Li Z, Collins S, Piers WE, Clegg W, Elsegood MRJ, Marder TB (1999) Activation of [Cp2ZrMe2] with new perfluoroaryl diboranes: solution chemistry and ethylene polymerization behavior in the presence of MeAl(BHT)2. Angew Chem Int Ed 38:3695–3698
Stapleton RA, Galan BR, Collins S, Simons RS, Garrison JC, Youngs WJ (2003) Bulky aluminum alkyl scavengers in olefin polymerization with Group 4 catalysts. J Am Chem Soc 125:9246–9247
Bochmann M, Jaggar AJ (1992) Cationic titanium alkyls as alkene polymerisation catalysts: solvent and anion dependence. J Organomet Chem 424:C5–C7
Deck PA, Beswick CL, Marks TJ (1998) Highly electrophilic olefin polymerization catalysts. Quantitative reaction coordinates for fluoroarylborane/alumoxane methide abstraction and ion-pair reorganization in Group 4 metallocene and “constrained geometry” catalysts. J Am Chem Soc 120:1772–1784
Beswick CL, Marks TJ (2000) Metal-alkyl group effects on the thermodynamic stability and stereochemical mobility of B(C6F5)3-derived Zr and Hf metallocenium ion-pairs. J Am Chem Soc 122:10358–10370
Beddie C, Hollink E, Wei P, Gauld J, Stephan DW (2004) Use of computational and synthetic chemistry in catalyst design: a new family of high-activity ethylene polymerization catalysts based on titanium tris(amino)phosphinimide complexes. Organometallics 23:5240–5251
Horton AD, Frijns JHG (1991) BPh −4 als ligand in kationischen zirconiumkomplexen: neuartiger bindungsmodus und fluktuationsprozesse. Angew Chem 103:1181–1183
Yang X, Stern C, Marks TJ (1991) Models for organometallic molecule-support complexes. Very large counterion modulation of cationic actinide alkyl reactivity. Organometallics 10:840–842
Chien JCW, Tsai WM, Rausch MD (1991) Isospecific polymerization of propylene catalyzed by rac-ethylenebis(indenyl)methylzirconium cation. J Am Chem Soc 113:8570–8571
Jia L, Yang X, Stern CL, Marks TJ (1997) Cationic metallocene polymerization catalysts based on tetrakis(pentafluorophenyl)borate and its derivatives. Probing the limits of anion “noncoordination” via a synthetic, solution dynamic, structural, and catalytic olefin polymerization study. Organometallics 16:842–857
Gillis DJ, Tudoret MJ, Baird MC (1993) Novel arene complexes of titanium(IV), zirconium(IV), and hafnium(IV). J Am Chem Soc 115:2543–2545
Lancaster SJ, Robinson OB, Bochmann M, Coles SJ, Hursthouse MB (1995) Synthesis and reactivity of new mono(cyclopentadienyl)zirconium and -hafnium alkyl complexes. Crystal and molecular structure of [{C5H3(SiMe3)2}HfMe2(η6-toluene)][BMe(C6F5)3]. Organometallics 14:2456–2462
Scollard JD, McConville DH (1996) Living polymerization of alpha-olefins by chelating diamide complexes of titanium. J Am Chem Soc 118:10008–10009
Beck S, Geyer A, Brintzinger H-H (1999) Diffusion coefficients of zirconocene-borate ion pairs studied by pulsed field-gradient NMR-evidence for ion quadruples in benzene solutions. Chem Commun 24:2477–2478
Stahl NG, Zuccaccia C, Jensen TR, Marks TJ (2003) Metallocene polymerization catalyst ion-pair aggregation by cryoscopy and pulsed field gradient spin-echo NMR diffusion measurements. J Am Chem Soc 125:5256–5257
Rodriguez-Delgado A, Hannant MD, Lancaster SJ, Bochmann M (2004) Anion effects on the activity and stereoselectivity in propene polymerizations catalyzed by C2-symmetric and “oscillating” catalysts. Macromol Chem Phys 205:334–346
Song F, Hannant MD, Cannon RD, Bochmann M (2004) Zirconocene-catalyzed propene polymerisation: kinetics, mechanism, and the role of the anion. Macromol Symp 213:173–185
Garratt S, Carr AG, Langstein G, Bochmann M (2003) Isobutene polymerization and isobutene–isoprene copolymerization catalyzed by cationic zirconocene hydride complexes. Macromolecules 36:4276–4287
Lancaster SJ, Rodriguez A, Lara-Sanchez A, Hannant MD, Walker DA, Hughes DH, Bochmann M (2002) [H2N{B(C6F5)3}2]-: a new, remarkably stable diborate anion for metallocene polymerization catalysts. Organometallics 21:451–453
Hannant MH, Wright JA, Lancaster SJ, Hughes DL, Horton PN, Bochmann M (2006) The synthesis of new weakly coordinating diborate anions: anion stability as a function of linker structure and steric bulk. Dalton Trans 20:2415–2426
Wu F, Dash AK, Jordan RF (2004) Structures and reactivity of Zr(IV) chlorobenzene complexes. J Am Chem Soc 126:15360–15361
Butts MD, Scott BL, Kubas GJ (1996) Syntheses and structures of alkyl and aryl halide complexes of the type [(PiPr3)2PtH(η1-XR)]BArf and analogues with Et2O, THF, and H2 ligands. Halide-to-metal π bonding in halocarbon complexes. J Am Chem Soc 118:11831–11843
Kulawiec RJ, Faller JW, Crabtree RH (1990) Binding and activation of halocarbons by iron(II) and ruthenium(II). Organometallics 9:745–755
Witte PT, Meetsma A, Hessen B, Budzelaar PHM (1997) Coordination of ethene and propene to a cationic d0 vanadium center. J Am Chem Soc 119:10561–10562
Doerrer LH, Green MLH, Haussinger D, Sassmannshausen J (1999) Evidence for cationic Group 4 zirconocene complexes with intramolecular phenyl co-ordination. J Chem Soc, Dalton Trans 13:2111–2118
Brookhart M, Green MLH, Wong L-L (1988) Carbon–hydrogen-transition metal bonds. Prog Inorg Chem 36:1–124
Scherer W, McGrady GS (2004) Agostic interactions in d0 metal alkyl complexes. Angew Chem Int Ed 43:1782–1806
Ijpeij EG, Coussens B, Zuideveld MA, van Doremaele GHJ, Mountford P, Lutz M, Spek AL (2010) Synthesis, solid state and DFT structure and olefin polymerization capability of a unique base-free dimeric methyl titanium dication. Chem Commun 46:3339–3341
Clot E, Eisenstein O (2004), Agostic interactions from a computational perspective: one name, many interpretations. In: principles and applications of density functional theory in inorganic chemistry II, Springer Berlin Heidelberg, vol 113
Jordan RF, Bradley PK, Baenziger NC, LaPointe RE (1990) β-agostic interactions in (C5H4Me)2Zr(CH2CH2R)(PMe3)+ complexes. J Am Chem Soc 112:1289–1291
Song F, Lancaster SJ, Cannon RD, Schormann M, Humphrey SM, Zuccaccia C, Macchioni A, Bochmann M (2005) Synthesis, ion aggregation, alkyl bonding modes, and dynamics of 14-electron metallocenium ion pairs [(SBI)MCH2SiMe3·X-] (M=Zr, Hf): inner-sphere (X=MeB(C6F5)3) versus outer-sphere (X=B(C6F5)4) structures and the implications for continuous or intermittent alkene polymerization mechanisms. Organometallics 24:1315–1328
Bolton PD, Clot E, Cowley AR, Mountford P (2005) Well-defined imidotitanium alkyl cations: agostic interactions, migratory insertion vs. [2+2] cycloaddition, and the first structurally authenticated AlMe3 adduct of any transition metal alkyl cation. Chem Commun 26:3313–3315
Bochmann M (2010) The chemistry of catalyst activation: the case of Group 4 polymerization catalysts. Organometallics 29:4711–4740
Bochmann M, Wilson LM, Hursthouse MB, Motevalli M (1988) Insertion reactions of nitriles in cationic alkylbis(cyclopentadienyl)titanium complexes—the facile synthesis of azaalkenylidene titanium complexes and the crystal and molecular-structure of [(Indenyl)2Ti(NCMePh)(NCPh)]BPh4. Organometallics 7:1148–1154
Alelyunas YW, Jordan RF, Echols SF, Borkowsky SL, Bradley PK (1991) Acetonitrile insertion reactions of zirconium complexes (C5H4R)2Zr(R)(L)+. Organometallics 10:1406–1416
Tatsumi K, Nakamura A, Hofmann P, Stauffert P, Hoffmann R (1985) Carbon monoxide activation by biscyclopentadienyl complexes of Group 4 metals and actinides: η2-acyl complexes. J Am Chem Soc 107:4440–4451
Cabrera L, Hollink E, Stewart JC, Wei P, Stephan DW (2005) Cationic methyl- and chlorotitanium phosphinimide complexes. Organometallics 24:1091–1098
Pellecchia C, Grassi A, Zambelli A (1994) Single insertion of an alpha-olefin into the cationic complex [Zr(CH2Ph)3]+ affording isolable [Zr(CH2Ph)2(CH2CHRCH2Ph)]+ adducts: a model for the insertion mechanism in Ziegler–Natta polymerisation. Organometallics 13:298–302
Horton AD, de With J (1997) Controlled alkene and alkyne insertion reactivity of a cationic zirconium complex stabilized by an open diamide ligand. Organometallics 16:5424–5436
Vatamanu M, Stojcevic G, Baird MC (2007) Detection of an η1-alkene intermediate of the type [Cp2Zr(Me)(η1-alkene)]+: the role of such species in metallocene catalyst deactivation to allylic species. J Am Chem Soc 130:454–456
Sauriol F, Sonnenberg JF, Chadder SJ, Dunlop-Brière AF, Baird MC, Budzelaar PHM (2010) Remarkable reactions and intermediates in titanocene(IV) chemistry: migratory insertion reactions of 2,2-disubstituted-1-alkenes, intramolecular 1,5-σ bond metathesis via ε-agostic interactions, and a rare example of a β-agostic alkyltitanocene complex. J Am Chem Soc 132:13357–13370
Dunlop-Brière AF, Budzelaar PHM, Baird MC (2012) α- and β-Agostic alkyl–titanocene complexes. Organometallics 31:1591–1594
Jordan RF, Bajgur CS, Dasher WE, Rheingold AL (1987) Hydrogenation of cationic dicyclopentadienylzirconium(IV) alkyl complexes. Characterization of cationic zirconium(IV) hydrides. Organometallics 6:1041–1051
Yamatsugu K, Yin L, Kamijo S, Kimura Y, Kanai M, Shibasaki M (2009) A synthesis of Tamiflu by using a barium-catalyzed asymmetric Diels–Alder-type reaction. Angew Chem Int Ed 48:1070–1076
Kato N, Mita T, Kanai M, Therrien B, Kawano M, Yamaguchi K, Danjo H, Sei Y, Sato A, Furusho S, Shibasaki M (2006) Assembly state of catalytic modules as chiral switches in asymmetric Strecker amino acid synthesis. J Am Chem Soc 128:6768–6769
Delferro M, Marks TJ (2011) Multinuclear olefin polymerization catalysts. Chem Rev 111:2450–2485
Li H, Marks TJ (2006) Nuclearity and cooperativity effects in binuclear catalysts and cocatalysts for olefin polymerization. Proc Natl Acad Sci USA 103:15295–15302
Li H, Li L, Marks TJ (2004) Polynuclear olefin polymerization catalysis: proximity and cocatalyst effects lead to significantly increased polyethylene molecular weight and comonomer enchainment levels. Angew Chem Int Ed 43:4937–4940
Li H, Stern CL, Marks TJ (2005) Significant proximity and cocatalyst effects in binuclear catalysis for olefin polymerization. Macromolecules 38:9015–9027
Li H, Li L, Schwartz DJ, Metz MV, Marks TJ, Liable-Sands L, Rheingold AL (2005) Coordination copolymerization of severely encumbered isoalkenes with ethylene: enhanced enchainment mediated by binuclear catalysts and cocatalysts. J Am Chem Soc 127:14756–14768
Motta A, Fragalà IL, Marks TJ (2009) Proximity and cooperativity effects in binuclear d(0) olefin polymerization catalysis. Theoretical analysis of structure and reaction mechanism. J Am Chem Soc 131:3974–3984
Guo N, Li L, Marks TJ (2004) Bimetallic catalysis for styrene homopolymerization and ethylene–styrene copolymerization. Exceptional comonomer selectivity and insertion regiochemistry. J Am Chem Soc 126:6542–6543
Guo N, Stern CL, Marks TJ (2008) Bimetallic effects in homopolymerization of styrene and copolymerization of ethylene and styrenic comonomers: scope, kinetics, and mechanism. J Am Chem Soc 130:2246–2261
Lee MH, Kim SK, Do Y (2005) Biphenylene-bridged dinuclear group 4 metal complexes: enhanced polymerization properties in olefin polymerization. Organometallics 312:3618–3620
Jungling S, Mullhaupt R, Plenio H (1993) Cooperative effects in binuclear zirconocenes: their synthesis and use as catalyst in propene polymerization. J Organomet Chem 460:191–195
Soga K, Ban HT, Uozumi T (1998) Synthesis of a dinuclear ansa-zirconocene catalyst having a biphenyl bridge and application to ethene polymerization. J Mol Catal A: Chem 128:273–278
Spaleck W, Kuber F, Bachmann B, Fritze C, Winter A (1998) New bridged zirconocenes for olefin polymerization: binuclear and hybrid structures. J Mol Catal A: Chem 128:279–287
Lee H-W, Ahn S-H, Park Y-H (2003) Copolymerization characteristics of homogeneous and in situ supported [(CH2)5(C5H4)2][(C9H7)ZrCl2]2 catalyst. J Mol Catal A: Chem 194:19–28
Noh SK, Lee J, Lee D-H (2003) Syntheses of dinuclear titanium constrained geometry complexes with polymethylene bridges and their copolymerization properties. J Organomet Chem 667:53–60
Xu S, Dai X, Wu T, Wang B, Zhou X, Weng L (2002) Synthesis, structure and polymerization catalytic properties of doubly bridged bis(cyclopentadienyl) dinuclear titanium and zirconium complexes. J Organomet Chem 645:212–217
Tian G, Wang B, Xu S, Zhou X, Liang B, Zhao L, Zou F, Li Y (2002) Ethylene polymerization with sila-bridged dinuclear zirconocene catalysts. Macromol Chem Phys 203:31–36
Noh SK, Kim S, Yang Y, Lyoo WS, Lee D-H (2004) Preparation of syndiotactic polystyrene using the doubly bridged dinuclear titanocenes. Eur Polym J 40:227–235
Salata MR, Marks TJ (2009) Catalyst nuclearity effects in olefin polymerization. Enhanced activity and comonomer enchainment in ethylene/olefin copolymerizations mediated by bimetallic Group 4 phenoxyiminato catalysts. Macromolecules 42:1920–1933
Abramo GP, Li L, Marks TJ (2002) Polynuclear catalysis: enhancement of enchainment cooperativity between different single-site olefin polymerization catalysts by ion pairing with a binuclear cocatalyst. J Am Chem Soc 124:13966–13967
Wang J, Li H, Guo N, Li L, Stern CL, Marks TJ (2004) Covalently linked heterobimetallic catalysts for olefin polymerization. Organometallics 23:5112–5114
Kuwabara J, Takeuchi D, Osakada K (2006) Early-late heterobimetallic complexes as initiator for ethylene polymerization. Cooperative effect of two metal centers to afford highly branched polyethylene. Chem Commun 36:3815–3817
Liu S, Motta A, Delferro M, Marks TJ (2013) Synthesis, characterization, and heterobimetallic cooperation in a titanium-chromium catalyst for highly branched polyethylenes. J Am Chem Soc 135:8830–8833
Green MLH, Popham NH (1999) Synthesis of heterobinuclear metallocenes containing bridging ansa-bis-eta-cyclopentadienyl ligands. J Chem Soc, Dalton Trans 1049–1059
Acknowledgments
We thank LANXESS Elastomers B.V. for their continued support of our work in the area of olefin polymerisation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Collins, R.A., Russell, A.F. & Mountford, P. Group 4 metal complexes for homogeneous olefin polymerisation: a short tutorial review. Appl Petrochem Res 5, 153–171 (2015). https://doi.org/10.1007/s13203-015-0105-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-015-0105-2