
Abstract
The endothelium has increasingly been recognized as a smart barrier and a key regulator of blood flow in micro- and macrovascular beds. Endothelial dysfunction marks a stage of atherosclerosis and is an important prognostic marker for cardiovascular disease. Yet, some people who tend to be slim and physically active and with rather low blood pressure show a propensity to respond to certain stimuli such as emotional stress with endothelial-mediated vascular dysregulation (Flammer syndrome). This leads to characteristic vascular symptoms such as cold hands but also a risk for vascular-mediated diseases such as normal-tension glaucoma. It is the aim of this review to delineate the differences between Flammer syndrome and its “counterpart” endothelial dysfunction in the context of cardiovascular diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The endothelium has increasingly been recognized as a smart barrier and a key regulator of blood flow in micro- and macrovascular circulation. Endothelial dysfunction is a key mediator in the development of atherosclerosis and is present long before atherosclerotic plaques or even cardiovascular events. Endothelial dysfunction in this context can be considered as the “risk of the risk factors” because it depicts the sum of all known, but also hitherto unknown cardiovascular risk factors together [1]. Once endothelial dysfunction is present, it predisposes the vessel to vascular lesions, inflammation, vasoconstriction, thrombosis, and finally plaque rupture. Furthermore, it is an important prognostic marker for cardiovascular events. Interestingly, cardiovascular-protective drugs, healthy nutrition, and lifestyle changes all ameliorate endothelial dysfunction.
On the other side, a dysfunctional endothelium must not necessarily have a connection with atherosclerotic disease. Blood supply to different organs or vascular beds is regulated by the vascular endothelium. A dysfunctional endothelium may in certain persons with a predisposition lead to an inadequate organ perfusion due to vascular dysregulation. Affected persons may respond to certain stimuli, e.g., emotional stress, with endothelial-mediated inadequate vascular constriction or dilatation (Flammer syndrome), leading to characteristic vascular symptoms such as cold hands or vascular-mediated diseases such as normal-tension glaucoma.
It is the aim of this review to first discuss endothelial dysfunction from the cardiologist’s perspective and then to delineate the differences between the Flammer syndrome and its “counterpart” endothelial dysfunction in the context of cardiovascular diseases.
What is endothelial function and how is blood flow regulated?
The concept of cardiovascular pathophysiology over the course of decades has been rewritten for several times by new insights into the function of the endothelium. An organ not easily recognized, this single layer of squamous cells in constant contact with our blood covers an area equivalent to a soccer field. Penicillin Nobel laureate Lord Florey has concluded in 1966 that his purely morphological investigations [2] in this previously assumed mere “sheet of nucleated cellophane” had left much to be desired.
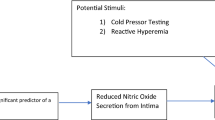
Today, the endothelium is considered a smart barrier and a key regulator of blood flow in micro- and macrovascular beds modulated by paracrine, autocrine, and endocrine response to mechanical and chemical stimuli (Fig. 1). Beyond vascular homeostasis and vascular tone, it orchestrates diverse functions: smooth muscle cell proliferation [3, 4], transendothelial leukocyte diapedesis [5, 6], and thrombosis and thrombolysis [7].

The endothelium actively sends relaxing/dilating and constrictive signals to the smooth muscle cells. Three central pathways are outlined: The constituent endothelial nitric oxide synthase (eNOS, NOS III) is regulated by endocrine and paracrine effects such as endothelin-1 (ETR, ET-1) and acetylcholine (ACh) as well as shear stress via pertussis toxin-sensitive Gq/i pathways, calcium, and calmodulin. Nitric oxide (NO) signals relaxation, but uncoupling can lead to increased oxidative stress (H 2 O 2 ). The endothelial cells also evoke hyperpolarization of the cell membrane of smooth muscle cells (endothelium-dependent hyperpolarization factor (EDHF)). Cyclooxygenase 1 (COX) produces eicosanoids that have in the case of prostacyclin (PGI 2 ) relaxing effects through cyclic AMP or constrictive effects particularly for thromboxane A2 (TXA 2 ). Angiotensin II (A II) has direct (by angiotensin receptor 1 (AT1)) or indirect constrictive effects through ET-1
Perfusion is determined by cardiac output and systemic and local resistance. Locally, blood flow is the result of vascular relaxation and contraction that is balanced by endothelium-derived vasodilatative and vasoconstrictive factors. In impaired function, the scale is tipped towards prevailing constrictive factors and/or downregulated vasodilatative factors. Among these factors, one signal molecule stands out as hub and target of many pathways and mechanisms: nitric oxide (NO).
Alfred Nobel invented dynamite, i.e., explosive nitroglycerin kept in check because it is absorbed in porous kieselguhr. Ironically, he refused to take nitroglycerin for his chest pain, as he rejected the already common knowledge of his time that nitroglycerin could ameliorate angina. It would take another century until 1998 that a Nobel prize was awarded for the discovery of NO’s role in endothelial function. Robert F. Furchgott and colleagues [8] had observed that acetylcholine dilated blood vessels only if the endothelium was intact. He and Louis J. Ignarro identified the endothelium-derived relaxation factor as identical to NO in 1986/1987 [9]. This event triggered a deluge of research about the first gaseous signal molecule ever discovered.
It is important to understand the biochemical foundations of NO for endothelial function. Nitric oxide synthase III (eNOS, encoded in the NOS3 gene) dimerizes in the presence of tetrahydrobiopterin as a cofactor. It exhibits a reductase and an oxidase domain that are conformationally cross-linked after calmodulin [10] binding. In this active conformation, eNOS reduces oxygen on its heme group through an electron chain of NADPH, FAD, and FMN to eventually insert it in l-arginin’s guadino-group to form l-citrullin and NO. The short-lived NO (5–10-s half-life) rapidly diffuses to the surrounding smooth muscle cells to signal via intracellular cGMP [11] resulting in calcium-mediated relaxation, thus vasodilation (Fig. 1). In absence of eNOS’ obligate cofactor tetrahydrobiopterin or its substrate l-arginine (e.g., through oxidative stress), eNOS uncouples to homodimers and instead of NO produces reactive oxygen species (ROS) diminishing NO, rapidly signaling through reactions with proteins and transcription factors a state of distress [12,13,14].
An important counterweight in the vascular balance is cyclooxygenase (COX). Mostly COX1 and if expressed [15] COX2 have a pivotal role in producing vasoconstrictive factors. COXs transform arachidonic acid into endoperoxides and further into thromboxane A2 (TXA2) and prostaglandins D2, E2, and/or F2 α. Prostacyclin [16] through prostacyclin synthase (CYP8A1) is, on the other hand, mainly a dilating factor, a potent antiaggregant, and a direct antagonist to thromboxane A2.
A not exclusively vasoregulatory function of NO is thrombogenesis involving von Willebrand factor. Local presence of thrombin evokes protective NO release. Platelet release of serotonin and ADP in turn increase NO synthesis and release in healthy endothelium to induce dilatation. Without a functional endothelium, thrombus formation is mechanically promoted by vasoconstriction by thromboxane A2 and effect of serotonin directly on the smooth muscle cells.
Various chemical signals (Fig. 1) work in concert to orchestrate contracting factors (e.g., metabolites of arachidonic acid, angiotensin) and vasodilation (foremost regulation of NO bioavailability, prostacyclin, endothelial hyperpolarization factor). Endothelin-1 (ET-1) is two-faced in this balance. A minor share is secreted intraluminally not exclusively by endothelial cells (and particularly in the pulmonary [17] vascular bed). Its primary function is para- and autocrine: On endothelial cells, it induces NO generation through ETB1 receptors, whereas directly acting on ETA and ETB2 receptors on smooth muscle cells (by diffusion or when endothelium is stripped), it acts as a vasoconstrictor about 100-fold more potent than noradrenaline [18]. It modulates cardiac function and propels (or is a marker of) hypertrophy, arrhythmia, chronic heart failure, myocardial infarction, and hypertension with implications of regulatory function for sympathetic innervation [19].
Shear stress, the frictional force generated by blood flow, is a potent mechanical inductor of NO. The molecular mechanisms are mainly calcium-independent [20] and involve posttranslational modification by phosphorylation of eNOS at regulatory sites [21], messenger RNA (mRNA) stabilization [22], e.g., through heat shock protein 90 (hsp-90), and translocation [23]. NO produced in response to shear stress triggers vascular smooth muscle relaxation, inhibition of apoptosis [24], and inhibition of thrombocyte or monocyte adhesion [25]. Repeated transient shear stress induces NOS3 (eNOS gene) transcription and leads thus to chronically higher NO production in response to stimuli. This is one explanation for the beneficial effect of physical exercise [26, 27]. Shear stress is also the key concept for testing endothelial dysfunction in vivo.
How can we measure endothelial function?
A toolbox of measuring methods has been established for research and to some extent for clinical practice. All of which are based on the principle of endothelium-dependent dilatation in response to hyperemia or pharmacological endothelial stimuli (e.g., acetylcholine, bradykinin, serotonin; see Fig. 1). Besides different advantages in technical ease, invasiveness, and reliability, a major difference is whether macro- or microvascular beds are intended to be examined. Biomarkers such as ET-1, circulating endothelial microparticles, and progenitor cells can be analyzed [28].
In 1986, Ludmer and colleagues [29] demonstrated endothelial dysfunction for the first time in vivo. The authors captured epicardial coronary artery dilatation after acetylcholine intracoronary infusion by quantitative coronary angiography in healthy coronaries with intact endothelium. Interestingly, in atherosclerotic arteries, they noticed the opposite effect, vasoconstriction. These findings laid the groundwork to understand the pathogenesis of atherosclerosis and widened the view from structure to function.
Several other techniques employ invasive quantitative coronary angiography or intravascular ultrasound with exercise [30] or mental stress (calculating) [31] as it allows for immediate observation of a clinically important vasculature. A test that relies on sympathetic mediation for NO release is the cold pressor test: A subject submerges her hand for 2 min in ice water. In healthy subjects, α 2-adrenoceptor-dependent vasodilatation as seen, e.g., in quantitative coronary angiography will prevail over α 1-adrenoceptor-mediated constriction (vice versa in dysfunctional endothelium) [32, 33].
In 1992, Science proclaimed NO “molecule of the year” [34] on a dedicated title. This very year, the first non-invasive method to assess endothelial function was published [35] by Celermajer and colleagues. The brachial artery response to shear stress is measured by ultrasound as surrogate for the epicardial coronary arteries, an association later confirmed to be correlated [36]. Flow-mediated dilatation (FMD) is measured sonographically as peak percent dilatation from brachial conduit artery baseline diameter. The measurement is derived from 10-min continuous 2D mode imaging after blood flow had been restricted by inflating an upper arm cuff for 5 min [37] above systolic blood pressure. Hyperemic shear stress is induced after sudden cuff deflation (Fig. 2). To ascertain measurement of endothelial-dependent function, usually the dilatation of the artery in response to a dose of glycerol trinitrate is measured [38, 39].

Method of flow-mediated dilation to measure endothelial dysfunction. The graph shows the diameter at baseline (A), during 5 min of cuff inflation (B), and the reactive hyperemic vasodilation with initial vasoconstriction (C). The diameters were measured on a longitudinal section of the brachial conduit artery in 2D mode. The measurements are automatically computed by FMD Studio (Quipu srl, Pisa)
FMD is probably the most popular method to measure endothelial dysfunction due to its non-invasiveness and correlation with coronary vasoreactivity [40], as well as being mainly NO-dependent [39] (though not exclusively [41]). Strain-gauge venous plethysmography of the forearm circulation resembles FMD but is semi-invasive. In contrast, pharmacological agents (e.g., acetylcholine) are administered by intra-arterial infusion in a dose-dependent manner to test vasoreactivity of the brachial artery. The other forearm serves as internal control due to minimal systemic effect of the infusion [42]. Generally, FMD and plethysmography are better suited for serial or longitudinal studies than between-group testing due to interindividual variability [43].
The microvasculature serves directly to regulate blood flow for tissue metabolic needs and is responsible for 70% of the systemic vascular resistance. Capillary venular interactions can become relevant, myogenic regulation less so, and endothelial anatomy differs in vascular beds (e.g., fenestration, innervation). It comes to no surprise that in two large cross-sectional cohort studies [44, 45], non-invasive FMD of the brachial conduit artery and digital vascular dysfunction only modestly correlated and differed in their relation to traditional cardiovascular risk factors [46]. Thus, they might reflect different aspects of vascular biology [47].
In peripheral pneumatic arterial tonometry (PAT), digital pulse wave amplitude at baseline (preocclusion) and changes during reactive hyperemia are recorded and the reactive hyperemia-PAT index (RHI) is calculated as a standardized post-/preocclusion ratio [47, 48]. RHI is correlated with coronary [49] endothelial dysfunction. The main advantages of the EndoPAT analysis are that interrater and test-retest reliability are high while being non-invasive [48]. The peripheral vascular bed at distal limbs is the major site of sympathetic α-adrenergic vasoconstrictor activity [50, 51]. The use of the specific NO antagonist l-NMMA has defined the contribution of NO to these vasomotor responses: Approximately 60% of the PAT response is mediated by NO release [52, 53]; the rest is probably mediated by the autonomous nervous system.
The eye as a direct window to vascular function has an appeal beyond structural assessment [54]. Static and dynamic retinal vessel analyses (RVA) are unique and elegant in several ways. The retina is the only vascular bed of which we are sure that responses to stimuli are independent of the autonomic innervation. In addition, due to the blood-retinal-barrier, most circulating vasoactive molecules do not get direct access to smooth muscle cells. In other words, it is the ideal place to study the function of the vascular endothelium.
Static RVA (Fig. 3) can be reproducibly and automatically performed on routine fundus photographs [56]: arteriovenous ratio is measured (semi-)automatically as the ratio of equivalents of arteriolar and venular diameters in a 0.5–1 optic disc diameter distance around the optic nerve head [57]. The smaller and larger diameters are approximately normally distributed which allows for calculation of the equivalents according to Parr and Hubbard [58,59,60]. The ratio is a cardiovascular risk factor and predictor of long-term outcome [61].

Static retinal vessel analysis by semi-automatically tracing all vessels in a range of 0.5 to 1 its diameter around the optic disc. By dividing the sum of diameters of arterioles and venules, the arteriovenous ratio is calculated. The “Atherosclerosis in Community Study” (ARIC) has widely validated association of the arteriovenous ratio with cardiovascular risk and outcome. Normal values are age-dependent and lie in a range of 0.75–1 [55]
Dynamic RVA (DVA) induces shear stress through flicker light stimulation. The retina is filmed by a charge-coupled camera to determine baseline diameters and while episodes of optoelectric diffuse flicker light alternate with 80-s constant illumination phases. Uniquely, shear stress is not induced by reperfusion reactive hyperemia but functional hyperemia by neurovascular coupling. The exact pathways involved in neuro-glia-vascular coupling are not fully understood. Likely, oxygen demand in response to flicker light increases [62] and stimulated neurons signal a calcium concentration surge in Müller glia that release vasodilatory prostaglandin E2, NO, and epoxyeicosatrienoic acids [63, 64]. The gliovascular unit, thus, relaxes the smallest arteriolar walls. Due to then less resistant smallest vessels, blood starts flowing faster, which increases shear stress in bigger arteriolar segments. An intact retinal arteriolar endothelium responds to the shear stress by vasodilatation to keep shear stress constant (Fig. 4). NO has been confirmed to play a major role in retinal flicker-induced vasodilatation [68]. However, it cannot be excluded that a disturbed or reduced neurovascular coupling itself is involved as well: neurovascular coupling depends on the function of the astrocytes [69] and these astrocytes are particularly often “activated” (and therefore altered in their function) in subjects with Flammer syndrome [70].

Description of dynamic retinal vessel analysis. a Video automated tracing of marked arteriole (red) and venula (blue) for tracing during flicker light. b Relative diameter over time: baseline, dilatation due to flicker light (during gray-shaded area) expressed as relative diameter compared to baseline (100%), and return to baseline. Of note, normal values are in a range of 2–12% and a 19% increment in radius will double the volume flow rate according to Poiseuille’s law. Also baseline amplitude, slope, and postdilatative contraction can be measured [65]. Importantly, both arterial and venous (c) dilatative response have been linked to disease [66, 67]. Green line: normal range, red/blue lines: normal flicker-induced arterial/venous dilatation, dark gray line: pathological, blunted response to flicker light
Endothelial dysfunction—a hallmark of cardiovascular disease
Loss of NO bioavailability is the salient feature of a dysfunctional endothelium, which in turn is the sentinel of systemic or focal vascular disease. Presence of classic cardiovascular risk factors is associated with deteriorated endothelial function, [1] and conversely, risk factor modification is connected with its improvement. That a considerable number of patients at risk cannot be identified based on conventional risk factors [71] as incorporated in the Framingham risk score prompted the quest for other relevant risk predictors.
The prognostic value of FMD as surrogate for endothelial function has first been shown for hypertension. Modena and colleagues [72] measured how FMD changed in 400 postmenopausal women before and 6 months after establishment of optimal blood pressure control. Women responding to therapy with >10% FMD improvement had significantly fewer events during 67 month follow-up. In the Multiethnic Study of Atherosclerosis, healthy elderly (>72 years) had a significantly longer event-free survival over a 5-year follow-up if they initially had normal endothelial function [73].
The Atherosclerosis Risk in Communities (ARIC) study generated much insight in the association of microvascular retinal endothelial dysfunction and systemic risks and showed predictive value for static RVA [74,75,76,77]. Over 16 years of follow-up in 10,470 initially healthy individuals, it was shown that narrower retinal arterioles and wider venules confer a significant risk for ischemic stroke and all-cause mortality [61].
The etiology of atherosclerosis is a complex process that starts long before structural manifestation and their debilitating sequelae. In fact, there is evidence for onset of the atherosclerotic process already in early adulthood and even childhood [35]. Dyslipidemia is a major factor in development and progression of atherosclerosis. Oxidated LDL (ox-LDL) downregulates eNOS/NOS3 expression [3]. Endothelin through ET-B receptor augments ox-LDL uptake in plaques and upregulates lectin-like ox-LDL receptor-1 mRNA that facilitates ox-LDL internalization [78, 79]. A process called “uncoupling” of eNOS is initiated triggering the production of superoxide anions (O2 −) [80, 81]. This potent free radical is a messenger molecule and, in a vicious cycle, propels the production of ox-LDL. The thus impaired endothelial cell signals proliferation to smooth muscle cells with subsequent extracellular matrix changes in the intima compartment. The inflammatory process starts with the attraction of monocytes. Transendothelial migration of monocytes and subsequent transformation in foam cells are the result. Importantly, exposition to undisturbed laminar flow enhances NO-forming capacity and is strongly protective [82].
A fundamental pathomechanistic insight can be gained by scrutiny of the endothelium. Takotsubo syndrome [83] often presents with symptoms of an acute coronary syndrome but is typically [84] characterized by apical ballooning of the heart that restitutes within weeks. No or no relevant stenoses of the epicardial coronaries are found, and biomarkers of cardiac muscle necrosis like troponin are only slightly elevated. In approximately two thirds of all cases, an intense (positive [85] or negative) emotional or physical stress precedes the transient, reversible, systolic dysfunction of the left ventricle (“broken heart syndrome”). About 90% of Takotsubo cardiopathy patients are female [86]. As evidently epicardial coronary artery stenosis is no explanation, hypotheses for the etiopathology of Takotsubo cardiopathy soon focused on transient vascular spasm, microvascular functional impairment, and autonomous nervous dysfunction. These hypotheses were supported by a recent study: FMD was found to be significantly reduced in Takotsubo patients in a stable phase whereas sympathetic activity in response to stress tended to be increased [43].
Endothelial dysfunction plays a role not only as a marker of disease progression but, conversely, as a response marker to interventions. Dietary interventions [4], e.g., with flavonoids such as in dark chocolate [87, 88], led to improved endothelial function, as did physical exercise [74, 89,90,91]. Quantification of endothelial dysfunction can predict therapeutic success of pharmacologic intervention, too. Angiotensin-converting enzyme inhibitors (ACE-I) showed clear improvement of endothelial dysfunction in several modalities [92,93,94]. Additional improvement could be shown for add-on spironolactone in chronic heart failure patients [95]. Calcium channel blockers are vasodilators acting on L-type calcium channels in vascular smooth muscle cells. Some calcium channel blockers activate endothelial NO synthase or have antioxidative properties, thus increasing NO bioavailability. Nifedipine improved coronary endothelial function in stable coronary artery disease patients in the ENCORE II study [96]. Endothelial dysfunction, thus, continues to be a relevant mechanistic explanation variable in recent and future intervention studies.
Endothelial dysregulation in Flammer syndrome
At first glimpse, the symptoms constituting Flammer syndrome (FS, primary vascular dysregulation) do not seem to fit together and lack a common pathophysiologic basis. Historically, the first two dots of FS’s diverse symptoms were connected by the observation that some glaucoma patients had conspicuously cold hands [97, 98] (please see article by to Prof. J. Flammer and Dr. K. Konieczka’s in this volume for a scientific detective story of the history and findings in Flammer syndrome).
Crucially, people with FS seem less prone to atherosclerosis but exhibit signs of endothelial dysfunction. Endothelial dysfunction appears to take on a different role in vascular dysregulation than in atherosclerotic etiology. The phenotype of FS is already suggestive of protective factors against atherosclerosis: People with FS tend to be more physically active and slimmer than patients with atherosclerosis particularly with metabolic syndrome [99]. As an overall principle underlying FS, an increased sensitivity to emotional or physical stress emerges as trigger of varying blood perfusion with transient malsupply [100]. This, in turn, causes oxidative stress and impaired endothelial function [101].
Identification of individuals with FS may be clinically relevant as systemic vascular dysregulation has been associated with several ocular diseases [102], including glaucoma [103,104,105,106,107,108,109,110], central serous chorioretinopathy [111], and central retinal vein occlusion. The role of the endothelium as major pathomechanistic red line connects many dots that outline a sketch of FS. Particularly, ET-1 (ET-1), autonomic dysfunction, and oxidative stress may link the differing vascular beds involved in FS. Consequently, FS may inflict heart [109] and brain [112, 113] as a systemic state.
Microvascular regulation and its failure may be a more crucial aspect to FS than macrovascular dysfunction that is also commonly found in atherosclerotic patients. Not only atherosclerosis but also microvascular dysfunction affects the heart. The link to Takotsubo syndrome or microvascular angina may yet to be substantiated, but evidence for transient hypoperfusion of the heart has been described [114, 115]. In 24-h EKG recordings of glaucoma patients, transient especially nocturnal ST elevations have been recorded, a typical sign of cardiac ischemia.
It has been hypothesized that an imbalance of ET-1 and endothelial NO due to impaired endothelium is a crucial pathomechanism in FS disturbing ocular perfusion [101].
The importance of ET-1 (or circulatory vasoconstrictors) arises from the extraordinary blood supply of and around the optic nerve head (ONH): superficial layers receive blood from central retinal artery branches with a blood-brain barrier. The prelaminar region (anterior to the lamina cribrosa), on the other hand, is a pure capillary circulation supplied by choroidal artery branches and the short posterior ciliary arteries, which do not possess a blood-brain barrier. The retinal vessels completely lack autonomic innervation. The regulators of the size of retinal vessels are, thus, the vascular endothelial cells. An effect from systemic ET-1 is likely if the blood-brain barrier is disrupted [66, 116]. The blood-brain barrier in the ONH is incomplete [117]. ET-1 locally produced in response to hypoxia could exert vasoconstrictive effects on and around the ONH by local spillover despite intact endothelium. One reason for this is that molecules can freely diffuse from the choroid into the ONH; another is the cross talk between arteries and veins in areas where they share a common adventitia.
In normal-tension glaucoma (NTG), for which FS is an important risk factor [97, 98], circulating ET-1 is elevated and even more so than in high-tension glaucoma (HTG) [118]. ET-1, on the other hand, reduces ocular blood flow but does not interfere with autoregulation. In FS, the capacity to avoid too little or too much perfusion is fundamentally disturbed. The glaucoma pathomechanism as explained by impaired autoregulation in FS is as follows: transient hypoxia leads to oxidative stress that further impairs not only endothelium but the ONH. The ONH damage explains the bundle-shaped damage and loss of retinal ganglion cells and their axons [107]. Hypoxia (e.g., by varying blood supply in FS) in the retina induces ET-1 production mainly by unoxidized hypoxia-induced transcription factor 1α (HIF-1α) that serves as a “master switch” [119] that activates (among others) transcription of ET-1 and VEGF [120].
Autonomic dysfunction is suggested by the observation of cold extremities (hands, feet, cornea) exacerbated by cold or emotional upset [97, 116]. Cold acra have been found in FS similar to Raynaud’s phenomenon (CREST) or even Raynaud’s disease, yet less pronounced [98]. The role of endothelial dysfunction in primary and secondary Raynaud’s phenomenon has been demonstrated with evidence from various modalities including autonomic nervous stress tests like the cold pressor test [121,122,123,124,125]. It may be regarded a heterogeneous syndrome involving endothelial dysfunction as a key player but not every form entails a propensity for atherosclerosis. Which pathway contributes most to malicious vasoconstriction in Raynaud’s disease remains open.
Conclusion: two endotheliopathies, two collectives at risk
Endothelial dysfunction is classically regarded as the first phase of atherosclerosis. Not only does it contribute to atherogenesis but it serves as a measurable surrogate marker. Much evidence has been gathered that it marks atherosclerotic progression and that its role as a marker holds true also in the reverse direction: Lifestyle changes like physical activity, dietary intervention, and drugs such as ACE inhibitors measurably improve endothelial dysfunction. Outcome studies be it in the realm of prevention medicine or pharmacotherapeutic interventions have established the association of ameliorated endothelial dysfunction with improved hard endpoints in morbidity and mortality.
Quite different facets of the endothelium’s important role in vascular health become apparent in FS. One concerns a core role particularly of the microvasculature: perfusion. Varying blood flow ensuing transient malperfusion is a risk factor for many ocular and other diseases. Increased reactivity to stimuli such as cold and emotional stress underlies, thus, a complex phenotype at heightened risk. The FS phenotype in many respects directly contrasts the typical patient with endothelial dysfunction in atherosclerosis (cf. Fig. 7 in Flammer, Konieczka: “The discovery of the Flammer syndrome” in this volume). Classically, endothelial dysfunction has been regarded as an integrative measure of cardiovascular risk comprising other traditional factors such as hypertension and metabolic syndrome, the integral of all risk factors. FS challenges our view in the presence of endothelial dysfunction in a collective that is diametrically opposed to the typical patient at risk for cardiovascular events: active, slim, normo- or hypotensive, and less prone to metabolic syndrome [126]. Is too much of good bad?
Understanding of endothelial dysfunction of small vessels aids to connect symptoms and signs that are not obviously related, at first sight. Many areas are yet to be explored, e.g., the contribution of metabolic processes as well as the role of genetic and epigenetic factors in either of the two different endotheliopathies. Many open questions warrant further studies to help delineate FS, identify FS individuals, and ideally enable us to offer preventive interventions to avoid sequelae and ameliorate symptoms.
This article, thus, conforms with the PPPM principles of preventive measures and patient stratification stated in the “EPMA White Paper” [127].
References
Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23(2):168–75.
Florey. The endothelial cell. Br Med J. 1966;2(5512):487–90.
Vanhoutte PM, Shimokawa H, Tang EHC, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol. 2009;196(2):193–222.
Sudano I, Spieker LE, Hermann F, Flammer A, Corti R, Noll G, et al. Protection of endothelial function: targets for nutritional and pharmacological interventions. J Cardiovasc Pharmacol. 2006;47(Suppl 2):S136–50. discussion S72-6
Speer T, Rohrer L, Blyszczuk P, Shroff R, Kuschnerus K, Krankel N, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of toll-like receptor-2. Immunity. 2013;38(4):754–68.
Mudau M, Genis A, Lochner A, Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. 2012;23(4):10.
Lüscher TFVP. The endothelium: modulator of cardiovascular function. Boca Ranton: CRC Press; 1990.
Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373–6.
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84(24):9265–9.
Busse R, Mulsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265(1–2):133–6.
Ignarro LJ, Harbison RG, Wood KS, Kadowitz PJ. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J Pharmacol Exp Ther. 1986;237(3):893–900.
Cosentino F, Katusic ZS. Tetrahydrobiopterin and dysfunction of endothelial nitric oxide synthase in coronary arteries. Circulation. 1995;91(1):139–44.
Harrison DG, Ohara Y. Physiologic consequences of increased vascular oxidant stresses in hypercholesterolemia and atherosclerosis: implications for impaired vasomotion. Am J Cardiol. 1995;75(6):75B–81B.
Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113(13):1708–14.
Camacho M, Lopez-Belmonte J, Vila L. Rate of vasoconstrictor prostanoids released by endothelial cells depends on cyclooxygenase-2 expression and prostaglandin I synthase activity. Circ Res. 1998;83(4):353–65.
Moncada S, Higgs EA, Vane JR. Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregation. Lancet. 1977;1(8001):18–20.
Tsutamoto T, Wada A, Maeda Y, Adachi T, Kinoshita M. Relation between endothelin-1 spillover in the lungs and pulmonary vascular resistance in patients with chronic heart failure. J Am Coll Cardiol. 1994;23(6):1427–33.
Miyauchi T, Tomobe Y, Shiba R, Ishikawa T, Yanagisawa M, Kimura S, et al. Involvement of endothelin in the regulation of human vascular tonus. Potent vasoconstrictor effect and existence in endothelial cells. Circulation. 1990;81(6):1874–80.
Lehmann LH, Stanmore DA, Backs J. The role of endothelin-1 in the sympathetic nervous system in the heart. Life Sci. 2014;118(2):165–72.
Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, et al. Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem. 2000;275(7):5179–87.
Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Phys Cell Phys. 2003;285(3):C499–508.
Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res. 2000;86(3):347–54.
Shaul PW. Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol. 2002;64:749–74.
Dimmeler S, Hermann C, Galle J, Zeiher AM. Upregulation of superoxide dismutase and nitric oxide synthase mediates the apoptosis-suppressive effects of shear stress on endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19(3):656–64.
De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96(1):60–8.
Endres M, Gertz K, Lindauer U, Katchanov J, Schultze J, Schrock H, et al. Mechanisms of stroke protection by physical activity. Ann Neurol. 2003;54(5):582–90.
Gertz K, Priller J, Kronenberg G, Fink KB, Winter B, Schrock H, et al. Physical activity improves long-term stroke outcome via endothelial nitric oxide synthase-dependent augmentation of neovascularization and cerebral blood flow. Circ Res. 2006;99(10):1132–40.
Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet JM, et al. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation. 2000;101(8):841–3.
Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315(17):1046–51.
Gordon JB, Ganz P, Nabel EG, Fish RD, Zebede J, Mudge GH, et al. Atherosclerosis influences the vasomotor response of epicardial coronary arteries to exercise. J Clin Invest. 1989;83(6):1946–52.
Yeung AC, Vekshtein VI, Krantz DS, Vita JA, Ryan TJ Jr, Ganz P, et al. The effect of atherosclerosis on the vasomotor response of coronary arteries to mental stress. N Engl J Med. 1991;325(22):1551–6.
Wood DL, Sheps SG, Elveback LR, Schirger A. Cold pressor test as a predictor of hypertension. Hypertension. 1984;6(3):301–6.
Sudano I, Spieker L, Binggeli C, Ruschitzka F, Luscher TF, Noll G, et al. Coffee blunts mental stress-induced blood pressure increase in habitual but not in nonhabitual coffee drinkers. Hypertension. 2005;46(3):521–6.
Koshland DE Jr. The molecule of the year. Science. 1992;258(5090):1861.
Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340(8828):1111–5.
Takase B, Uehata A, Akima T, Nagai T, Nishioka T, Hamabe A, et al. Endothelium-dependent flow-mediated vasodilation in coronary and brachial arteries in suspected coronary artery disease. Am J Cardiol. 1998;82(12):1535–9. A7-8
Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39(2):257–65.
Sudano I, Flammer AJ, Periat D, Enseleit F, Hermann M, Wolfrum M, et al. Acetaminophen increases blood pressure in patients with coronary artery disease. Circulation. 2010;122(18):1789–96.
Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, et al. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91(5):1314–9.
Anderson TJ, Uehata A, Gerhard MD, Meredith IT, Knab S, Delagrange D, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26(5):1235–41.
Parker BA, Tschakovsky ME, Augeri AL, Polk DM, Thompson PD, Kiernan FJ. Heterogenous vasodilator pathways underlie flow-mediated dilation in men and women. Am J Physiol Heart Circ Physiol. 2011;301(3):H1118–26.
Linder L, Kiowski W, Buhler FR, Luscher TF. Indirect evidence for release of endothelium-derived relaxing factor in human forearm circulation in vivo. Blunted response in essential hypertension. Circulation. 1990;81(6):1762–7.
Naegele M, Flammer AJ, Enseleit F, Roas S, Frank M, Hirt A, et al. Endothelial function and sympathetic nervous system activity in patients with Takotsubo syndrome. Int J Cardiol. 2016;224:226–30.
Schnabel RB, Schulz A, Wild PS, Sinning CR, Wilde S, Eleftheriadis M, et al. Noninvasive vascular function measurement in the community: cross-sectional relations and comparison of methods. Circ Cardiovasc Imaging. 2011;4(4):371–80.
Hamburg NM, Palmisano J, Larson MG, Sullivan LM, Lehman BT, Vasan RS, et al. Relation of brachial and digital measures of vascular function in the community: the Framingham heart study. Hypertension. 2011;57(3):390–6.
Hamburg NM, Keyes MJ, Larson MG, Vasan RS, Schnabel R, Pryde MM, et al. Cross-sectional relations of digital vascular function to cardiovascular risk factors in the Framingham Heart Study. Circulation. 2008;117(19):2467–74.
Dhindsa M, Sommerlad SM, DeVan AE, Barnes JN, Sugawara J, Ley O, et al. Interrelationships among noninvasive measures of postischemic macro- and microvascular reactivity. J Appl Physiol (1985). 2008;105(2):427–32.
Patvardhan EA, Heffernan KS, Ruan JM, Soffler MI, Karas RH, Kuvin JT. Assessment of vascular endothelial function with peripheral arterial tonometry: information at your fingertips? Cardiol Rev. 2010;18(1):20–8.
Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Kuvin JT, Lerman A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J Am Coll Cardiol. 2004;44(11):2137–41.
Burton AC. The range and variability of the blood flow in the human fingers and the vasomotor regulation of body temperature. Am J Phys. 1939;127:437–53.
Celermajer DS. Reliable endothelial function testing: at our fingertips? Circulation. 2008;117(19):2428–30.
Nohria A, Gerhard-Herman M, Creager MA, Hurley S, Mitra D, Ganz P. Role of nitric oxide in the regulation of digital pulse volume amplitude in humans. J Appl Physiol (1985). 2006;101(2):545–8.
Noon JP, Haynes WG, Webb DJ, Shore AC. Local inhibition of nitric oxide generation in man reduces blood flow in finger pulp but not in hand dorsum skin. J Physiol. 1996;490(Pt 2):501–8.
Flammer J, Konieczka K, Bruno RM, Virdis A, Flammer AJ, Taddei S. The eye and the heart. Eur Heart J. 2013;34(17):1270–8.
Wong TY, Klein R, Couper DJ, Cooper LS, Shahar E, Hubbard LD, et al. Retinal microvascular abnormalities and incident stroke: the Atherosclerosis Risk in Communities Study. Lancet. 2001;358(9288):1134–40.
Neubauer AS, Ludtke M, Haritoglou C, Priglinger S, Kampik A. Retinal vessel analysis reproducibility in assessing cardiovascular disease. Optom Vis Sci. 2008;85(4):247–54.
Heitmar R, Summers RJ. Assessing vascular function using dynamic retinal diameter measurements: a new insight on the endothelium. Thromb Haemost. 2012;107(6):1019–26.
Hubbard LD, Brothers RJ, King WN, Clegg LX, Klein R, Cooper LS, et al. Methods for evaluation of retinal microvascular abnormalities associated with hypertension/sclerosis in the Atherosclerosis Risk in Communities Study. Ophthalmology. 1999;106(12):2269–80.
Parr JC, Spears GF. Mathematic relationships between the width of a retinal artery and the widths of its branches. Am J Ophthalmol. 1974;77(4):478–83.
Parr JC, Spears GF. General caliber of the retinal arteries expressed as the equivalent width of the central retinal artery. Am J Ophthalmol. 1974;77(4):472–7.
Seidelmann SB, Claggett B, Bravo PE, Gupta A, Farhad H, Klein BE, et al. Retinal vessel calibers in predicting long-term cardiovascular outcomes: the Atherosclerosis Risk in Communities Study. Circulation. 2016;134(18):1328–38.
Hammer M, Vilser W, Riemer T, et al. Retinal venous oxygen saturation increases by flicker light stimulation. Invest Ophthalmol Vis Sci. 2017;52(1):274–7.
Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. 2006;26(11):2862–70.
Newman EA. Functional hyperemia and mechanisms of neurovascular coupling in the retinal vasculature. J Cereb Blood Flow Metab. 2013;33(11):1685–95.
Mroczkowska S, Ekart A, Sung V, Negi A, Qin L, Patel SR, et al. Coexistence of macro- and micro-vascular abnormalities in newly diagnosed normal tension glaucoma patients. Acta Ophthalmol. 2012;90(7):e553–9.
Flammer J, Konieczka K. Retinal venous pressure: the role of endothelin. EPMA J. 2015;6(1):21.
Nguyen TT, Kawasaki R, Wang JJ, Kreis AJ, Shaw J, Vilser W, et al. Flicker light–induced retinal vasodilation in diabetes and diabetic retinopathy. Diabetes Care. 2009;32(11):2075–80.
Lakshminarayanan S, Gardner TW, Tarbell JM. Effect of shear stress on the hydraulic conductivity of cultured bovine retinal microvascular endothelial cell monolayers. Curr Eye Res. 2000;21(6):944–51.
Filosa JA, Morrison HW, Iddings JA, Du W, Kim KJ. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience. 2016;323:96–109.
Grieshaber MC, Orgul S, Schoetzau A, Flammer J. Relationship between retinal glial cell activation in glaucoma and vascular dysregulation. J Glaucoma. 2007;16(2):215–9.
Greenland P, Smith SC Jr, Grundy SM. Improving coronary heart disease risk assessment in asymptomatic people: role of traditional risk factors and noninvasive cardiovascular tests. Circulation. 2001;104(15):1863–7.
Modena MG, Bonetti L, Coppi F, Bursi F, Rossi R. Prognostic role of reversible endothelial dysfunction in hypertensive postmenopausal women. J Am Coll Cardiol. 2002;40(3):505–10.
Yeboah J, Folsom AR, Burke GL, Johnson C, Polak JF, Post W, et al. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation. 2009;120(6):502–9.
Tikellis G, Anuradha S, Klein R, Wong TY. Association between physical activity and retinal microvascular signs: the Atherosclerosis Risk in Communities (ARIC) Study. Microcirculation. 2010;17(5):381–93.
Lesage SR, Mosley TH, Wong TY, Szklo M, Knopman D, Catellier DJ, et al. Retinal microvascular abnormalities and cognitive decline: the ARIC 14-year follow-up study. Neurology. 2009;73(11):862–8.
Pellanda LC, Duncan BB, Vigo A, Rose K, Folsom AR, Erlinger TP, et al. Low birth weight and markers of inflammation and endothelial activation in adulthood: the ARIC study. Int J Cardiol. 2009;134(3):371–7.
Miller EA, Pankow JS, Millikan RC, Bray MS, Ballantyne CM, Bell DA, et al. Glutathione-S-transferase genotypes, smoking, and their association with markers of inflammation, hemostasis, and endothelial function: the atherosclerosis risk in communities (ARIC) study. Atherosclerosis. 2003;171(2):265–72.
Masaki T. Endothelial dysfunction and LOX-1: forty years from muscle to endothelium. Circ Res. 2003;92(8):819–20.
Mehta JL, Basnakian AG. Interaction of carbamylated LDL with LOX-1 in the induction of endothelial dysfunction and atherosclerosis. Eur Heart J. 2014;35(43):2996–7.
Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem. 2001;276(18):14533–6.
Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res. 1999;43(3):562–71.
Gimbrone MA Jr, Garcia-Cardena G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. 2013;22(1):9–15.
Ghadri JR, Ruschitzka F, Luscher TF, Templin C. Takotsubo cardiomyopathy: still much more to learn. Heart. 2014;100(22):1804–12.
Ghadri JR, Cammann VL, Napp LC, Jurisic S, Diekmann J, Bataiosu DR, et al. Differences in the clinical profile and outcomes of typical and atypical Takotsubo syndrome: data from the International Takotsubo Registry. JAMA Cardiol. 2016;1(3):335–40.
Ghadri JR, Sarcon A, Diekmann J, Bataiosu DR, Cammann VL, Jurisic S, et al. Happy heart syndrome: role of positive emotional stress in Takotsubo syndrome. Eur Heart J. 2016;37(37):2823–9.
Templin C, Ghadri JR, Diekmann J, Napp LC, Bataiosu DR, Jaguszewski M, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929–38.
Flammer AJ, Hermann F, Sudano I, Spieker L, Hermann M, Cooper KA, et al. Dark chocolate improves coronary vasomotion and reduces platelet reactivity. Circulation. 2007;116(21):2376–82.
Flammer AJ, Sudano I, Wolfrum M, Thomas R, Enseleit F, Periat D, et al. Cardiovascular effects of flavanol-rich chocolate in patients with heart failure. Eur Heart J. 2012;33(17):2172–80.
Schmidt A, Pleiner J, Bayerle-Eder M, Wiesinger GF, Rodler S, Quittan M, et al. Regular physical exercise improves endothelial function in heart transplant recipients. Clin Transpl. 2002;16(2):137–43.
Gielen S, Hambrecht R. Treatment strategies in endothelial dysfunction: physical exercise versus pharmacological therapy. Eur J Cardiovasc Prev Rehabil. 2005;12(4):318–20.
Ghisi GL, Durieux A, Pinho R, Benetti M. Physical exercise and endothelial dysfunction. Arq Bras Cardiol. 2010;95(5):e130–7.
Shahin Y, Khan JA, Samuel N, Chetter I. Angiotensin converting enzyme inhibitors effect on endothelial dysfunction: a meta-analysis of randomised controlled trials. Atherosclerosis. 2011;216(1):7–16.
Cannon RO 3rd. Potential mechanisms for the effect of angiotensin-converting enzyme inhibitors on endothelial dysfunction: the role of nitric oxide. Am J Cardiol. 1998;82(10A):8S–10S.
Flammer AJ, Hermann F, Wiesli P, Schwegler B, Chenevard R, Hurlimann D, et al. Effect of losartan, compared with atenolol, on endothelial function and oxidative stress in patients with type 2 diabetes and hypertension. J Hypertens. 2007;25(4):785–91.
Bauersachs J, Heck M, Fraccarollo D, Hildemann SK, Ertl G, Wehling M, et al. Addition of spironolactone to angiotensin-converting enzyme inhibition in heart failure improves endothelial vasomotor dysfunction: role of vascular superoxide anion formation and endothelial nitric oxide synthase expression. J Am Coll Cardiol. 2002;39(2):351–8.
Luscher TF, Pieper M, Tendera M, Vrolix M, Rutsch W, van den Branden F, et al. A randomized placebo-controlled study on the effect of nifedipine on coronary endothelial function and plaque formation in patients with coronary artery disease: the ENCORE II study. Eur Heart J. 2009;30(13):1590–7.
Saner H, Wurbel H, Mahler F, Flammer J, Gasser P. Microvasculatory evaluation of vasospastic syndromes. Adv Exp Med Biol. 1987;220:215–8.
Konieczka K, Ritch R, Traverso CE, Kim DM, Kook MS, Gallino A, et al. Flammer syndrome. EPMA J. 2014;5(1):11.
Flammer J, Konieczka K. Glaukom. Swiss Med Forum. 2017;17(5):105–12.
Flammer J, Pache M, Resink T. Vasospasm, its role in the pathogenesis of diseases with particular reference to the eye. Prog Retin Eye Res. 2001;20(3):319–49.
Grieshaber MC, Mozaffarieh M, Flammer J. What is the link between vascular dysregulation and glaucoma? Surv Ophthalmol. 2007;52(Suppl 2):S144–54.
Flammer J, Pache M, Resink T. Vasospasm, its role in the pathogenesis of diseases with particular reference to the eye. Prog Retin Eye Res. 2001;20(3):319–49.
Hafez AS, Bizzarro R, Descovich D, Lesk MR. Correlation between finger blood flow and changes in optic nerve head blood flow following therapeutic intraocular pressure reduction. J Glaucoma. 2005;14(6):448–54.
Buckley C, Hadoke PW, Henry E, O’Brien C. Systemic vascular endothelial cell dysfunction in normal pressure glaucoma. Br J Ophthalmol. 2002;86(2):227–32.
Findl O, Rainer G, Dallinger S, Dorner G, Polak K, Kiss B, et al. Assessment of optic disk blood flow in patients with open-angle glaucoma. Am J Ophthalmol. 2000;130(5):589–96.
Flammer J, Haefliger IO, Orgul S, Resink T. Vascular dysregulation: a principal risk factor for glaucomatous damage? J Glaucoma. 1999;8(3):212–9.
Flammer J, Mozaffarieh M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv Ophthalmol. 2007;52:S162–S73.
Flammer J, Orgul S, Costa VP, Orzalesi N, Krieglstein GK, Serra LM. The impact of ocular blood flow in glaucoma. Prog Retin Eye Res. 2002;21(4):359-93.
Henry E, Newby DE, Webb DJ, O’Brien C. Peripheral endothelial dysfunction in normal pressure glaucoma. Invest Ophthalmol Vis Sci. 1999;40(8):1710–4.
Neufeld AH. Nitric oxide: a potential mediator of retinal ganglion cell damage in glaucoma. Surv Ophthalmol. 1999;43:S129–S35.
Gugleta K, Orgul S, Hasler PW, Picornell T, Gherghel D, Flammer J. Choroidal vascular reaction to hand-grip stress in subjects with vasospasm and its relevance in glaucoma. Invest Ophthalmol Vis Sci. 2003;44(4):1573–80.
Huemer KH, Garhofer G, Zawinka C, Golestani E, Litschauer B, Schmetterer L, et al. Effects of dopamine on human retinal vessel diameter and its modulation during flicker stimulation. Am J Phys Heart Circ Phys. 2003;284(1):H358–H63.
Gugleta K, Zawinka C, Rickenbacher I, Kochkorov A, Katamay R, Flammer J, et al. Analysis of retinal vasodilation after flicker light stimulation in relation to vasospastic propensity. Invest Ophthalmol Vis Sci. 2006;47(9):4034–41.
Kaiser HJ, Flammer J, Burckhardt D. Silent myocardial ischemia in glaucoma patients. Ophthalmologica. 1993;207(1):6–7.
Waldmann E, Gasser P, Dubler B, Huber C, Flammer J. Silent myocardial ischemia in glaucoma and cataract patients. Graefes Arch Clin Exp Ophthalmol. 1996;234(10):595–8.
Flammer J, Konieczka K, Flammer AJ. The primary vascular dysregulation syndrome: implications for eye diseases. EPMA J. 2013;4(1):14.
Hofman P, Hoyng P, van der Werf F, Vrensen GF, Schlingemann RO. Lack of blood-brain barrier properties in microvessels of the prelaminar optic nerve head. Invest Ophthalmol Vis Sci. 2001;42(5):895–901.
Kaiser HJ, Flammer J, Wenk M, Luscher T. Endothelin-1 plasma levels in normal-tension glaucoma: abnormal response to postural changes. Graefes Arch Clin Exp Ophthalmol. 1995;233(8):484–8.
Arjamaa O, Nikinmaa M. Oxygen-dependent diseases in the retina: role of hypoxia-inducible factors. Exp Eye Res. 2006;83(3):473–83.
Desai D, He S, Yorio T, Krishnamoorthy RR, Prasanna G. Hypoxia augments TNF-alpha-mediated endothelin-1 release and cell proliferation in human optic nerve head astrocytes. Biochem Biophys Res Commun. 2004;318(3):642–8.
Latuskiewicz-Potemska J, Chmura-Skirlinska A, Gurbiel RJ, Smolewska E. Nailfold capillaroscopy assessment of microcirculation abnormalities and endothelial dysfunction in children with primary or secondary Raynaud syndrome. Clin Rheumatol. 2016;35(8):1993–2001.
Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol. 2008;35(9):1801–8.
Easter MJ, Marshall JM. Contribution of prostanoids to endothelium-dependent vasodilatation in the digital circulation of women with primary Raynaud’s disease. Clin Sci (Lond). 2005;109(1):45–54.
Czupryniak A, Kaluzynska A, Nowicki M, Wiecek B, Bald E, Owczarek D. Raynaud’s phenomenon and endothelial dysfunction in end-stage renal disease patients treated with hemodialysis. Kidney Blood Press Res. 2005;28(1):27–31.
Cooke JP, Marshall JM. Mechanisms of Raynaud’s disease. Vasc Med. 2005;10(4):293–307.
Park AY, Cha S. Effects of cold sensitivity in the extremities on circulating adiponectin levels and metabolic syndrome in women. BMC Complement Altern Med. 2017;17(1):150.
Golubnitschaja O, Costigliola V, EPMA. General report & recommendations in predictive, preventive and personalised medicine 2012: white paper of the European Association for Predictive, Preventive and Personalised Medicine. EPMA J. 2012;3(1):14.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Andreas Flammer, MD, FESC, has received speaker honoraria and travel compensation from Imedos, GmbH, Jena, Germany. Otherwise, the authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barthelmes, J., Nägele, M.P., Ludovici, V. et al. Endothelial dysfunction in cardiovascular disease and Flammer syndrome—similarities and differences. EPMA Journal 8, 99–109 (2017). https://doi.org/10.1007/s13167-017-0099-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13167-017-0099-1