
Abstract
The xerogel and aerogel Ni(20 wt %)/Al2O3 catalysts were prepared through incipient-wetness-impregnation and co-precipitation-supercritical drying (CP-SCD), respectively. All the fresh and used catalysts were well characterised using FESEM, BET, XRD, H2-TPR and H2-TPD techniques. Their properties and catalytic performance in dry reforming of CH4 in a fluidised bed reactor were comparatively investigated. In comparison with the xerogel catalyst, the aerogel catalyst possessed smaller crystallite size of nickel, larger specific surface area, higher nickel dispersion, lower bulky density and better fluidization quality. More importantly, the aerosol catalyst showed higher catalytic activity, stability and less carbon deposition in dry reforming, due to the excellent physicochemical properties of the aerogel catalyst and its enhanced fluidization quality.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dry reforming of CH4 by CO2 is a promising technology for energy and environment, in which two of the most abundant greenhouse gases can be converted into invaluable syngas, a mixture for synthesis of advanced chemicals, clean fuels or fuel cells [1, 2]. However, the practical applications of dry reforming are still facing great challenges with respect to the catalysts stability and process engineering [2–4]. Among a variety of reforming catalysts, Ni-based catalysts have comparable activity to costly noble metal supported catalysts but are more sensitive to surface carbon deposition (also called coking), leading to either catalyst deactivation or blocking the reactor [1, 2, 5]. Generally, coking can be largely depressed through using highly dispersed nano-sized Ni on porous supports, tuning the surface acidity of the catalysts or utilising new reformers [2, 6, 7].
In practice, high dispersion of Ni active sites can be realised through innovative synthesis methodology. For example, Ni/Al2O3 catalysts synthesised by sol–gel method own smaller particle sizes than the catalysts synthesised through conventional impregnation methodology, while the synthesis necessitates expensive and moisture-sensitive organic precursors [8]. Reverse-emulsion synthesis offers large surface areas and smaller particle sizes of the resultant catalysts, though it is costly and complicated to fabricate catalyst in the large scale [9]. The combustion-synthesis has been well explored for preparing Ni-based nanocatalysts, however, the great concerns on safety and environment are raised [10]. Ni-based aerogel catalysts prepared by supercritical drying (SCD) of the catalysts precursor hydrogel represent a simple and cost-effective technique to synthesise the finite Ni-based catalysts with high surface area and excellent performance in dry reforming [11]. However, the aerosol itself could not fully overcome the issue of deactivation due to coking in the fixed bed reformer [12]. In addition, the application of aerogel catalysts in large-scale fixed bed reactors is limited by high pressure drop, which can be partially resolved through using fluidised bed reformer or monolithic catalysts with open channels [2, 6]. The monolithic catalysts would inevitably increase the cost of the catalysts and, more importantly, reduce the efficiency of the accessibility to the active metal sites.
The fluidised bed reformer may maximise the benefits of nanoscale catalysts via directly loading the catalysts into the reformer without needs of further manipulation [12, 13]. In addition, the fluidised bed reactor is more stable than the fixed bed reformer in operation because the pressure drop is independent of the particle size in fluidised bed reactor [14]. The motion of the catalyst particles in the fluidization bed would increase the contact of the nanoparticles with the reactant gases, leading to more efficient utilisation of the catalysts. Despite that the benefits of aerogel particles have been evidenced in the previous work employing cool models [6, 15], the dry reforming on the aerosol Ni-based catalysts in a fluidised bed reformer has seldom been explored, especially its performance in resistance to carbon deposition.
In this paper, we disclose the benefits of fluidised bed dry reforming on the Ni(20 wt %)/Al2O3 aerogel catalyst, which was prepared by the ethanol supercritical drying (SCD) of the hydrogel precursor obtained from co-precipitation synthesis, in comparison to the xerogel Ni(20 wt %)/Al2O3 prepared by incipient-wetness-impregnation. The physicochemical properties of the catalysts were characterised via FESEM, XRD, BET and H2-TPR/D techniques.
Experimental
Materials
All reagents, Al(NO3)3·9H2O, Ni(NO3)2·6H2O, concentrated ammonia solution and ethanol, were purchased from Sigma-Aldrich and used directly without pre-purification.
Catalyst preparation
Ni(20 wt %)/Al2O3 aerogel catalyst was prepared via hydrogen reduction of the NiO/Al2O3 aerogel nanoparticles obtained from the combinational technique of co-precipitation and the supercritical drying (CP-SCD). Briefly, under continuous vigorous stirring at room temperature, dilute ammonia solution (5.0 wt %) was added slowly into the mixed solution containing Al(NO3)3·9H2O and Ni(NO3)2·6H2O, in the similar stoichiometry to Ni(20 wt %)/Al2O3. The pH of the synthetic system was maintained at constant pH within 8–9. The resultant hydrogel was aged at room temperature, following with washing alternatively with distilled water and absolute ethanol. The as-obtained gel was then transferred into an autoclave filled with ethanol for supercritical drying (SCD), operating at 260 °C, 8.0 MPa for 2 h. After the release of the ethanol vapour at 260 °C, the powder was cooled down to room temperature. Before and after the SCD, continuous N2 flow was introduced into the autoclave to remove oxygen. The obtained aerogel was then calcined at 650 °C for 4 h in air before reduction to receive the Ni(20 wt %)/Al2O3 aerogel Ni-based catalyst, denoted as AG-20NiAl.
For comparison purpose, a xerogel Ni(20 wt %)/Al2O3 catalyst was prepared by impregnating a Puriss γ-Al2O3 (Sigma-Aldrich) with Ni(NO3)2 solution. The impregnated catalyst was dried overnight at 110 °C, followed by calcinations at 650 °C for 4 h, designated as XG-20NiAl.
Catalyst characterisation
The crystal phases of the fresh catalysts and spent catalysts were determined using X-ray diffraction on an X’Pert PRO, PANalytical X-Ray Diffractometer, Cu Ka1 (λ = 1.5408 Å) radiator, 40 kV 40 mA. The specific surface areas (SSA) of the samples were measured by N2 adsorption at −196 °C on an Autosorb-1 apparatus (Quantachrom, USA). The morphology of the catalysts was recorded through a field-emission scanning electron microscopy (FESEM, JEOL JSM-6700F).
Temperature-programmed reduction (TPR) was performed on chemisorption machine (ChemBet 3000, Quantachrom, USA), in which 30 mg of fresh catalyst was loaded in the thermostatic zone. Prior to TPR test, the catalyst was heated up to 150 °C under an Ar flow (30 mL/min) and remained for 30 min. After Ar purging, the reactor was cooling down to room temperature in the same Ar flow. The purging gas was then switched to a 30 mL/min H2/Ar (H2 vol % = 5 %) mixed flow as soon as the TPR test started, where the catalyst was heated up to 900 °C and held there for more than 30 min to complete the reduction, the heating rate was 10 °C/min. The H2 consumption was monitored by a thermal conductivity detector (TCD). Temperature-programmed desorption of hydrogen (H2-TPD) was performed on the same system as TPR. After reduction at 900 °C, the sample was cooled down to room temperature in the H2 flow and remained in the H2 flow for 30 min before switching to argon flow, and heating up to 900 °C at a rate of 10 °C/min. The desorbed hydrogen was analysed using TCD.
Catalytic dry reforming
Dry reforming was conducted over the catalysts in a quartz micro-reactor (internal diameter of 18 mm) under atmospheric pressure. Before dry reforming reactions, the catalysts were reduced in situ at 800 °C/1 h in the H2/N2 flow (20 vol % H2, 150 mL/min). For the fluidised bed operation at fixed temperature of 800 °C, the reduced catalysts (200 mg) were fluidised by introducing the pre-mixed gases of CH4, CO2, N2 (300 mL/min, molar ratio of 1:1:1) through a porous quartz distributor sintering at the reactor bottom. The reactants and products were analysed online by gas chromatographer (TP260, Tianpu, China) equipped with 13× and GDC-104 columns. An ice-cold trap was set between the reactor exit and the GC sampling valve to avoid the influence of by-product, water.
Upon completion of the dry reforming reactions, the catalysts were cooled down to room temperature under inert gas flow. The spent catalysts were discharged from the reactor and analysed by thermogravimetry (TG, STA 449C, Netzsch, Germany) to study the surface carbon deposition. In the TG experiments, 10 mg of the used catalysts was heated in an air flow (20 mL/min) from room temperature to 900 °C at ramp of 10 °C/min.
Results and discussion
XRD analysis of the samples
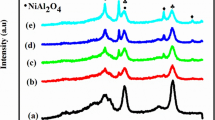
Figure 1A shows the XRD patterns of the AG-20NiAl and XG-20NiAl oxides calcined at 650 °C for 4 h. For the aerogel AG-20NiAl oxide sample, the Bragg diffraction peaks at 19°, 37°, 45°, 60° and 65° can be well defined as (111), (311), (400), (511) and (440) crystal faces of the cubic phase NiAl2O4 spinel, with space group of Fd3m (JCPDS: 10–0339), while the diffraction peaks of γ-Al2O3 support are very broad or masked by the spinel diffraction peaks [16]. There is no significant diffraction of NiO, indicating that Ni species are well bonded in the spinel with high dispersion. Despite that the spinel is a thermodynamically stable species, [16] all the NiO, NiAl2O4 spinel and γ-Al2O3 support can be well distinguished in the XRD pattern of the XG-NiAl oxide. The coexistence of NiO and NiAl2O4 phases suggests that the Ni species were in poor dispersion in the XG-NiAl. It was the IM preparation and the high Ni-loading leading to the isolated NiO phase from NiAl2O4, because high Ni-loading favours faster nucleation in calcinations to generate large NiO crystallites [17], which are difficult to diffuse and incorporate into the Al2O3 matrix to form spinel NiAl2O4.

XRD patterns of the A oxides calcined at 650 °C/4 h and B the catalysts reduced at 800 °C/1 h: (a) AG-20NiAl and (b) XG-20NiAl
XRD patterns of the reduced catalysts were presented in Fig. 1B, in which the strong diffraction peaks centred at 44.5°, 51.7° and 76.2° can be assigned to metallic Ni. It can be seen that such diffraction peaks of Ni in the AG-20NiAl are much broader and less intensive than those of XG-20NiAl aerogel catalyst, suggesting the Ni particles are much smaller than those on XG-20NiAl. As shown in Table 1, the mean crystallite size of metallic Ni in AG-20NiAl is much smaller than that of XG-20NiAl. The smaller Ni crystallite size in the AG-20NiAl can be associated to more NiAl2O4 spinel phase existed in the aerosol catalyst, whose matrix limits the over-growth of Ni metal during reduction. However, in the xerogel catalyst, Ni species were mainly hosted in NiO which was overlapped with NiAl2O4 leading to aggregation in hydrogen thermal reduction [16].
Morphologies and texture properties of the samples
Figure 2 shows the SEM images of the AG-NiAl and XG-20NiAl oxides (Fig. 2a) and corresponding reduced catalysts (Fig. 2b). The as-prepared aerogel AG-NiAl oxide calcined at 650 °C comprises a number of nanoplates (Fig. 2a) with diameters in the range of 15–80 nm. After reduction at 800 °C for 1 h, there were no significant morphology changes observed for the AG-20NiAl aerogel catalyst as shown in Fig. 2b, but the particle sizes became even smaller. However, the SEM images of the oxide and reduced XG-NiAl samples (Fig. 2c, d) show that the samples contained a number of big particles, larger than 200 nm in diameter and the reduction did not significantly destroy their morphology either. Despite that the aerogel and xerogel catalysts were both slightly aggregated during reduction, the SEM observations evidenced that the CP-SCD method is a more effective methodology to prepare nanoscale aerogel catalysts. In addition, the aerogel catalyst has much lighter bulk density, only 1/17 of XG-20NiAl density. The smaller particle sizes and lighter density of AG-20NiAl indicate that it would have larger surface area (SSA) than XG-20NiAl catalyst. Indeed, the SSA of the aerogel catalyst (190 m2/g) is about 1.5 times of that of XG-20NiAl (135 m2/g).

SEM images of the oxide (a) and reduced (b) AG-20NiAl (b), oxide(c) and reduced (d) XG-20NiAl samples
The excellent textural properties of the aerogel AG-20NiAl can be attributed to the advantages of the supercritical drying treatment. Before the SCD, there was no doubt that the structure-captured water supports the skeleton of the hydrogel. Most of the surface water will be partly replaced by the ethanol during ethanol washing, while the rest hydrogel-captured water can be further substituted by the ethanol during the SCD treatment. More importantly, the precursors’ surface hydroxyl groups would be etherified under the SCD conditions. Therefore, the intrinsic skeleton of the hydrogel can be maintained or expended, leading to large surface area of the aerogel sample [18]. The morphology and the high SSA remained even after hydrogen reduction at 800 °C. In contrast, the structural water and surface hydroxyl groups on the XG-20NIAl experienced direct thermal dehydration and dehydroxylation of surface hydroxyl groups, rather than ethanol exchanging and etherisation, during the sequential thermal treatments. This would result in particle aggregation and thus sintering as a result of thermal losses of structural water and surface hydroxyl groups.
H2-TPR/D analysis
Figure 3 displays the TPR profiles of the AG-20NiAl and XG-20NiAl oxides after calcination at 650 °C for 4 h. There are two major reduction peaks observed for AG-20NiAl oxide, the maximum reduction at 730 °C can be assigned to reduction of surface NiAl2O4, while the second at 865 °C due to the reduction of bulk NiAl2O4 [12, 17, 19]. It was the large SSA leading to the more surface rather than bulky NiAl2O4 existing in the aerogel oxide. In contrast, three reduction peaks centring at 445, 670 and 805 °C were observed and associated to the highly dispersed NiO, bulky NiO and NiAl2O4 spinel, respectively. It was well documented that the reducibility of Ni2+ ions was closely related to the particle sizes of the Ni species which were determined by the synthesis methodology. The aerosol AG-20NiAl oxide synthesised by CP-SCD possessed smaller particle size and larger surface areas, which would have more reducible surface NiAl2O4 species and thus higher Ni metal dispersion than XG-20NiAl oxide.

TPR profiles of the a AG-20NiAl and b XG-20NiAl oxides calcined at 650 °C
The Ni reduction degree and Ni dispersion degree of the catalysts were retrieved by analyses of the integration areas of H2 consumption in TPR and desorption in H2-TPD. As listed in Table 1, the aerogel and the Xerogel oxides have almost the similar reducibility (93–95 %) while the Ni dispersion of the AG-20NiAl is 2.5 times that of XG-NiAl. The high dispersion of Ni in the reduced AG-20NiAl was also evidenced by the smaller Ni crystallite size than that of reduced XG-20NiAl catalyst.
Catalytic performance of the fluidization dry reforming
The fluidization quality of the reduced AG-20NiAl is much better than that of XG-20NiAl, in terms of the bed expansion ratio of fluidization to fixed beds, where the expansion ratio was defined as the ratio of fluidised bed height to that of fixed bed (Hfluid/Hfix). The bed expansion ratio of AG-20NiAl is 4.0 (Hfluid = 40 mm, Hfix = 10 mm) while only 1.2 for the XG-20NiAl (Hfluid = 6 mm, Hfix = 4 mm). The different fixed bed heights between AG-20NiAl and XG-20NiAl were due to their bulk density.
Figure 4 compared the performance of dry reforming over the AG-20NiAl and XG-20NiAl catalysts in the fluidised bed reactor for 30 h. In terms of the methane and CO2 conversions, the initial catalytic activities of the AG-20NiAl catalyst were only 5 % better than those of XG-20NiAl. The slight difference can be attributed to the worse fluidization state and less active sites on the XG-20NiAl than AG-20NiAl catalyst. As reaction time increased, the conversions of CH4 and CO2 on the XG-20NiAl dropped in only 2 h, whereas the conversions on the aerogel only showed a slight decrease (<5 %) in 30 h. Moreover, the selectivity of H2 and CO remained the same for the AG-20NiAl and the XG-20NiAl catalysts in 5 h, whereas rapid drops of the H2 and CO selectivity started after 5 h on the XG-20NiAl catalysts. The results suggest that AG-20NiAl possesses higher stability than the XG-20NiAl catalyst in the fluidised bed reactor. The higher activity and stability of the aerogel catalyst can be attributed to its excellent fluidization quality and physicochemical properties, including the reducibility and large surface area.

Time in stream of conversion and selectivity of the fluidization dry reforming on aerogel and xerogel Ni/Al2O3 catalysts at 800 °C
Analysis of the catalyst deactivation
From the mechanistic point of view, the performance of the dry reforming reaction is determined by two key elementary reactions, i.e., methane decomposition (Eq. 1) and the sequential carbon gasification by CO2 (Eq. 2) [1, 2]. It has been well evidenced that the smaller Ni crystallites would be more active in both the methane decomposition and carbon gasification [22]. As shown in Tables 1 and 2, the Ni crystallite of AG-20NiAl catalyst (9.2 nm) is much smaller than that of XG-20NiAl, leading to superior initial activity on the AG-20NiAl. After 30 h time on stream, the Ni crystallite sizes are much close, however, their stability is distinct and requires further investigation.
It has been well documented that the Ni catalyst deactivation in dry reforming can be induced by the sintering of Ni crystallite and the carbon deposition on the catalysts [2, 3, 5]. The sintering of active phase can be characterised by XRD measurements of the used catalysts. The Ni crystallite sizes of the used catalysts were calculated using the Scherrer equation on the (1 1 1) diffractions and were listed in Table 2. It can be observed that the Ni crystallite sizes of the used AG-20NiAl and XG-20NiAl catalysts were both increased to a comparable level after 30 h of dry reforming, indicating sintering occurred for the two catalysts. The sintering of active phase in dry reforming is unavoidable for both pristine and modified Ni/Al2O3 catalysts under high reaction temperature [2, 20–22]. For example, some promoters, such as La2O3 or CeO2, may inhibit the crystallite growth of Ni to some extent only [23]. In this work, after 30 h dry reforming, there is only a marginal difference of the sintering between aerogel and xerogel catalysts, but their activities and stabilities are distinctive: 40 % higher conversion of the reactants and nearly 30 % higher selectivity were observed on the aerogel than the xerogel catalyst, suggesting sintering is not the major reason responsible for the catalysts deactivation.
Surface carbon deposition is a major cause for deactivation of Ni-based catalysts [7, 21, 24]. We investigated the carbon deposition on used catalyst employing SEM and TGA. Figure 5 compared the SEM images of the used AG-20NiAl and AG-20NiAl catalysts. It can be clearly seen that more fibres, due to the graphitic carbon [9, 21], deposited on the XG-20NiAl, suggesting it was suffering from heavier coking than the AG-20NiAl. In this study, as shown in Table 2, we found the diameters of the filamentous carbon fall in the range of 20–30 nm, which are consistent with the crystallite sizes of Ni on the aerogel and xerogel catalysts. This finding is well consistent with the previous studies in fixed bed reactor that the diameters of the deposited carbon fibres depend on the crystallite size of the metal catalysts [21].
The coking-induced deactivation of Ni catalyst is also closely related to the type, amount, and location of the carbon formed [2, 3, 5]. It was accepted that there are two types of carbon deposited on the Ni catalysts in dry reforming: amorphous and filamentous carbon. The amorphous carbon would not lead to serious deactivation because it can be gasified at high temperature by CO2, whereas the fibrous carbon is difficult to be gasified and results in permanent deactivation. Therefore, the fibrous carbon deposition necessitates catalyst regeneration with extra energy penalty [24]. In this study, the aerogel AG-20NiAl had less filamentous carbon deposition and thus exhibited better performance (activity and stability) than xerogel catalyst in dry reforming. Although SEM measurement may observe the filamentous, it failed to identify the amorphous carbon and the overall coking amount. Temperature-programmed-oxidation (TPO) of the deactivated catalysts has been shown great potential to solve the issue indirectly [6, 12, 13].

SEM images of used catalysts a AG-20NiAl and b XG-20NiAl in dry reforming
Temperature-programmed-oxidation (TPO) experiments of the used catalysts were performed on a TG. The obtained TPO profiles of the aerogel and xerogel catalysts were presented by DTG signals (Fig. 6). There are two peaks between 400 and 650 °C, a strong TPO peak along with a shoulder peak can be observed for the spent AG-20NiAl and XG-20NiAl catalysts, indicating two types of deposited carbon were formed on the active metal sites. On the used AG-20NiAl, the major peak centres at 530 °C, corresponding to the amorphous carbon, while the shoulder peak centring at 580 °C may be attributed to the fibrous graphitic carbon [9, 21, 25]. The weight ratio between the amorphous and filamentous carbon on the AG-20NiAl catalyst is 75:25 (%). However, the major TPO peak of the used XG-20NiAl located at 580 °C is assigned to the fibrous carbon, and the small shoulder peak centres at 450 °C corresponding to the amorphous carbon deposited on the used XG-20NiAl catalyst. The ratio between the amorphous and filamentous is 30:70 (%). In addition, the integrated areas of the carbon TPO peaks, corresponding to the overall carbon deposition, on the used AG-20NiAl catalysts are nearly half (14.6:26.7) of that on XG-20NiAl. Accordingly, the filamentous carbon (TPO peak at higher temperature) deposited on the aerosol AG-20NiAl catalyst is only one-fifth (14.6 × 25 %:26.7 × 70 %) of that on xerogel XG-20NiAl catalyst, while their calculated surface amorphous carbon amount is almost the same. The TPO results revealed that the aerosol catalyst is more resistant to coking, in particular the filamentous carbon, and thus owns higher stability than the xerogel catalyst in the long-term tests.

Temperature-programmed-oxidation (TPO) profiles of the carbon deposited on the used a AG-20NiAl and b XG-20NiAl catalysts
Besides the intrinsic physicochemical properties of the aerogel catalyst discussed in the characterisation section, the better fluidization quality of aerogel catalyst also plays important roles on restricting carbon deposition in the dry reforming. The two major elementary reactions, methane decomposition and CO2 gasification of C (Eqs. 1, 2), compete with each other in the dry reforming and determine the carbon deposition level. In principle, the methane decomposition is thermodynamically favourable in dry reforming and is responsible for carbon formation at low temperature, while the carbon gasification is thermodynamically unfavourable which can be realised in high concentration of CO2 or applying higher temperature to shift the carbon diminishing reaction [26]. It has been acknowledged that the fluidization of the catalyst particles offers them alternative environments of rich-CO2 and lean-CO2, where the CO2-rich zones would facilitate removal of deposited carbon on Ni/Al2O3 [15]. In comparison with xerogel, the better fluidization of aerogel catalyst with larger bed expansion entitled them longer contact time with CO2, which would facilitate the gasification of the deposited amorphous carbon and reducing the possibility to form refractory filamentous carbon [2, 22]. The less filamentous carbons on the used AG-20NiAl than XG-20NiAl were really evidenced by the TPO and SEM measurements, better stability and activity in the fluidization dry reforming reactions. Therefore, the combination of the aerogel catalysts with the fluidised bed reactor is a novel and very promising mode in the syngas production from methane reforming with CO2.
Conclusions
Nano-sized Ni(20 wt %)/Al2O3 aerogel (AG-20NiAl) was prepared using co-precipitation and supercritical drying (CP-SCD), and compared with the corresponding xerogel (XG-20NiAl). The as-prepared and reduced aerogel catalysts possessed larger specific surface area, lighter mass density, smaller Ni crystallite size and higher Ni dispersion than those of the xerogels.
With calcinations at 650 °C/4 h, the Ni species were homogeneously dispersed in the aerogel via forming the Al2O3 supported NiAl2O4 spinel structure, whereas the xerogel catalyst owned poor Ni dispersion with isolated NiO besides NiAl2O4 spinel supported on Al2O3. The aerogel and xerogel showed distinctive reducibility as evidenced by H2-TPR. It was the supercritical drying (SCD) and the stronger interaction between NiAl2O4 spinel and Al2O3 support giving rise to higher dispersion of metallic Ni in aerogel than in xerogel catalyst. H2-TPD results confirmed the higher dispersion of Ni in the aerogel reduced at 800 °C/1 h.
The reduced aerogel AG-20NiAl catalyst showed more significant bed expansion and better fluidization quality, than xerogel XG-20NiAl.In terms of the conversion of reactants (methane and CO2) and selectivity of products (CO and hydrogen), the AG-20NiAl catalyst exhibited superior catalytic performance to xerogel XG-20NiAl catalyst, in particular, the long-term stability. Despite that initial catalytic performance of XG-20NiAl catalyst was only slightly lower than AG-20NiAl, the xerogel started deactivation in a very short duration, whereas no significant activity loss observed on the AG-20NiAl catalyst in the 30 h time in streams of dry reforming.
The full characterisations of the fresh and used catalysts revealed that carbon deposition, rather than the sintering, is the major reason for catalyst deactivation. The aerogel AG-20NiAl catalyst had much less carbon deposition than xerogel, especially the filamentous carbon formed on the aerogel is only 20 % of that on xerogel. The intrinsic physicochemical properties and the better fluidization quality of aerogel endow it the outstanding performance in dry reforming.
References
Jiang Z, Xiao T, Kuznetsov V, Edwards P (2010) Turning carbon dioxide into fuel. Philos Trans Royal Soc A Math Phys Eng Sci 368:3343
Fan M-S, Abdullah AZ, Bhatia S (2009) Catalytic technology for carbon dioxide reforming of methane to synthesis gas. ChemCatChem 1:192
Budiman A, Song S-H, Chang T-S, Shin C-H, Choi M-J (2012) Dry reforming of methane over cobalt catalysts: a literature review of catalyst development. Catal Surv Asia 16:183
Shi L, Yang G, Tao K, Yoneyama Y, Tan Y, Tsubaki N (2013) An introduction of CO2 conversion by dry reforming with methane and new route of low-temperature methanol synthesis. Acc Chem Res
Gharibi M, Zangeneh FT, Yaripour F, Sahebdelfar S (2012) Nanocatalysts for conversion of natural gas to liquid fuels and petrochemical feedstocks. Appl Catal A 443–444:8
Hao Z, Zhu Q, Jiang Z, Li H (2008) Fluidization characteristics of aerogel Co/Al2O3 catalyst in a magnetic fluidized bed and its application to CH4-CO2 reforming. Powder Technol 183:46
Walker DM, Pettit SL, Wolan JT, Kuhn JN (2012) Synthesis gas production to desired hydrogen to carbon monoxide ratios by tri-reforming of methane using Ni-MgO-(Ce,Zr)O2 catalysts. Appl Catal A 445–446:61
Natesakhawat S, Oktar O, Ozkan US (2005) Effect of lanthanide promotion on catalytic performance of sol-gel Ni/Al2O3 catalysts in steam reforming of propane. J Mol Catal A Chem 241:133
Hayashi H, Murata S, Tago T, Kishida M, Wakabayashi K (2001) Methane-steam reforming over Ni/Al2O3 catalyst prepared using W/O microemulsion. Chem Lett 34
Li H, Wang J (2004) Study on CO2 reforming of methane to syngas over Al2O3-ZrO2 supported Ni catalysts prepared via a direct sol-gel process. Chem Eng Sci 59:4861
Osaki T, Tanaka T, Horiuchi T, Sugiyama T, Suzuki K, Mori T (2000) Application of NiO–Al2O3 aerogels to the CO2‐reforming of CH4. Appl Organomet Chem 14:789
Hao Z, Zhu Q, Lei Z, Li H (2008) CH4-CO2 reforming over Ni/Al2O3 aerogel catalysts in a fluidized bed reactor. Powder Technol 182:474
Hao Z, Zhu Q, Jiang Z, Hou B, Li H (2009) Characterization of aerogel Ni/Al2O3 catalysts and investigation on their stability for CH4-CO2 reforming in a fluidized bed. Fuel Process Technol 90:113
Ji Y, Li W, Xu H, Chen Y (2001) A study of carbon deposition on catalysts during the catalytic partial oxidation of methane to syngas in a fluidized bed. React Kinet Catal Lett 73:27
Effendi A, Hellgardt K, Zhang ZG, Yoshida T (2003) Characterisation of carbon deposits on Ni/SiO2 in the reforming of CH4-CO2 using fixed- and fluidised-bed reactors. Catal Commun 4:203
Reyes-Rojas A, Esparza-Ponce HE, Reyes-Gasga J (2006) Study of the Ni–NiAl2O4–YSZ cermet for its possible application as an anode in solid oxide fuel cells. J Phys Condens Matter 18:4685
Li C, Chen Y-W (1995) Temperature-programmed-reduction studies of nickel oxide/alumina catalysts: effects of the preparation method. Thermochim Acta 256:457
Zangouei M, Moghaddam AZ, Arasteh M (2010) The influence of nickel loading on reducibility of NiO/Al2O3 catalysts synthesized by sol-gel method. Chem Eng Res Bull 14:97–102
Zangouei M, Moghaddam AZ, Arasteh M (2010) The influence of nickel loading on reducibility of NiO/Al2O3 catalysts synthesized by sol-gel method. Chem Eng Res Bull 14:97–102
Goula MA, Lemonidou AA, Efstathiou AM (1996) Characterization of carbonaceous species formed during reforming of CH4 with CO2 over Ni/CaO–Al2O3 catalysts studied by various transient techniques. J Catal 161:626
Kim JH, Suh DJ, Park TJ, Kim KL (2000) Effect of metal particle size on coking during CO2 reforming of CH4 over Ni-alumina aerogel catalysts. Appl Catal A 197:191
York AE, Xiao T, Green MH (2003) Brief overview of the partial oxidation of methane to synthesis gas. Top Catal 22:345
De Freitas Silva T, Dias JAC, MacIel CG, Assaf JM (2013) Ni/Al2O3 catalysts: effects of the promoters Ce, La and Zr on the methane steam and oxidative reforming reactions. Catal Sci Technol 3:635
Tomishige K, Chen YG, Fujimoto K (1999) Studies on carbon deposition in CO2 reforming of CH4 over nickel-magnesia solid solution catalysts. J Catal 181:91
Zhang WD, Liu BS, Zhu C, Tian YL (2005) Preparation of La2NiO4/ZSM-5 catalyst and catalytic performance in CO2/CH4 reforming to syngas. Appl Catal A 292:138
Chen X, Huang J, Kong Z, Zhong D (2005) ensitive liquid chromatography–tandem mass spectrometry method for the simultaneous determination of paracetamol and guaifenesin in human plasma. J Chromatogr B Anal Technol Biomed Life Sci 817:263
Acknowledgments
The paper dedicates to the second KACST-Oxford Petrochemical Forum (2012) and ZJ appreciates the lecture invitation from KACST, Saudi Arabia. ZJ acknowledges the financial supports from Shell Foundation, the principal’s major grant at Oxford Jesus College, the grants for international collaborations from British Royal Society (TG092414 and TG101750). ZJ and YZ thank the international collaboration grant (2013DFA40460) from China Ministry of Science and Technology (MOST).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Jiang, Z., Liao, X. & Zhao, Y. Comparative study of the dry reforming of methane on fluidised aerogel and xerogel Ni/Al2O3 catalysts. Appl Petrochem Res 3, 91–99 (2013). https://doi.org/10.1007/s13203-013-0035-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-013-0035-9